

Abstract
The biological interpretation of genetic interactions is a major challenge. Recently, Kelley and Ideker proposed a method to analyze together genetic and physical networks, which explains many of the known genetic interactions as linking different pathways in the physical network. Here, we extend this method and devise novel analytic tools for interpreting genetic interactions in a physical context. Applying these tools on a large-scale Saccharomyces cerevisiae data set, our analysis reveals 140 between-pathway models that explain 3765 genetic interactions, roughly doubling those that were previously explained. Model genes tend to have short mRNA half-lives and many phosphorylation sites, suggesting that their stringent regulation is linked to pathway redundancy. We also identify ‘pivot' proteins that have many physical interactions with both pathways in our models, and show that pivots tend to be essential and highly conserved. Our analysis of models and pivots sheds light on the organization of the cellular machinery as well as on the roles of individual proteins.
Keywords: essential genes, genetic interactions, pathway analysis, protein interactions, S. cerevisiae
Introduction
Gene knockout studies have shown that only ∼18% of Saccharomyces cerevisiae genes are essential for growth on a rich medium (Giaever et al, 2002). Consequently, buffering on the genetic level is believed to be abundant in eukaryotes (Hartman et al, 2001). To better understand the role of nonessential genes, several large-scale studies performed double knockouts (Pan et al, 2004; Tong et al, 2004) and identified many events of synthetic lethality, where a mutant carrying deletions of two nonessential genes is lethal, and synthetic sickness, where the mutant shows a weaker phenotype. We will use here the term genetic interaction (GI) for the interaction of two genetic perturbations in affecting the phenotype, whether lethal or sick. The graph that has genes as nodes and edges corresponding to GIs is called the GI network.
Recent technologies also enable a systematic mapping of protein–protein (Ito et al, 2001) and protein–DNA (Lee et al, 2002) interactions (physical interactions (PIs)), yielding large PI networks. As the networks get larger, the need for computational tools for dissecting them is mounting. The integrated analysis of PI and GI networks is a compelling challenge, as they carry important and complementary biological signals. Initial studies have shown that proteins in the same region of the GI network are slightly more likely to interact physically (Tong et al, 2001, 2004), and that a protein with many PIs is likely to have also many GIs (Ozier et al, 2003).
The modular nature of the cellular organization has been widely recognized (Hartman et al, 2001). Many methods have been developed for detecting functional modules within PI networks. Such modules, often termed pathways, represent physically interacting proteins involved in carrying out a particular function. Depending on the detection method, pathways may represent molecular complexes (Bader and Hogue, 2003) or signaling cascades (Rives and Galitski, 2003). Kelley and Ideker (2005) defined a pathway as a group of proteins that are densely interconnected in the PI network, and studied the frequency of GIs within and between such pathways. In a systematic analysis of large-scale GI and PI data, they concluded that between-pathway explanations of GIs are ∼3.5 times more abundant than within-pathway explanations, and concluded that GIs mostly bridge redundant processes. Further arguments for the prevalence of between-pathway GIs were given by Ye et al (2005), who postulated that genes in the same pathway are expected to share common GI partners, and used similarity of GI patterns in a successful function prediction. Similar results were established recently on the DNA damage system (Pan et al, 2006).
Results and discussion
Assembly of GI and PI networks
We assembled a GI network (Figure 1A) by taking from the BioGRID database version 2.0.19 (Stark et al, 2006) 13 632 synthetic lethality and synthetic fitness interactions for S. cerevisiae, covering 2682 genes. By focusing on genes with at least two interactions, we obtained a GI network of 1869 genes and 12 850 interactions. Our PI network, consisting of protein–protein and protein–DNA interactions from multiple sources (Supplementary information 1), contained 68 172 interactions covering 6184 proteins.
Figure 1.

Study outline and methodology. (A) Overview of the analysis methods and the reported results. (B) A BPM constructed from two dense pathways in the PI network. (C) A BPM constructed from two connected pathways in the PI network. (D, E) Examples of two biological explanations for pivot proteins. In (D), the pivots correspond to shared members of two linear pathways, where the signal flow is indicated by the arrow directions, and in (E), they correspond to shared complex members. Note that the pivots in (E) are not redundant, as they are densely connected to both pathways with physical interactions and do not have a genetic interaction between them.
Pathway definitions and between-pathway models
Our starting point was the computational framework of Kelley and Ideker (2005) for detection of between-pathway interpretations for GIs. Kelley and Ideker define a ‘pathway' as a densely connected set of proteins in the PI network, and a ‘between-pathway model' as a disjoint pair of pathways that are densely interconnected in the GI network (Figure 1B). Models are defined probabilistically and are found using a greedy algorithm. While the requirement of high PI density is appropriate for complexes, many other known biological pathways (e.g. linear signaling cascades) do not induce dense subnetworks in the physical network. We therefore chose to employ an alternative definition, in which a pathway is simply a connected subnetwork in the PI network (a connected pathway, described in Materials and methods). An example of two sparse pathways is presented in Figure 2A. The buffering between the mechanism of DNA repair through homologous recombination and the response to oxidative stress is indeed only partially recovered when using the dense pathway definition (not shown). We define a between-pathway model (BPM) as in Kelley and Ideker (2005), but using the new notion of a pathway (Figure 1C). The scoring of models and the model detection algorithm are defined in Materials and methods. A comparison of our models with those found using dense pathways on the same interaction data (Supplementary information A) shows that we construct more between-pathway explanations of GIs (3765 versus 3117), while maintaining the significant functional content of models. Our models also allow more direct interpretation of specific buffering cases than congruence methods (Supplementary information B).
Figure 2.

Model examples. Rectangles represent genes, and the two pathways are shown in different colors. The blue ovals are the pivot nodes. Essentials genes are drawn with thicker border. See main text for discussion of each of the BPMs (A–E). In (A), for clarity, 52 genetic interactions between the pathways are not shown.
A comprehensive model map in S. cerevisiae
Our BPM finding approach generated 140 models and provided between-pathway explanations for 3765 GIs, a 2.7-fold increase from the 1377 interactions explained in Kelley and Ideker (2005). This is mainly due to the incorporation of more GIs (12 850 instead of 4125) and to a lesser extent due to using more PIs (68 172 versus 27 604), as we use those only to create ‘scaffolds' of pathways and not for scoring models. The gene content of the models is available in a supplementary archive. A full description and an interactive visualization of the models are available at http://acgt.cs.tau.ac.il/bpm.
Functional enrichment of models
We utilized the TANGO algorithm (Shamir et al, 2005) in order to test the functional enrichment of the models in GO categories (Ashburner et al, 2000). A total of 71.4 and 69.3% of the BPMs were enriched with at least one functional category from the ‘biological process' and the ‘cellular compartment' ontologies, respectively (Supplementary Table S1). Of the complexes annotated in SGD GO-slim (Cherry et al, 1998), 46.3% were enriched in at least one BPM. Despite the low coverage of the GI network, the coverage of complexes is comparable to that achievable by direct analysis of the PI network (Supplementary information C).
Phenotypic coherence within and between pathways
To what extent do the two pathways in a BPM have the same function? To answer this question, we used the phenotypic contribution of non-essential genes in S. cerevisiae, as measured by the fitness of deletion mutants in diverse treatments (Brown et al, 2006). We found that a BPM pathway is significantly more coherent in its phenotypic response pattern than random connected groups of the same size in the physical network (Pearson correlation of 0.384 versus an average of 0.0382, P<0.001; Supplementary Figure S2). The correlation between the pathways in a BPM is also higher than expected (0.112 versus 0.0378 expected, P<0.001; Supplementary Figure S2).
Identification of pivot proteins
As we established that the pathways within models frequently represent coherent functional units, and as functional units within the cell sometimes share components (Krause et al, 2004), we tested the possibility of computationally detecting such shared components within the PI network. To this end, for each model, we sought proteins that are densely connected to both of its pathways (Figure 1D and E, Materials and methods), and called them the pivot proteins for the model. Altogether, we identified 124 distinct pivots in 40 models. On average, 1.09 pivots were found in each model, and each pivot appeared in 1.22 models.
We systematically analyzed the representation of proteins that are known to take part in several distinct processes in the group of pivots. To this end, we identified proteins participating in several complexes or pathways (see Materials and methods), and also used a curated set of multicomplexed genes (Krause et al, 2004). As summarized in Table I, the pivots were enriched in all three sets. One example of such overlap is in BPM 96 (Figure 2B). In a model containing as pathways parts of the SWR1 and Ino80 complexes involved in chromatin remodeling, we identified the pivot proteins Arp4, Rvb1 and Rvb2, three out of the four proteins known to participate in both the SWR1 and the Ino80 complexes (Shen et al, 2000; Krogan et al, 2003). In BPM 97 (Figure 2C), Sus1, which has been shown to take part both in the nuclear pore and the SAGA complex (Rodriguez-Navarro et al, 2004) was identified as a pivot in a model representing GIs between the two pathways. When pivots do not correspond to known complex or pathway overlaps, they frequently represent general purpose genes cooperating with multiple pathways. For example, in BPM 87, the general transcription factor Spt15 was identified as a pivot of a model that contains components of the distinct transcription-related complexes RSC, SWR1 and SAGA (Figure 2D).
Table 1. Multiple roles of pivot proteins.
| No. of proteins | Pivots | Expected | Significance | |
|---|---|---|---|---|
| GO complexes | 206 | 21 | 4.35 | P=1.27 × 10−9 |
| KEGG pathways | 390 | 11 | 8.24 | P=0.200 |
| Filtered KEGG pathways | 71 | 6 | 1.50 | P=3.68 × 10−3 |
| Known complex overlaps | 39 | 8 | 0.55 | P=7.49 × 10−9 |
The enrichment of proteins known to participate in multiple physical pathways within the set of pivot proteins. GO complexes are taken from SGD ‘macromolecular complex GO-slim' ontology. The filtered KEGG pathways are KEGG pathways in which at least 50% genes formed a connected component in our physical network. Known complex overlaps are taken from Krause et al (2004). Significance was evaluated using hypergeometric distribution.
Essentiality and evolutionary retention of pivot proteins
The two pathways that form a model are often partially redundant in function, and as the pivots represent proteins that are active in both pathways, we hypothesized that the pivots will frequently correspond to essential genes. Indeed, 72 of the pivots were found to be essential, a highly significant fraction given the total number of essential genes in the network (22.6 essentials expected, P=1.42 × 10−23). Although we observed a general strong correlation between degree and essentiality (P=5.87 × 10−85 using rank-sum test), as previously reported (Jeong et al, 2001), the high prevalence of essential genes among pivots is far beyond what can be explained by their degrees alone (39.98 essentials expected, P<10−5). The essential pivots also tend to have closer functions to their BPMs (Supplementary information D). Essentiality was recently shown to be connected to the evolutionary retention of genes in eukaryotes (Gustafson et al, 2006). Using the data from Gustafson et al (2006) we found that the pivots are significantly retained in evolution (P=9.79 × 10−9), even when controlling for the large fraction of essential genes (P=0.029).
Example: the spindle checkpoint model
An interesting example of using models and pivots for understanding cellular mechanisms is BPM 66 (Figure 2E). This model represents buffering between different components of the kinetochore, a complex bound to the centromeres of chromosomes during mitosis. Together with its pivots, the model is composed of members of three known subcomplexes of the inner kinetochore: Ndc80, COMA and MIND (De Wulf et al, 2003). Specifically, the pivots Mtw1 and Dsn1 correspond to a distinct unit of the MIND complex, which bridges different kinetochore subcomplexes (De Wulf et al, 2003; Westermann et al, 2003). The two subunits of the Ndc80 complex, Nuf2/Spc24 and Tid3/Spc25, which were shown to be at least partially redundant (McCleland et al, 2003), are placed in different pathways. Note that even though the four subunits of the Ndc80 complex show all possible GIs and PIs between pairs, the biologically correct partition of this complex into pathways was obtained by taking into consideration the other GIs in the model. For example, Mad1 physically connects only with the Tid3/Spc25 subunit and genetically interacts only with the Nuf2/Spc24 subunit. These additional external interactions cause the biologically correct partition to score higher than other alternatives.
Mad1, a highly conserved protein with a specific function in the spindle cell cycle checkpoint, is of additional interest. At the spindle checkpoint, the cells are arrested in metaphase until all chromosomes successfully attach to microtubules. Tid3 and Spc25, members of the Ndc80 complex, which appear in the pathway with Mad1, were specifically linked to the spindle checkpoint in several organisms (summarized in Bharadwaj et al, 2004). Moreover, the recruitment of Mad1 was shown to be dependent on Spc25 and Tid3 in Xenopus and human cells (McCleland et al, 2003; Bharadwaj et al, 2004) and the spindle checkpoint was shown to be defective in spc25 mutants (Wigge and Kilmartin, 2001). How exactly does Mad1 attach to the kinetochore is currently not known. Although Mad1 shows a yeast two-hybrid interaction with Spc25 in S. cerevisiae (Newman et al, 2000) and with Tid3 in human cells (Martin-Lluesma et al, 2002), attempts to demonstrate a biochemical interaction between Ndc80 and Mad1 have been reported unsuccessful (Martin-Lluesma et al, 2002; McCleland et al, 2003).
In our model, Mad1 is linked to the pivot Smc1, a member of the cohesin complex, required for sister chromatid cohesion during mitosis. Smc1 was shown to localize to the kinetochore during meiosis and to interact with Tid3 in yeast and human cells (Zheng et al, 1999; Gregson et al, 2002). Furthermore, Smc1 was shown to be required for proper assembly of the mitotic spindle in human cells (Gregson et al, 2001), but its exact function in the metaphase is unknown. Our findings suggest that the connection of Mad1 to the kinetochore in general and to the Ndc80 complex in particular, is mediated through Smc1. Note how the use of pivots provides additional clues to BPM annotation and to the understanding of inter-pathway organization.
Physiological properties of models
Recently, the physiological properties of the PI network hubs were extensively analyzed (Batada et al, 2006). As many proteins in our models were such hubs, we asked whether their physiological properties differed from those of other hubs. Here, we focus on the analysis of mRNA stability (Wang et al, 2002) and the number of putative phosphorylation sites (Obenauer et al, 2003), two properties manifesting the turnover and regulation of the gene. Detailed analysis, covering additional properties, is described in Supplementary information E and Supplementary Table S2.
Our tests show that BPM genes behave significantly unlike other genes. The average mRNA half-life of BPM genes was 21.4, versus 26.2 in all other genes (P=9.97 × 10−10, rank-sum test). The average number of phosphorylation sites was 5.1, versus 4.0 for all other genes (P=3.13 × 10−9). Both parameters were significantly correlated between the two pathways that constitute a BPM (intraclass correlations of 0.408 and 0.189, P<10−4 and P=0.0184, respectively). This finding cannot be explained by the high degrees of model genes (Supplementary Table S2).
Note that the mRNA half-lives are experimentally derived, whereas the phosphorylation sites are computationally inferred from sequence. On all the 3552 genes for which both parameters are available, they are not correlated (ρ=−0.0182 P=0.276). However, the parameters are significantly correlated on the genes within BPMs (566 genes, ρ=−0.132, P=1.6 × 10−3). These results remain highly significant when controlling for key functions enriched in the model participants (Supplementary Figure S4). These findings suggest that genes in BPMs might experience more stringent regulation. A possible hypothesis is that in pathways for which redundant mechanisms are available, a tighter regulation allows the cell to switch between the alternatives faster. However, this conclusion has to be revalidated when GIs covering a wider functional range become available.
Our results indicate that despite the limitations of today's GI and PI networks, their integrated analysis is a powerful approach for understanding the organization of the yeast cellular system. We expect that such analysis will provide insights into the large picture of genetic redundancy in higher eukaryotes as well.
Materials and methods
Scoring models
We build upon the probabilistic score described in Kelley and Ideker (2005) and Sharan et al (2005) to identify between-pathway explanations for GIs by finding BPMs within the GI and the PI networks. Let GG=(V, EG) be the GI network and let GP=(V, EP) be the PI network. Note that nodes in V represent both the genes and their products, depending on the context. A BPM is a pair of disjoint sets V1, V2, such that (a) ∣V1∣, ∣V2∣⩾2, (b) each Vi induces a connected subgraph in GP and (c) there are unusually many GIs between V1 and V2 (Figure 1C). We call each Vi a pathway. Property (c) reflects the assumption that genetic buffering implies a dense set of GIs between the pathways.
We now quantify property (c). We derive a log-odds score reflecting the likelihood that the density of GIs between the two pathways is unusually high. We compare two hypotheses: under the BPM hypothesis, every pair of genes, one from V1 and the other from V2, genetically interact with a high probability β, independently of all other gene pairs, and the likelihood of a model (V1, V2) is thus Π(a, b)∈(V1 × V2)βI(a, b)+(1−β)(1−I(a, b)), where I(a, b) equals 1 if there exists a GI between a and b and otherwise equals 0; in the null hypothesis, every pair (a, b) is connected with probability ra, b, representing the chance of observing this interaction at random, given the degrees of a and b in GG. We estimate ra, b by generating a random ensemble of networks with the same degree sequence and counting what fraction of them contain an interaction between a and b. The log-odds score is then
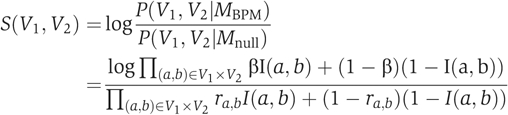 |
The main difference between this score and the score described in Kelley and Ideker (2005) is in the structure imposed by the BPMs in the PI network. Here we do not score the model for density of PIs within each pathway, and instead require that each is connected in GP.
Model finding algorithm
The model finding procedure described in Kelley and Ideker (2005) is a greedy network search algorithm that uses as seeds single GIs. We improve this procedure by initializing the algorithm with better seeds that are maximal bicliques in the GI network (a biclique is a disjoint pair of node sets such that every node in each set has edges to all nodes in the other set). The following procedure is performed for each u, v such that (u, v)∈EP
Identify B—the set of nodes adjacent to both u and v in GG. Proceed only if ∣B∣⩾kmin.
Partition B into connected components in GP: B1, B2, …, Bl.
For each Bi such that ∣Bi∣⩾kmin, identify the set Ai of nodes adjacent to all the nodes in Bi in GG.
For each Ai, partition it into the connected components it induces in GP, Ai1, Ai2, …, Aim and add (Bi, Aij) to the set of seeds if ∣Aij∣⩾kmin.
This algorithm produces maximal bicliques (V1, V2) in GG, such that each Vi induces a connected component in GP and has size ⩾kmin. The produced set of seeds is then filtered for overlaps. We used kmin=2 in all tests. Owing to the relatively sparse nature of both interaction networks, this method is very efficient in practice.
The optimization phase starts with each seed as a candidate model, and greedily tries to improve the score of the current model through addition, removal or exchange of nodes between the two pathways while keeping each pathway connected in the PI network. In order to efficiently keep track of the connectivity requirement, we use the notion of articulation nodes. An articulation node in a connected graph is a node whose removal disconnects the graph. Articulation nodes can be efficiently detected during a depth-first search traversal of the graph, by calculating the ‘lowpoint' values of every node (cf. Even, 1979). The algorithm maintains the connectivity of each pathway by dynamically updating, for each pathway, its set of articulation nodes. This set is used to ban optimization moves which disrupt connectivity. After the optimization, a filtering step removes models that overlap by >50% in both pathways to higher scoring models. An additional model filtering step computes an empiric P-value by sampling 1000 random gene groups of the same size, and retains only BPMs with P<0.05 (see Supplementary information F).
Identification of pivot proteins
Given a model (V1, V2), we seek nodes that are densely connected to both pathways in the physical network. Specifically, for every v∈V, denote by Ni(v) the nodes in Vi that are adjacent to v in GP. We call v a pivot if, for i=1, 2, ∣Ni(v)∣⩾lmin and ∣Ni(v)∣ is significant, given the degree of v in GP (P<Pmax, using hypergeometric test). In the actual analysis described in this paper, we used lmin=2 and Pmax=0.05. In addition, to filter master regulator genes that are involved in many processes, such as protein folding chaperons, we only considered as pivots proteins with degree <250 in GP.
Statistical analysis
Correlation analysis was performed using the non-parametric Spearman test, unless otherwise indicated. All P-values reported when controlling for a specific gene class (e.g. essential genes) were obtained by random sampling of a large number of gene groups with the same fraction of genes from that class. The P-values reported when controlling for the degrees were calculated by first binning all the genes into 40 equal-size bins based on their degree, and then sampling genes from the bins, while maintaining the proportion of genes from each bin.
Supplementary Material
Supplementary Dataset
Supplementary Figures and Tables
Acknowledgments
We thank the members of the Shamir laboratory for helpful discussions. IU is a fellow of the Edmond J Safra Bioinformatics Program at Tel-Aviv University. RS was supported in part by the Wolfson foundation, and by the EMI-CD project that is funded by the European Commission within its FP6 Programme, under the thematic area ‘Life Sciences, genomics and biotechnology for health', contract number LSHG-CT-2003-503269.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Hogue CW (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batada NN, Hurst LD, Tyers M, Russell R (2006) Evolutionary and physiological importance of hub proteins. PLoS Comput Biol 2: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R, Qi W, Yu H (2004) Identification of two novel components of the human NDC80 kinetochore complex. J Biol Chem 279: 13076–13085 [DOI] [PubMed] [Google Scholar]
- Brown JA, Sherlock G, Myers CL, Burrows NM, Deng C, Wu HI, McCann KE, Troyanskaya OG, Brown JM (2006) Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol 2: 2006.0001 16738548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, Weng S, Botstein D (1998) SGD: Saccharomyces genome database. Nucleic Acids Res 26: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wulf P, McAinsh AD, Sorger PK (2003) Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev 17: 2902–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even S (1979) Graph Algorithms. Potomac, MD: Computer Science Press [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391 [DOI] [PubMed] [Google Scholar]
- Gregson HC, Schmiesing JA, Kim JS, Kobayashi T, Zhou S, Yokomori K (2001) A potential role for human cohesin in mitotic spindle aster assembly. J Biol Chem 276: 47575–47582 [DOI] [PubMed] [Google Scholar]
- Gregson HC, Van Hooser AA, Ball AR Jr, Brinkley BR, Yokomori K (2002) Localization of human SMC1 protein at kinetochores. Chromosome Res 10: 267–277 [DOI] [PubMed] [Google Scholar]
- Gustafson AM, Snitkin ES, Parker SC, DeLisi C, Kasif S (2006) Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genomics 7: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman JLt, Garvik B, Hartwell L (2001) Principles for the buffering of genetic variation. Science 291: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Yoshida M (2001) Exploring the protein interactome using comprehensive two-hybrid projects. Trends Biotechnol 19: S23–S27 [DOI] [PubMed] [Google Scholar]
- Jeong H, Mason SP, Barabasi AL, Oltvai ZN (2001) Lethality and centrality in protein networks. Nature 411: 41–42 [DOI] [PubMed] [Google Scholar]
- Kelley R, Ideker T (2005) Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol 23: 561–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause R, von Mering C, Bork P, Dandekar T (2004) Shared components of protein complexes—versatile building blocks or biochemical artefacts? BioEssays 26: 1333–1343 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H, Haw RA, Pootoolal J, Tong A, Canadien V, Richards DP, Wu X, Emili A, Hughes TR, Buratowski S, Greenblatt JF (2003) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12: 1565–1576 [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, Zeitlinger J, Jennings EG, Murray HL, Gordon DB, Ren B, Wyrick JJ, Tagne JB, Volkert TL, Fraenkel E, Gifford DK, Young RA (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804 [DOI] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297: 2267–2270 [DOI] [PubMed] [Google Scholar]
- McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT (2003) The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev 17: 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JR, Wolf E, Kim PS (2000) A computationally directed screen identifying interacting coiled coils from Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97: 13203–13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31: 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozier O, Amin N, Ideker T (2003) Global architecture of genetic interactions on the protein network. Nat Biotechnol 21: 490–491 [DOI] [PubMed] [Google Scholar]
- Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD (2006) A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell 124: 1069–1081 [DOI] [PubMed] [Google Scholar]
- Pan X, Yuan DS, Xiang D, Wang X, Sookhai-Mahadeo S, Bader JS, Hieter P, Spencer F, Boeke JD (2004) A robust toolkit for functional profiling of the yeast genome. Mol Cell 16: 487–496 [DOI] [PubMed] [Google Scholar]
- Rives AW, Galitski T (2003) Modular organization of cellular networks. Proc Natl Acad Sci USA 100: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Navarro S, Fischer T, Luo MJ, Antunez O, Brettschneider S, Lechner J, Perez-Ortin JE, Reed R, Hurt E (2004) Sus1, a functional component of the SAGA histone acetylase complex and the nuclear pore-associated mRNA export machinery. Cell 116: 75–86 [DOI] [PubMed] [Google Scholar]
- Shamir R, Maron-Katz A, Tanay A, Linhart C, Steinfeld I, Sharan R, Shiloh Y, Elkon R (2005) EXPANDER—an integrative program suite for microarray data analysis. BMC Bioinformatics 6: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ideker T, Kelley B, Shamir R, Karp RM (2005) Identification of protein complexes by comparative analysis of yeast and bacterial protein interaction data. J Comput Biol 12: 835–846 [DOI] [PubMed] [Google Scholar]
- Shen X, Mizuguchi G, Hamiche A, Wu C (2000) A chromatin remodelling complex involved in transcription and DNA processing. Nature 406: 541–544 [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535–D539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368 [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C (2004) Global mapping of the yeast genetic interaction network. Science 303: 808–813 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO (2002) Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA 99: 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Cheeseman IM, Anderson S, Yates JR 3rd, Drubin DG, Barnes G (2003) Architecture of the budding yeast kinetochore reveals a conserved molecular core. J Cell Biol 163: 215–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigge PA, Kilmartin JV (2001) The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol 152: 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Peyser BD, Pan X, Boeke JD, Spencer FA, Bader JS (2005) Gene function prediction from congruent synthetic lethal interactions in yeast. Mol Syst Biol 1: 2005.0026 16729061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Chen Y, Lee WH (1999) Hec1p, an evolutionarily conserved coiled-coil protein, modulates chromosome segregation through interaction with SMC proteins. Mol Cell Biol 19: 5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Dataset
Supplementary Figures and Tables