

Abstract
Innate immunity is critical for clearing Pseudomonas aeruginosa from the lungs. In response to P. aeruginosa infection, a central transcriptional regulator of innate immunity—NF-κB—is translocated within 15 min to the nuclei of respiratory epithelial cells expressing wild-type (WT) cystic fibrosis (CF) transmembrane conductance regulator (CFTR). P. aeruginosa clearance from lungs is impaired in CF, and rapid NF-κB nuclear translocation is defective in cells with mutant or missing CFTR. We used WT and mutant P. aeruginosa and strains of transgenic mice lacking molecules involved in innate immunity to identify additional mediators required for P. aeruginosa-induced rapid NF-κB nuclear translocation in lung epithelia. We found neither Toll-like receptor 2 (TLR2) nor TLR4 nor TLR5 were required for this response. However, both MyD88-deficient mice and interleukin-1 receptor (IL-1R)-deficient mice failed to rapidly translocate NF-κB to the nuclei of respiratory epithelial cells in response to P. aeruginosa. Cultured human bronchial epithelial cells rapidly released IL-1β in response to P. aeruginosa; this process was maximized by expression of WT-CFTR and dramatically muted in cells with ΔF508-CFTR. The IL-1R antagonist blocked P. aeruginosa-induced NF-κB nuclear translocation. Oral inoculation via drinking water of IL-1R knockout mice resulted in higher rates of lung colonization and elevated P. aeruginosa-specific antibody titers in a manner analogous to that of CFTR-deficient mice. Overall, rapid IL-1 release and signaling through IL-1R represent key steps in the innate immune response to P. aeruginosa infection, and this process is deficient in cells lacking functional CFTR.
One component of the hypersusceptibility of cystic fibrosis (CF) patients to chronic Pseudomonas aeruginosa infection is due to the lack of functional CF transmembrane conductance regulator (CFTR) protein in the cell. In individuals with wild-type CFTR (WT-CFTR), binding of the first extracellular domain of this protein to the outer core of the lipopolysaccharide (LPS) of P. aeruginosa (36, 57) occurs rapidly following lung infection and leads to high levels of resistance to infection. We and others have shown that this interaction between WT-CFTR and P. aeruginosa leads to internalization of the bacterium by respiratory epithelial cells via lipid raft formation (25, 56), induction of nuclear translocation of NF-κB (42), activation of Src-like tyrosine kinases p59Fyn and p60Src followed by tyrosine phosphorylation of additional epithelial cell proteins (12), transcriptional activation of genes for interleukin-6 (IL-6), IL-8, CXCL1, and intercellular adhesion molecule 1 (40), and rapid apoptosis to resolve the infection (8). Multiple groups have additionally shown that binding of Salmonella enterica serovar Typhi pili and/or LPS to CFTR also mediates intestinal epithelial cell uptake of this organism (21, 28, 35, 50). Overall, the lack of functional CFTR has been shown to affect a number of responses of the lung to P. aeruginosa, which appears to lead to a dampened early innate immune response allowing the organisms to eventually establish a chronic infection (34).
In the presence of WT-CFTR, a central mediator of the lung epithelial cell response to P. aeruginosa infection is the transcription factor NF-κB, which is translocated to the nuclei of respiratory epithelial cells within 15 min, whereas cells with mutant CFTR alleles such as the ΔF508-CFTR allele, or cells completely lacking CFTR, fail to make this rapid response (42). NF-κB regulates the transcription of genes for numerous inflammatory factors, including IL-6, IL-8, and CXCL1. The production of these three cytokines is reduced during the earliest stages of infection in cells with mutant CFTR (40) but increased following prolonged infection of either cultured transformed cells or primary epithelial cells isolated from CF lungs (10, 23). Both IL-8 and CXCL1 are neutrophil chemoattractants (16, 22, 41), indicating that neutrophil recruitment may be impaired in the early stages of P. aeruginosa infection in CF patients, but such recruitment is clearly extensive during chronic infection of CF patients (10, 24, 45). A reduced inflammatory response at an early stage of infection may lead to the establishment of chronic colonization by P. aeruginosa, which could subsequently overwhelm the CF patient's ability to clear the pathogen effectively.
In addition to CFTR-dependent responses, other bacteria-host interactions have been shown to be involved in airway signaling during P. aeruginosa infection. Several reports indicate that binding of P. aeruginosa flagellin to Toll-like receptor 5 (TLR5) induces NF-κB-dependent epithelial cell responses (1, 30, 58), and TLR2 has also been reported to mediate NF-κB nuclear translocation in airway epithelial cells in response to P. aeruginosa (48, 58). However, none of these studies has measured responses in cells with mutant CFTR, and all of the measured NF-κB-dependent responses occur hours after infection as opposed to the 15-min time point postinfection which, we have reported, leads to maximal NF-κB nuclear translocation in both cultured epithelial cells and lungs with WT, but not mutant, CFTR (42). P. aeruginosa flagella have been reported to activate respiratory epithelial cell responses via TLR2 and TLR5 (1), and P. aeruginosa LPS has been reported to activate respiratory epithelial cells via TLR4 (19, 29). Yet neither TLR2 nor TLR4 nor TLR5 single-knockout mice have enhanced resistance or susceptibility to P. aeruginosa lung infection (14, 17, 39, 47), just changes in cytokine responses, indicating that cellular activation mediated individually by these TLRs is not a major factor in the acute host response to this pathogen. Various combined deficiencies in two TLRs can result in some alterations in the inflammatory phenotype but do not seem to impact the overall host resistance to P. aeruginosa infection in a significant way (14, 39, 47). Moreover, a recent study shows that, following recognition of bacterial pathogen-associated molecules that bind to TLRs, dendritic cell activation occurs not on the cell surface but inside phagosomes following endocytosis or phagocytosis of bacteria (5). This finding indicates that, even when TLRs are involved in the cellular responses to P. aeruginosa, the CFTR-dependent bacterial internalization that we and others (12, 36) have shown to occur in airway epithelial cells could potentially be a key factor in activating these cells via TLR signaling in response to infection with this pathogen.
In view of the important role of NF-κB in the early innate immune response to P. aeruginosa and the potential role of delayed NF-κB activation in the initiation of the early phases of infection in CF patients, we investigated the pathways that are involved in CFTR-dependent or CFTR-modulated activation of NF-κB within the first few minutes of P. aeruginosa infection. We identified the IL-1 receptor (IL-1R), which transduces its response via MyD88, as an important component of this system by testing for NF-κB translocation in the lung epithelia of several strains of knockout mice in response to P. aeruginosa. We found that rapid release of IL-1β in response to P. aeruginosa was enhanced in the presence of functional CFTR in respiratory epithelial cells, although levels of preformed IL-1β were comparable for cells with WT and mutant CFTR genes. We also found that IL-1R knockout mice were susceptible to chronic P. aeruginosa lung infection, as are CF mice, following oral exposure to the pathogen in drinking water. These results suggest that IL-1β release and signaling through IL-1R and MyD88 play important roles in the response to P. aeruginosa in respiratory epithelial cells and that this process is deficient in the setting of nonfunctional CFTR.
MATERIALS AND METHODS
Bacterial strains.
PAO1 and N6 are LPS-smooth, nonmucoid strains of P. aeruginosa. PAO1 was originally obtained from M. Vasil (University of Colorado, Denver, CO). N6, a clinical isolate from a CF patient early in the course of infection, was provided by J. Burns (University of Washington, Seattle, WA). PAO1 fliC::gent, a nonmotile mutant unable to produce the major flagellin protein that binds to TLR5, was provided by R. Ramphal (University of Florida, Gainesville, FL) (15).
Cell lines.
The construction and growth of CFT1 cell lines used in this study have been described previously (37). CFT1-ΔF508 (ΔF508-CFTR) and CFT1-LCFSN (WT-CFTR) cells are both human bronchial epithelial cell lines with an identical starting genetic background containing two ΔF508 alleles of CFTR. The ΔF508-CFTR (CFT1-ΔF508) cells have a third allele of ΔF508-CFTR. The WT-CFTR (CFT1-LCFSN) cells have a WT allele for human CFTR expressed in the ΔF508 background (37).
Mouse strains.
IL-1R knockout mice of strain B6;129S-IL1r1<tm1Rom1> and controls of strain B6.129SF2/J were obtained from The Jackson Laboratory. TLR2 knockout mice and MyD88 knockout mice were kindly provided by M. Freeman (Massachusetts General Hospital, Harvard Medical School, Boston, MA) (20, 49). C3H/HeJ (TLR4 dominant-negative) and C57BL/6 mice were obtained from The Jackson Laboratory, as were tumor necrosis factor receptor 1 (TNF-R1) knockout mice. All animal protocols were approved by the Harvard Medical Area Institutional Animal Care and Use Committee.
Acute infection of mice.
Mice were infected by intranasal application of P. aeruginosa as described previously (2, 43). For determination of NF-κB nuclear translocation, mice were killed by CO2 asphyxiation at various time points after infection, lungs were inflated by instillation of 4% paraformaldehyde after visualization of the trachea, and the tissues were then removed and processed and stained as described for identification of CFTR, NF-κB, nucleic acid, and translocation of NF-κB p65 into the nucleus (42).
Chronic infection of mice.
Chronic infection of mice was established as described previously (9, 43). We used strain N6, a clinical isolate from a CF patient early in the course of infection, because prior studies had shown that this strain, in contrast to strain PAO1, is highly efficient at chronically infecting transgenic CF mice (9). We also carried out a comparable experiment, but for a shorter time course, with strain PAO1. Briefly, P. aeruginosa was grown overnight at 37°C on tryptic soy agar and then resuspended in sterile water to an optical density at 650 nm (OD650) of 0.4 (∼2 × 109 CFU/ml) and diluted in sterile water to achieve 5 × 106 to 7 × 106 CFU/ml. IL-1R knockout mice and controls were initially treated for 5 to 7 days with oral levofloxacin and gentamicin as described previously (9) to clear any preexisting oropharyngeal Enterobacter spp. (which interfere with the detection of P. aeruginosa) or trace levels of P. aeruginosa, and then the mice were given the defined P. aeruginosa strain in their drinking water for five days. After exposure to the bacteria, mice were given sterile water containing 0.2 M sodium acetate at pH 4.0 with 0.5 mg nitrofurantoin/ml to prevent growth of the Enterobacter spp. in the drinking water and transmission of Enterobacter colonization among the mice housed in the same cage. We have maintained transgenic CF mice on such water for up to 2 years with no obvious compromise to the animals' health. Orally nonabsorbable antibiotics were also added to the water (100 μg gentamicin/ml; 100 μg ceftazidime/ml), which not only contributed to the prevention of P. aeruginosa growth in drinking water but also cleared mere oropharyngeal colonization by P. aeruginosa. This treatment is essential in order to allow us to distinguish colonization confined to the oral cavity, a nonmeaningful outcome, from true lung infection, which cannot be cleared by these antibiotics when given orally, as we have previously documented (9).
Infected mice were monitored for persistent infection by weekly or biweekly throat culture. Throat swabs were obtained using sterile calcium alginate swabs (Puritan Medical Products Company, Guilford, ME) inserted deep into the oropharynxes of mice that were anesthetized by inhalation of isoflurane gas. Throat swabs were incubated in tryptic soy broth for five hours, and then 1 mg nitrofurantoin/ml was added to eliminate any contaminating Enterobacter spp., and the culture was incubated overnight at 37°C. Cultures were then plated on Pseudomonas isolation medium and grown overnight at 37°C. Cultures yielding oxidase-positive, green-fluorescent colonies were scored as positive for the presence of P. aeruginosa.
Measurements of IgG responses to P. aeruginosa in mouse sera.
Sera were obtained from the tails of mice before exposure to P. aeruginosa and at several intervals after oral infection. Immunoglobulin G (IgG) antibody was evaluated by enzyme-linked immunosorbent assay (ELISA) using azide-killed whole bacteria to coat ELISA plates. Serial dilutions of sera were reacted with the whole cells, and absorbance readings from diluted sera drawn before experimental exposure to P. aeruginosa, although negligible, were subtracted from all postexposure measurements. Secondary antibody to mouse whole IgG molecule was used, and the final absorbance readings were taken at OD405. To calculate titers, power or exponential best-fit lines were derived for plots of absorbance (abscissa) versus serum dilution (ordinate), and titers for each mouse were calculated using an OD absorbance of 0.15 as the cutoff value; this value was the OD reading that exceeded all background levels measured in sera before the mice were experimentally exposed to P. aeruginosa.
Confocal microscopy for CFTR expression and NF-κB nuclear translocation.
Fixed lung tissues and cells were processed for immunohistochemical analysis of NF-κB nuclear translocation and CFTR expression as described previously (42). Tissue sections were first deparaffinized and then rehydrated for staining. Fixed cells and tissue sections were next incubated with rabbit antibody to human (p65) NF-κB (Santa Cruz Bio-technology, Santa Cruz, CA) for 90 min at 37°C, washed, and then incubated with Alexa Fluor 594 dye-labeled goat anti-rabbit IgG for 90 min at 37°C. After extensive washing, the nucleic acid-staining dye 4,6-diamidino-2-phenyindole dihydrochloride (DAPI) was added to visualize the nucleus. Deparaffinized, rehydrated tissue sections and fixed and permeabilized cells in dishes were stained for CFTR expression by incubation with a mouse IgM anti-CFTR monoclonal antibody, CF3 (54), for 90 min at room temperature and then counterstained with fluorescein isothiocyanate-conjugated anti-mouse IgM. Sections were visualized by confocal laser microscopy.
To visualize NF-κB entry into the nuclei of cells, confocal laser microscopic images were processed to find the locations in the images where red pixels, showing the location of NF-κB, overlapped with blue pixels, showing the location of DAPI-stained nucleic acid. All of the pixels in the micrographs where this overlap occurred were depicted as a magenta color, indicative of the colocalization of NF-κB and nucleic acid at the resolution of 1 pixel (within ∼0.8 μm).
To determine the effect of IL-1 receptor antagonist (IL-1ra) on NF-κB nuclear translocation, 10 μg IL-1ra/ml was added to cultured WT-CFTR cells 10 min prior to the addition of P. aeruginosa strain PAO1. Fifteen minutes later, IL-1ra-treated cells and untreated control cells were fixed and stained for nuclear NF-κB as described above.
Quantitative analysis of the entry of NF-κB into the nucleus following P. aeruginosa infection was carried out using a TransAm NF-κB p65 transcription factor assay kit (Active Motif, Carlsbad, CA). The manufacturer's instructions were followed for the assay, which involved exposure of WT- or ΔF508-CFTR cells to P. aeruginosa strain PAO1 (multiplicity of infection of 30:1, bacteria to cells) for 15 min in the presence or absence of IL-1ra, harvesting of cells, preparation of nuclear extracts using a nuclear extract kit (Active Motif), and addition of the extracts to wells with an immobilized oligonucleotide containing a NF-κB consensus binding site. Specificity of binding was confirmed by adding either a WT or mutated NF-κB consensus oligonucleotide to the extracts at 20 pM/ml. After the addition of primary antibody to NF-κB, cells were incubated at room temperature for 1 h followed by washing and then addition of the horseradish peroxidase-conjugated secondary antibody provided in the kit for 1 h at room temperature again followed by washing; color development was measured at 450 nm 10 min after the addition of the developing solution. Activity in each extract was measured in triplicate, and results were averaged.
IL-1β detection in cell supernatants by ELISA.
P. aeruginosa was grown on tryptic soy agar plates overnight at 37°C, resuspended in antibiotic-free cell media to an OD650 of 0.4 (∼2 × 109 CFU/ml), and then diluted 1:15 in the same medium. Cells were washed with sterile 1× phosphate-buffered saline (PBS) (1.05 mM KH2PO4, 154 mM NaCl, 2.97 mM Na2HPO4), and then antibiotic-free cell culture medium was added with or without P. aeruginosa (multiplicity of infection of 30:1 to 50:1) as described previously (40). Supernatants were harvested at set time points, sterile filtered with low-protein-binding, 0.2-μm cutoff syringe filters (Pall Corporation, East Hills, NY), and dialyzed in 3,500-Da MWCO dialysis cassettes (Pierce Biotechnology, Inc., Rockford, IL) against 0.04× PBS (0.042 mM KH2PO4, 6.16 mM NaCl, 0.119 mM Na2HPO4). Supernatants were then frozen, lyophilized, and resuspended in water in a volume 1/25th that of the original supernatant solution. Total protein concentrations were determined by a Bradford assay. IL-1β was detected in concentrated supernatants by using a human IL-1β Quantikine immunoassay (R&D Systems, Minneapolis, MN). The manufacturer's instructions for testing cell culture supernatants were followed. Results are expressed as pg of IL-1 detected per mg of total cellular protein to normalize for differences in cell numbers when comparing cultures from cells of different CFTR genotypes grown in individual flasks.
Detection of preformed IL-1 in uninfected cell lysates.
Lysates were made as described previously (40). Briefly, uninfected cells were washed once with 1× PBS and lysed using lysis buffer (20 mM Tris, 1 mM EDTA, and 0.05% Triton X-100, pH 7.4 chilled) containing Complete Mini protease inhibitor cocktail (Roche Diagnostics Corp., Indianapolis, IN). The protein concentration in each sample was measured by the Bradford assay. IL-1 levels were measured by using a human IL-1β Quantikine immunoassay (R&D Systems).
Statistics.
Unpaired, two-tailed t tests were used to make two-group comparisons. Multigroup comparisons were analyzed by analysis of variance followed by Tukey's multiple comparisons test for determining significant differences between pairs of observations. The marginal probability of oropharyngeal colonization of mice chronically infected with P. aeruginosa was estimated as previously described (9) by solving generalized estimating equations (27) using R software for statistical computing (www.r-project.org). Slopes and confidence intervals of curves generated by ELISA were determined using Statview 5 software.
RESULTS
NF-κB nuclear translocation in lungs of P. aeruginosa-infected mice.
We first investigated whether MyD88 might be involved in activation and nuclear translocation of NF-κB in response to infection by P. aeruginosa, by analyzing the kinetics of NF-κB translocation in lung epithelial cells of MyD88 knockout mice. MyD88 is involved in signal transduction from cell surface receptors such as TLRs, TNF-R, and IL-1R. In Fig. 1, we stained for CFTR (green color) to ensure that the mice had apparently normal levels of production of this receptor for P. aeruginosa, as well as NF-κB (red color) and nucleic acid (blue color). Depicted in the first row of Fig. 1 is the overlap of CFTR and NF-κB in the cytoplasm, which appeared as a yellow to red-orange color, the intensity of which varied due to some differences in the levels of these two proteins in the cytoplasm of individual lung epithelial cells. Areas within the cells where one or the other protein was more concentrated appeared more green or more red. The second row of figures shows only NF-κB and nuclear staining. In the third and fourth rows of Fig. 1, the translocation of NF-κB was depicted in pseudocolored images as a magenta color in the nucleus, based on colocalization (within 0.8 μm) of pixels produced from fluorescence of the Alexa Fluor 594 and DAPI molecules, indicative of NF-κB entry into the nucleus.
FIG. 1.

NF-κB nuclear translocation in sections of small airways from either WT or MyD88 knockout C57BL/6 mice determined at the indicated times after intratracheal infection with P. aeruginosa strain PAO1. Sections were stained for NF-κB (red), CFTR (green), and nuclear DNA (blue). First row: yellow/orange color represents overlap between cytoplasmic CFTR and cytoplasmic NF-κB. Second row: cytoplasmic NF-κB (red) and nuclear DNA (blue) only. Third and fourth rows: colocalization of translocated nuclear NF-κB (red) and nuclear DNA (blue), represented as pseudocolored magenta at two different magnifications. Magnifications are indicated on the right-hand side of each row.
Overall, the images in Fig. 1 show that the lung epithelial cells of WT C57BL/6 mice readily translocated NF-κB to the nucleus within 15 min of infection, whereas at all time points examined, there was no evidence of NF-κB nuclear translocation in the lung epithelia of MyD88-deficient mice. Uninfected WT mice did not have any detectable NF-κB in the nuclei of lung epithelial cells (Fig. 1), nor did uninfected MyD88-deficient mice (not shown). In addition to the time points shown in Fig. 1, sections were examined at intermediary time points 30, 60, and 90 min postinfection, with similar negative results for NF-κB nuclear translocation by Myd88-deficient mice (data not shown).
We next hypothesized that signaling in response to P. aeruginosa, leading to NF-κB nuclear translocation, was likely mediated by a cellular factor that signals through the MyD88 adaptor molecule. To determine possible cell surface pattern recognition molecules that could be involved in this response to P. aeruginosa, lungs were taken from strains of mice deficient in TLR2 or from C3H/HeJ mice that express a dominant-negative TLR4, as well as from mice deficient in TNF-R1 or IL-1R. Time points from 15 min to 180 min after intranasal inoculation with P. aeruginosa strain PAO1 were examined. We also tested the ability of a fliC-deficient strain of P. aeruginosa (15) to induce NF-κB nuclear translocation in WT mice in order to evaluate the potential contribution of TLR5 to the response to this pathogen. As we saw for mice with WT-CFTR, TLR2 knockout and TLR4 dominant-negative mice translocated NF-κB to the nucleus at 15 min postinfection, whereas TNF-R1 knockout mice showed a delayed but detectable NF-κB nuclear translocation 75 min postinfection (Fig. 2). Infection of WT C57BL/6 mice with the fliC::gent mutant of strain PAO1 (15) also resulted in readily detectable NF-κB nuclear translocation 15 min following infection, indicating that TLR5 is not involved in this early signaling (Fig. 2); this result was consistent with the findings of others that fliC-deficient P. aeruginosa activate inflammation in WT mice (47).
FIG. 2.

NF-κB nuclear translocation in lung sections from the indicated strains of mice in response to intratracheal infection with P. aeruginosa strain PAO1 or strain PAO1 fliC::gent (ΔfliC) where indicated. Samples were taken 15 min postinfection for the WT mice infected with either PAO1 or the ΔfliC strain (second two columns) and for the TLR2 knockout (KO) and TLR4 dominant-negative (DN) mouse strains (C3H/HeJ) infected with P. aeruginosa PAO1. Maximal NF-κB nuclear translocation was seen at 75 min postinfection for the TNF-R1 knockout mouse strain infected with PAO1. Sections were stained for NF-κB (red), CFTR (green), and nuclear DNA (blue). First row: yellow/orange color represents overlap between cytoplasmic CFTR and cytoplasmic NF-κB. Second row: cytoplasmic NF-κB (red) and nuclear DNA (blue) only. Third and fourth rows: colocalization of translocated nuclear NF-κB (red) and nuclear DNA (blue), represented as pseudocolored magenta at two different magnifications. Magnifications are indicated on the right-hand side of each row.
In contrast, IL-1R knockout mice failed to translocate NF-κB in response to P. aeruginosa infection (Fig. 3); in these mice, NF-κB translocation appeared no different from that in uninfected IL-1R knockout mice. NF-κB nuclear translocation was evaluated at 15, 30, 45, 60, 75, 90, 120, 150, and 180 min postinfection; representative sections from infected lungs taken over the first hour are shown in Fig. 3, but the lack of detectable nuclear NF-κB was also evident at all additional time points (data not shown).
FIG. 3.

NF-κB nuclear translocation in small airway sections of WT or IL-1R knockout (KO) mice at the indicated times in response to intratracheal infection with P. aeruginosa strain PAO1. Sections were stained for NF-κB (red), CFTR (green), and nuclear DNA (blue). First row: yellow/orange color represents overlap between cytoplasmic CFTR and cytoplasmic NF-κB. Second row: cytoplasmic NF-κB (red) and nuclear DNA (blue) only. Third and fourth rows: colocalization of translocated nuclear NF-κB (red) and nuclear DNA (blue), represented as pseudocolored magenta at two different magnifications. Magnifications are indicated on the right-hand side of each row.
Production of IL-1 by cultured cells in response to P. aeruginosa infection.
The above findings led us to hypothesize that IL-1 release and signaling through IL-1R were involved in the rapid NF-κB nuclear translocation in response to P. aeruginosa infection in vivo and that this release could be affected by the functional state of CFTR. To study this process in more detail, we examined whether IL-1β is released by human bronchial epithelial cells following exposure to P. aeruginosa and whether IL-1β release is altered by the absence of functional CFTR. When we infected isogenic WT-CFTR and ΔF508-CFTR human bronchial epithelial cells with P. aeruginosa strain PAO1 over a time course of 0 to 60 min, we found that IL-1β was rapidly released by cells with WT-CFTR in response to P. aeruginosa, whereas cells with mutant CFTR showed a muted IL-1β release (Fig. 4A). Replicate experiments (Fig. 4B) consistently showed a more-robust release of IL-1β by cells with WT-CFTR, although the kinetics differed slightly among experiments; the peak of IL-1β release occurred in some cases at the 5- or 10-min time points (as in Fig. 4A) and in other cases at the 15- or 30-min points (Fig. 4B).
FIG. 4.
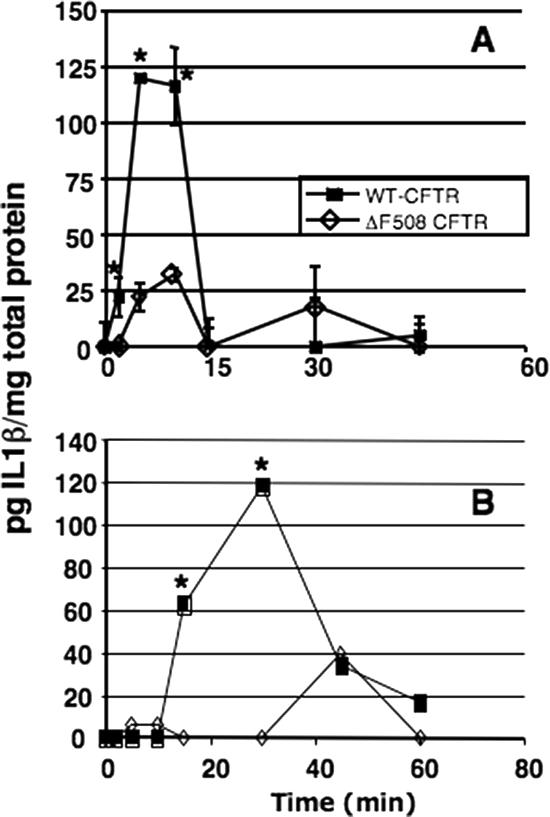
IL-1β release in supernatants of WT-CFTR and ΔF508-CFTR human bronchial epithelial cells in response to P. aeruginosa strain PAO1. Panels A and B show results from two different experiments conducted under identical conditions but yielding somewhat different kinetics of IL-1β release. Asterisks indicate a significant difference (P < 0.05, t tests) in the levels of IL-1 released at the same time point when comparing WT-CFTR and ΔF508-CFTR cells.
Because the rapid release of IL-1β seemed more likely to represent preformed cytokine rather than newly synthesized protein, we tested cell lysates in order to evaluate the level of preformed IL-1β present within uninfected epithelial cells. Uninfected cells with WT-CFTR contained 97.5 ± 0.05 pg of IL-1β per mg total protein, whereas uninfected ΔF508-CFTR cells contained 91.2 ± 0.02 pg of IL-1β per mg total protein. Thus, there was very little difference in the amounts of preformed IL-1β produced by WT and CF bronchial epithelial cells. This result suggested that the presence of WT-CFTR in bronchial epithelial cells did not affect the amount of preformed, stored IL-1β, but it did affect the release of IL-1β in response to bacterial infection.
To further support the link between the engagement of IL-1R by IL-1 and the NF-κB translocation induced by P. aeruginosa in WT-CFTR cells, we treated WT-CFTR bronchial epithelial cells with IL-1ra, cultured these cells with P. aeruginosa strain PAO1, and used confocal microscopy to visualize translocation of NF-κB in the cells 15 min postinfection. In the absence of IL-1ra, P. aeruginosa induced significant NF-κB nuclear translocation (Fig. 5), as has been reported previously (42). In contrast, when WT-CFTR cells were infected with P. aeruginosa in the presence of IL-1ra, no NF-κB translocation was observed (Fig. 5, upper panels). This was confirmed by a quantitative ELISA analysis of the effect of IL-1ra on NF-κB nuclear translocation: when either 10 μg or 4 μg of nuclear protein obtained from WT-CFTR cells exposed to P. aeruginosa strain PAO1 was added to an immobilized NF-κB-binding oligonucleotide, there was strong binding observed (Fig. 5, lower graph). Binding of the 10-μg sample of nuclear protein was specifically inhibited by a WT oligonucleotide probe containing a cognate NF-κB binding site but not by a probe with a mutated NF-κB binding site. Addition of 1 to 10 μg of IL-1ra to the WT-CFTR cells inhibited NF-κB nuclear translocation in a dose-dependent manner. In cells with the ΔF508-CFTR allele there was a slightly, but statistically significant, increased binding of 10 μg of nuclear protein to the oligonucleotide with the NF-κB binding site. NF-κB binding was reduced by the addition of 10 μg of IL-1ra, but binding was not significantly inhibited by an oligonucleotide with a cognate NF-κB binding site, indicating that the reaction was nonspecific. This was further confirmed by showing that in the presence of an oligonucleotide with a mutant NF-κB binding site, the nuclear extract from CF cells exposed to P. aeruginosa did not show significantly more binding than the control from uninfected cells. Thus, the apparent signal of NF-κB nuclear translocation in the CF cells cannot be considered to fulfill the criteria for specificity in this assay.
FIG. 5.

Inhibition of NF-κB nuclear translocation by addition of IL-1ra to cultures of P. aeruginosa-infected human bronchial epithelial cells that express WT-CFTR. The left upper panel shows colocalization of red-stained NF-κB and blue-stained DNA in nuclei (pseudocolored magenta) 15 min postinfection with P. aeruginosa strain PAO1. In the presence of IL-1ra, NF-κB is present in the cytoplasm (middle upper panel, red) but does not translocate to the nucleus (right upper panel). Magnifications are ×400. The lower graph indicates the quantitative analysis of NF-κB nuclear translocation in isogenic cell lines with WT- or ΔF508-CFTR 15 min following P. aeruginosa infection. IL-1ra was added at the indicated amount of protein to triplicate assay wells (200-μl volume each). Specificity of binding of the nuclear NF-κB was assessed by inhibition with either a WT NF-κB probe or a mutant NF-κB probe added to the nuclear extract. P values represent both overall analysis of variance and pair-wise comparisons using Tukey's multiple comparisons test.
Chronic infection of IL-1R knockout mice with P. aeruginosa.
We next used a chronic infection model to investigate whether the absence of IL-1R would lead to increased respiratory tract colonization and P. aeruginosa-specific immune responses in the sera of orally infected mice, a response that we have shown occurs in transgenic CF, but not WT, mice given this organism in their drinking water (9). IL-1R knockout and WT mice were infected via their drinking water with P. aeruginosa clinical isolate N6, which has previously been shown to induce a robust chronic lung infection in transgenic CF mice (9). The mice (six per group) were monitored for lung colonization by throat swabs on a weekly or biweekly basis. Throat swab cultures confirmed that all mice were free of P. aeruginosa after the initial 5 to 7 days of levofloxacin/gentamicin treatment and that all mice were colonized by P. aeruginosa after five days of exposure to P. aeruginosa in the drinking water. Figure 6 shows the percentage of mice that were colonized from 22 to 46 weeks after initial exposure to P. aeruginosa. IL-1R knockout mice had a much higher rate of colonization (83 to 100% weekly positive cultures through week 46) than WT mice (50% initial positive cultures, decreasing to 0 to 17% by week 34) (Fig. 6). The probability of colonization for the IL-1R knockout mice throughout the study period was 93%, whereas that for the WT mice was 17% (P < 0.001, generalized estimating equations). As a control, P. aeruginosa isolates obtained from the mice were tested for development of resistance to the nonabsorbable gentamicin and ceftazidime antibiotics that had been added to the drinking water to suppress colonization limited to the oral cavity. All strains recovered from the mice were found to be susceptible to these antibiotics just as the initial infecting strain had been.
FIG. 6.

Oropharyngeal colonization of WT and IL-1R knockout mice after exposure to P. aeruginosa strain N6 in sterile drinking water. Mice (six per group) were administered nonabsorbable antibiotics (100 μg gentamicin/ml and 100 μg ceftazidime/ml) along with orally absorbable nitrofurantoin (100 μg/ml) in sterile drinking water prior to week 20 and after week 24 in order to distinguish between oropharyngeal colonization and lung infection. The probability of colonization for the IL-1R knockout mice throughout the study period was 93% whereas that for the WT mice was 17% (P < 0.001, generalized estimating equations).
To distinguish mice with actual lung infections from those with only upper oropharyngeal colonization, serum samples were obtained periodically from the exposed IL-1R knockout and WT mice and tested by ELISA for the presence of P. aeruginosa-specific antibodies (9). IL-1R knockout mice exposed to P. aeruginosa strain N6 in their drinking water had much higher levels of P. aeruginosa-specific antibody than did WT mice in the same study (Table 1), and these differences were manifest as early as 6 weeks postinfection. This difference in antibody titer was maintained throughout the study.
TABLE 1.
Antibody titers in sera of IL-1R knockout or WT mice in a chronic colonization model using P. aeruginosa strain N6
| Week postinfection | Antibody titera
|
|
|---|---|---|
| IL-1R knockout mice | WT mice | |
| 6 | 2,761 ± 456 | 381 ± 41 |
| 18 | 7,551 ± 2,108 | 457 ± 68 |
| 31 | 15,957 ± 2,617 | 174 ± 71 |
| 49 | 13,698 ± 6,585 | 97 ± 40 |
Means ± standard errors of the means of the antibody titers calculated after subtraction of readings from preinfection sera. All titers in IL-1R knockout mice were significantly different from the corresponding titers in WT mice by a t test at a P value of <0.001.
WT and IL-1R knockout mouse serum antibody responses to chronic colonization with strain PAO1 (six mice per group) also showed marked differences at 6 and 21 weeks postinfection (see Fig. S1 in the supplemental material). This result indicated that strain PAO1, like strain N6, was capable of causing chronic lung infection in IL-1R knockout mice as measured by serum antibody responses. However, like many clinical CF isolates (4, 7, 13, 26), P. aeruginosa strain PAO1 was more difficult than strain N6 to culture from an oropharyngeal swab—a finding we had previously validated in transgenic CF mice (9)—and this measure of infection gave a greater degree of variability in positive cultures over the 21-week period of observation (data not shown).
After sacrifice, lungs from the WT and IL-1R knockout mice were analyzed for pathological changes and none were seen. This result indicated that, in spite of the apparent chronic lung colonization, the IL-1R knockout mice were able to contain the infection sufficiently to minimize the development of significant lung pathology. In this respect, the IL-1R knockout mice displayed a less-severe phenotype in response to infection than did transgenic mice lacking either functional CFTR or expressing ΔF508-CFTR (9). IL-1R knockout mice are likely to lack other significant factors that are present in CF mice and that contribute to pathology in CF lung disease, such as production of dehydrated mucus secretions which create an anaerobic environment needed for the emergence of the more-destructive mucoid phenotype of P. aeruginosa (55).
DISCUSSION
The innate immune response is critical for successful clearance of P. aeruginosa from the lungs. The recent finding that NF-κB is rapidly translocated to the nuclei of airway epithelial cells in response to P. aeruginosa infection in mice with WT-CFTR but not in CF mice (42) led us to investigate additional potential mediators of NF-κB translocation participating in the response to P. aeruginosa. Lack of MyD88 clearly affected the nuclear translocation of NF-κB in the lung epithelium following P. aeruginosa infection, but neither TLR2, TLR4, nor TLR5 was individually required for this early response. These results are consistent with studies demonstrating that TLR2, -4, and -5 are not individually involved in the resistance of mice to acute P. aeruginosa lung infection (14, 39, 47), whereas MyD88-deficient mice have increased susceptibility to P. aeruginosa lung infection (38, 39, 46). However, Feuillet et al. (14) did find cooperativity of TLR5 and TLR4 for generating a full response to P. aeruginosa infection in lung cells, consistent with the well-known redundant nature of innate immunity. Of note, the findings from several investigators conducting in vitro cell activation studies, in which P. aeruginosa flagella activate airway cells via binding to asialo GM1 and TLR5 (1, 30, 58), do not appear to reflect the in vivo infection situation, where flagella (14) or TLR5 (47) are dispensable for a WT response to P. aeruginosa lung infection. Furthermore, transgenic CF mice infected for 18 h with P. aeruginosa strain PAO1 do manifest the ability to translocate NF-κB to the nuclei of respiratory epithelial cells, indicative of a CFTR-independent late response (see Fig. S2 in the supplemental material); this late response is likely a component of the enhanced inflammatory response observed in CF mice following hours of P. aeruginosa infection (52, 53).
We also found that lack of TNF-R1 resulted in delayed, but nonetheless detectable, NF-κB nuclear translocation in murine airway epithelial cells, consistent with the findings that TNF-R1 is not required for elimination of P. aeruginosa lung infection (46) and that instillation of TNF-α into the lungs of P. aeruginosa-infected mice does not affect bacterial clearance (31). Therefore, our key finding was that CFTR-modulated rapid IL-1 release and signaling through IL-1R are key components of the very early innate immune response to P. aeruginosa infection.
Because elevated levels of IL-1β have also been found previously in CF patient sputum (6, 45), we examined the kinetics of release of this cytokine by respiratory epithelial cells. Cultured bronchial epithelial cells expressing WT-CFTR rapidly released IL-1β, supporting the hypothesis that an early IL-1-dependent signal plays an important role in the innate immune response to P. aeruginosa. The level of IL-1β release was significantly greater in WT-CFTR than in ΔF508-CFTR cells, again suggesting that the release is modulated by WT-CFTR, although the amount of preformed IL-1β was comparable in both cell lines. While at this point, we do not know the molecular basis for the CFTR-modulated release of IL-1β in response to P. aeruginosa infection, it may relate to the ion channel conductance property of CFTR. We have previously shown that P. aeruginosa rapidly activates Cl ion mobilization by CFTR (51), and we are currently investigating whether this property of CFTR is needed for release of IL-1β from airway epithelial cells.
An important role for IL-1 release and signaling through IL-1R in controlling P. aeruginosa lung infection was primarily substantiated by analyzing the responses of IL-1R knockout and WT mice to P. aeruginosa in a chronic lung colonization model. The IL-1R knockout mice had clear evidence of chronic P. aeruginosa lung colonization with strain N6, as measured by persistently positive throat cultures and significant serum antibody responses; in contrast, the WT mice appeared either to clear the infection or to confine the organisms to a tissue space that did not lead to inflammation and immune responses. Along similar lines, robust immune responses of IL-1R knockout mice to chronic infection with strain PAO1 were also observed, although culture results for this strain were not particularly informative. This combination of results is commonly observed in CF patients, where several studies have documented that serum antibody responses to P. aeruginosa are a more-sensitive measure than oropharyngeal culture for detecting infection early in the course of disease (4, 7, 13, 26). Nonetheless, given the large differences in serum IgG activity postinfection between IL-1R knockout and WT mice infected with either strain N6 or PAO1, it appears that a lack of signaling through IL-1R allows for the persistence of P. aeruginosa in an anatomical site where acquired immune responses are engendered. Serum antibody responses were not observed in any of the WT mice despite the fact that some of the mice had persistent or intermittent colonization with P. aeruginosa, indicating that in WT mice the microbes remained confined to the oropharynx at a low enough level to avoid induction of acquired immunity or that any immune response engendered in WT mice did not persist.
The lack of overt pathology in the lungs of the IL-1R knockout mice at the end of the chronic colonization trial, which contrasted with the lung pathology seen in a similar model using transgenic CF mice (9), indicates that factors other than aberrant signaling through IL-1R contribute to the development of the severe lung pathology seen in the setting of CF. Alternately, due to the relatively short life span of mice and the constraints that impact the length of time one can reasonably study a chronic infection in a laboratory setting, the IL-1R knockout mice may simply not have been infected long enough for more-severe pathology to develop. Indeed, CF patients are clearly exposed to P. aeruginosa at an early age and virtually all patients have detectable immune responses to P. aeruginosa by 3 years of age (7), long before significant lung pathology and other clinical manifestations of the disease arise in most patients (13, 26). Notably, a recent, well-conducted study showed that an average of almost 10 years is required for P. aeruginosa in CF patients to convert from the relatively benign nonmucoid form to the more-virulent mucoid form (26), and only when infection with mucoid P. aeruginosa emerges do significant lung function deterioration and pathology develop (11, 24, 32, 33). There is no reason to believe that, simply because mice have a shorter life expectancy than humans, the development of lung pathology in response to chronic P. aeruginosa lung infection will progress more rapidly or mucoid forms of the organism will emerge more quickly. Overall, the IL-1R knockout mice, like transgenic CF mice (9), are likely representative of a young CF patient during the first year of infection with nonmucoid P. aeruginosa, where little if any lung pathology is likely to be seen, and in whom clinical signs are mostly absent.
The role of IL-1 in P. aeruginosa infection has been studied previously in the context of acute infection. In these studies, IL-1 has been reported to have deleterious effects on the host. Schultz et al. (44) demonstrated that a lack of IL-1R had a protective effect in response to acute infection with P. aeruginosa; these investigators found fewer CFU of P. aeruginosa in the lungs of IL-1R knockout mice than the number of bacteria in the lungs of wild-type mice (44). In another study, IL-1β neutralizing antibodies administered immediately after acute P. aeruginosa infection protected the mice from sepsis and lethal pneumonia (18). Here, we found that a lack of IL-1R leads to increased colonization and P. aeruginosa-specific antibody production in a chronic infection model. The host response to a modest bacterial exposure in our chronic infection model suggests that a moderate amount of IL-1β release and signaling through IL-1R may play a beneficial role for the host. In contrast, in acute infection models, wherein the bacterial inoculum is large because this is needed to overcome host innate immunity, exaggerated IL-1β production may under some circumstances be detrimental to the host. This conclusion is supported by data from Amura et al. (3), who used a granulocytopenic mouse model to demonstrate that pretreatment with IL-1 before acute infection leads to enhanced protection from P. aeruginosa challenge. The beneficial effect of IL-1 in the latter model was likely due to the low dose of P. aeruginosa to which the granulocytopenic mice were exposed, allowing them to benefit from the early IL-1 exposure. Overall, the role of IL-1 and signaling through IL-1R in response to P. aeruginosa infection appears to be typical of the role of many inflammatory cytokines and their receptors in response to infection: properly timed release of physiologic amounts of the cytokine, usually early in the infectious process, leads to enhancement of innate immunity and effective microbial clearance, whereas continued cytokine release when infection persists leads to pathological responses that inhibit bacterial clearance and instead contribute to tissue pathology.
Together, our results indicate that release of IL-1 and signaling through IL-1R on respiratory epithelial cells are necessary for rapid NF-κB translocation in response to P. aeruginosa and this process is maximal in cells with WT-CFTR. Another component of the NF-κB response is the CFTR-mediated internalization of P. aeruginosa via lipid rafts (18, 25). Both IL-1 release and bacterial internalization may be central to the development of effective innate immunity, leading to clearance of this microbe from the respiratory tract. Signaling through other NF-κB activation pathways—such as TLRs—has also been shown to occur in airway epithelial cells, but none of the studies published to date have evaluated the effect of mutant CFTR on these responses, and the NF-κB-dependent responses have all been measured 4 h or more after infection. Therefore, the contribution of TLR pathways to host resistance or pathology may be occurring primarily at later time points in the infection. Of note, Blander and Medzhitov (5) have recently shown that TLR2 and TLR4 responses to bacterial activators occur not by binding to bacterial molecules on cell surfaces but instead occur within phagosomes following endocytic or phagocytic internalization of the microbes. If this result can be generalized to the airway epithelial cell responses to P. aeruginosa LPS, flagella, and other structures, then the ability of WT-CFTR to mediate P. aeruginosa internalization could represent a key component of TLR-based cellular innate responses to bacterial factors that are presented by the whole bacterium. Blander and Medzhitov's findings (5) also suggest that activation of TLRs by purified bacterial products may not fully represent the actual response that occurs with a TLR stimulus, which may be confined to interactions inside a phagosome. Overall, our findings suggest that WT P. aeruginosa must induce at least two responses in lung epithelial cells—IL-1 release and signaling, and entry of CFTR into lipid rafts and bacterial internalization (25)—in order to activate NF-κB nuclear translocation and that these responses are all key components of the CFTR-dependent resistance of the lung to P. aeruginosa infection.
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI HL58398 and HL32854 and the Albert J. Ryan Foundation.
Editor: A. D. O'Brien
Footnotes
Published ahead of print on 5 February 2007.
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1.Adamo, R., S. Sokol, G. Soong, M. I. Gomez, and A. Prince. 2004. Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and Toll-like receptor 2 as well as Toll-like receptor 5. Am. J. Respir. Cell Mol. Biol. 30:627-634. [DOI] [PubMed] [Google Scholar]
- 2.Allewelt, M., F. T. Coleman, M. Grout, G. P. Priebe, and G. B. Pier. 2000. Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect. Immun. 68:3998-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amura, C. R., P. A. Fontan, N. Sanjuan, and D. O. Sordelli. 1994. The effect of treatment with interleukin-1 and tumor necrosis factor on Pseudomonas aeruginosa lung infection in a granulocytopenic mouse model. Clin. Immunol. Immunopathol. 73:261-266. [DOI] [PubMed] [Google Scholar]
- 4.Banwart, B., M. L. Splaingard, P. M. Farrell, M. J. Rock, P. L. Havens, J. Moss, M. E. Ehrmantraut, D. W. Frank, and J. T. Barbieri. 2002. Children with cystic fibrosis produce an immune response against exoenzyme S, a type III cytotoxin of Pseudomonas aeruginosa. J. Infect. Dis. 185:269-270. [DOI] [PubMed] [Google Scholar]
- 5.Blander, J. M., and R. Medzhitov. 2006. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440:808-812. [DOI] [PubMed] [Google Scholar]
- 6.Bonfield, T. L., J. R. Panuska, M. W. Konstan, K. A. Hilliard, J. B. Hilliard, H. Ghnaim, and M. Berger. 1995. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 152:2111-2118. [DOI] [PubMed] [Google Scholar]
- 7.Burns, J. L., R. L. Gibson, S. McNamara, D. Yim, J. Emerson, M. Rosenfeld, P. Hiatt, K. McCoy, R. Castile, A. L. Smith, and B. W. Ramsey. 2001. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 183:444-452. [DOI] [PubMed] [Google Scholar]
- 8.Cannon, C. L., M. P. Kowalski, K. S. Stopak, and G. B. Pier. 2003. Pseudomonas aeruginosa-induced apoptosis is defective in respiratory epithelial cells expressing mutant cystic fibrosis transmembrane conductance regulator. Am. J. Respir. Cell Mol. Biol. 29:188-197. [DOI] [PubMed] [Google Scholar]
- 9.Coleman, F. T., S. Mueschenborn, G. Meluleni, C. Ray, V. J. Carey, S. O. Vargas, C. L. Cannon, F. M. Ausubel, and G. B. Pier. 2003. Hypersusceptibility of cystic fibrosis mice to chronic Pseudomonas aeruginosa oropharyngeal colonization and lung infection. Proc. Natl. Acad. Sci. USA 100:1949-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courtney, J. M., M. Ennis, and J. S. Elborn. 2004. Cytokines and inflammatory mediators in cystic fibrosis. J. Cyst. Fibros. 3:223-231. [DOI] [PubMed] [Google Scholar]
- 11.Demko, C. A., P. J. Byard, and P. B. Davis. 1995. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J. Clin. Epidemiol. 48:1041-1049. [DOI] [PubMed] [Google Scholar]
- 12.Esen, M., H. Grassme, J. Riethmuller, A. Riehle, K. Fassbender, and E. Gulbins. 2001. Invasion of human epithelial cells by Pseudomonas aeruginosa involves Src-like tyrosine kinases p60Src and p59Fyn. Infect. Immun. 69:281-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farrell, P. M., Z. Li, M. R. Kosorok, A. Laxova, C. G. Green, J. Collins, H. C. Lai, L. M. Makholm, M. J. Rock, and M. L. Splaingard. 2003. Longitudinal evaluation of bronchopulmonary disease in children with cystic fibrosis. Pediatr. Pulmonol. 36:230-240. [DOI] [PubMed] [Google Scholar]
- 14.Feuillet, V., S. Medjane, I. Mondor, O. Demaria, P. P. Pagni, J. E. Galan, R. A. Flavell, and L. Alexopoulou. 2006. Involvement of Toll-like receptor 5 in the recognition of flagellated bacteria. Proc. Natl. Acad. Sci. USA 103:12487-12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleiszig, S. M., S. K. Arora, R. Van, and R. Ramphal. 2001. FlhA, a component of the flagellum assembly apparatus of Pseudomonas aeruginosa, plays a role in internalization by corneal epithelial cells. Infect. Immun. 69:4931-4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiser, T., B. Dewald, M. U. Ehrengruber, I. Clark-Lewis, and M. Baggiolini. 1993. The interleukin-8-related chemotactic cytokines GRO alpha, GRO beta, and GRO gamma activate human neutrophil and basophil leukocytes. J. Biol. Chem. 268:15419-15424. [PubMed] [Google Scholar]
- 17.George, S. E., M. J. Kohan, M. I. Gilmour, M. S. Taylor, H. G. Brooks, J. P. Creason, and L. D. Claxton. 1993. Pulmonary clearance and inflammatory response in C3H/HeJ mice after intranasal exposure to Pseudomonas spp. Appl. Environ. Microbiol. 59:3585-3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grassme, H., V. Jendrossek, A. Riehle, G. von Kurthy, J. Berger, H. Schwarz, M. Weller, R. Kolesnick, and E. Gulbins. 2003. Host defense against Pseudomonas aeruginosa requires ceramide-rich membrane rafts. Nat. Med. 9:322-330. [DOI] [PubMed] [Google Scholar]
- 19.Greene, C. M., T. P. Carroll, S. G. Smith, C. C. Taggart, J. Devaney, S. Griffin, S. J. O'Neill, and N. G. McElvaney. 2005. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 174:1638-1646. [DOI] [PubMed] [Google Scholar]
- 20.Henneke, P., O. Takeuchi, R. Malley, E. Lien, R. R. Ingalls, M. W. Freeman, T. Mayadas, V. Nizet, S. Akira, D. L. Kasper, and D. T. Golenbock. 2002. Cellular activation, phagocytosis, and bactericidal activity against group B streptococcus involve parallel myeloid differentiation factor 88-dependent and independent signaling pathways. J. Immunol. 169:3970-3977. [DOI] [PubMed] [Google Scholar]
- 21.Hoare, A., M. Bittner, J. Carter, S. Alvarez, M. Zaldivar, D. Bravo, M. A. Valvano, and I. Contreras. 2006. The outer core lipopolysaccharide of Salmonella enterica serovar Typhi is required for bacterial entry into epithelial cells. Infect. Immun. 74:1555-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jorens, P. G., J. B. Richman-Eisenstat, B. P. Housset, P. P. Massion, I. Ueki, and J. A. Nadel. 1994. Pseudomonas-induced neutrophil recruitment in the dog airway in vivo is mediated in part by IL-8 and inhibited by a leumedin. Eur. Respir. J. 7:1925-1931. [PubMed] [Google Scholar]
- 23.Joseph, T., D. Look, and T. Ferkol. 2005. NF-kappaB activation and sustained IL-8 gene expression in primary cultures of cystic fibrosis airway epithelial cells stimulated with Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. 288:L471-L479. [DOI] [PubMed] [Google Scholar]
- 24.Kosorok, M. R., L. Zeng, S. E. West, M. J. Rock, M. L. Splaingard, A. Laxova, C. G. Green, J. Collins, and P. M. Farrell. 2001. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr. Pulmonol. 32:277-287. [DOI] [PubMed] [Google Scholar]
- 25.Kowalski, M. P., and G. B. Pier. 2004. Localization of cystic fibrosis transmembrane conductance regulator to lipid rafts of epithelial cells is required for Pseudomonas aeruginosa-induced cellular activation. J. Immunol. 172:418-425. [DOI] [PubMed] [Google Scholar]
- 26.Li, Z., M. R. Kosorok, P. M. Farrell, A. Laxova, S. E. West, C. G. Green, J. Collins, M. J. Rock, and M. L. Splaingard. 2005. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 293:581-588. [DOI] [PubMed] [Google Scholar]
- 27.Liang, K. Y., and S. L. Zeger. 1986. Longitudinal data analysis using generalized linear models. Biometrika 73:13-22. [Google Scholar]
- 28.Lyczak, J. B., T. S. Zaidi, M. Grout, M. Bittner, I. Contreras, and G. B. Pier. 2001. Epithelial cell contact-induced alterations in Salmonella enterica serovar Typhi lipopolysaccharide are critical for bacterial internalization. Cell. Microbiol. 3:763-772. [DOI] [PubMed] [Google Scholar]
- 29.MacRedmond, R., C. Greene, C. C. Taggart, N. McElvaney, and S. O'Neill. 2005. Respiratory epithelial cells require Toll-like receptor 4 for induction of human beta-defensin 2 by lipopolysaccharide. Respir. Res. 6:116-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNamara, N., M. Gallup, A. Sucher, I. Maltseva, D. McKemy, and C. Basbaum. 2006. ASIALO GM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate ERK1/2. Am. J. Respir. Cell Mol. Biol. 34:653-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morissette, C., C. Francoeur, C. Darmond-Zwaig, and F. Gervais. 1996. Lung phagocyte bactericidal function in strains of mice resistant and susceptible to Pseudomonas aeruginosa. Infect. Immun. 64:4984-4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parad, R. B., C. J. Gerard, D. Zurakowski, D. P. Nichols, and G. B. Pier. 1999. Pulmonary outcome in cystic fibrosis is influenced primarily by mucoid Pseudomonas aeruginosa infection and immune status and only modestly by genotype. Infect. Immun. 67:4744-4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedersen, S. S., N. Hoiby, F. Espersen, and C. Koch. 1992. Role of alginate in infection with mucoid Pseudomonas aeruginosa in cystic fibrosis. Thorax 47:6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pier, G. B. 2002. CFTR mutations and host susceptibility to Pseudomonas aeruginosa lung infection. Curr. Opin. Microbiol. 5:81-86. [DOI] [PubMed] [Google Scholar]
- 35.Pier, G. B., M. Grout, T. Zaidi, G. Meluleni, S. S. Mueschenborn, G. Banting, R. Ratcliff, M. J. Evans, and W. H. Colledge. 1998. Salmonella typhi uses CFTR to enter intestinal epithelial cells. Nature 392:79-82. [DOI] [PubMed] [Google Scholar]
- 36.Pier, G. B., M. Grout, and T. S. Zaidi. 1997. Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc. Natl. Acad. Sci. USA 94:12088-12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pier, G. B., M. Grout, T. S. Zaidi, J. C. Olsen, L. G. Johnson, J. R. Yankaskas, and J. B. Goldberg. 1996. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science 271:64-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Power, M. R., Y. Peng, E. Maydanski, J. S. Marshall, and T. J. Lin. 2004. The development of early host response to Pseudomonas aeruginosa lung infection is critically dependent on myeloid differentiation factor 88 in mice. J. Biol. Chem. 279:49315-49322. [DOI] [PubMed] [Google Scholar]
- 39.Ramphal, R., V. Balloy, M. Huerre, M. Si-Tahar, and M. Chignard. 2005. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J. Immunol. 175:3927-3934. [DOI] [PubMed] [Google Scholar]
- 40.Reiniger, N., J. K. Ichikawa, and G. B. Pier. 2005. Influence of cystic fibrosis transmembrane conductance regulator on gene expression in response to Pseudomonas aeruginosa infection of human bronchial epithelial cells. Infect. Immun. 73:6822-6830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richman-Eisenstat, J. B., P. G. Jorens, C. A. Hebert, I. Ueki, and J. A. Nadel. 1993. Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am. J. Physiol. 264:L413-L418. [DOI] [PubMed] [Google Scholar]
- 42.Schroeder, T. H., M. M. Lee, P. W. Yacono, C. L. Cannon, A. A. Gerceker, D. E. Golan, and G. B. Pier. 2002. CFTR is a pattern recognition molecule that extracts Pseudomonas aeruginosa LPS from the outer membrane into epithelial cells and activates NF-kappa B translocation. Proc. Natl. Acad. Sci. USA 99:6907-6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schroeder, T. H., N. Reiniger, G. Meluleni, M. Grout, F. T. Coleman, and G. B. Pier. 2001. Transgenic cystic fibrosis mice exhibit reduced early clearance of Pseudomonas aeruginosa from the respiratory tract. J. Immunol. 166:7410-7418. [DOI] [PubMed] [Google Scholar]
- 44.Schultz, M. J., A. W. Rijneveld, S. Florquin, C. K. Edwards, C. A. Dinarello, and T. van der Poll. 2002. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am. J. Physiol. Lung Cell. Mol. Physiol. 282:L285-L290. [DOI] [PubMed] [Google Scholar]
- 45.Schuster, A., A. Haarmann, and V. Wahn. 1995. Cytokines in neutrophil-dominated airway inflammation in patients with cystic fibrosis. Eur. Arch. Otorhinolaryngol. 252(Suppl. 1):S59-S60. [DOI] [PubMed] [Google Scholar]
- 46.Skerrett, S. J., H. D. Liggitt, A. M. Hajjar, and C. B. Wilson. 2004. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J. Immunol. 172:3377-3381. [DOI] [PubMed] [Google Scholar]
- 47.Skerrett, S. J., C. B. Wilson, H. D. Liggitt, and A. M. Hajjar. 2006. Redundant Toll-like receptor signaling in the pulmonary host response to Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell. Mol. Physiol. [DOI] [PubMed]
- 48.Soong, G., B. Reddy, S. Sokol, R. Adamo, and A. Prince. 2004. TLR2 is mobilized into an apical lipid raft receptor complex to signal infection in airway epithelial cells. J. Clin. Investig. 113:1482-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K. Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443-451. [DOI] [PubMed] [Google Scholar]
- 50.Tsui, I. S., C. M. Yip, J. Hackett, and C. Morris. 2003. The type IVB pili of Salmonella enterica serovar Typhi bind to the cystic fibrosis transmembrane conductance regulator. Infect. Immun. 71:6049-6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ullrich, S., S. Berchtold, C. Boehmer, S. Fillon, V. Jendrossek, M. Palmada, T. H. Schroeder, G. B. Pier, and F. Lang. 2003. Pseudomonas aeruginosa activates Cl- channels in host epithelial cells. Pflugers Arch. 447:23-28. [DOI] [PubMed] [Google Scholar]
- 52.van Heeckeren, A., J. Tscheikuna, R. W. Walenga, M. W. Konstan, P. B. Davis, B. Erokwu, M. A. Haxhiu, and T. W. Ferkol. 2000. Effect of Pseudomonas infection on weight loss, lung mechanics, and cytokines in mice. Am. J. Respir. Crit. Care Med. 161:271-279. [DOI] [PubMed] [Google Scholar]
- 53.van Heeckeren, A. M., M. D. Schluchter, W. Xue, and P. B. Davis. 2006. Response to acute lung infection with mucoid Pseudomonas aeruginosa in cystic fibrosis mice. Am. J. Respir. Crit. Care Med. 173:288-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walker, J., J. Watson, C. Holmes, A. Edelman, and G. Banting. 1995. Production and characterisation of monoclonal and polyclonal antibodies to different regions of the cystic fibrosis transmembrane conductance regulator (CFTR): detection of immunologically related proteins. J. Cell Sci. 108:2433-2444. [DOI] [PubMed] [Google Scholar]
- 55.Worlitzsch, D., R. Tarran, M. Ulrich, U. Schwab, A. Cekici, K. C. Meyer, P. Birrer, G. Bellon, J. Berger, T. Weiss, K. Botzenhart, J. R. Yankaskas, S. Randell, R. C. Boucher, and G. Doring. 2002. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 109:317-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaas, D. W., M. J. Duncan, G. Li, J. R. Wright, and S. N. Abraham. 2005. Pseudomonas invasion of type I pneumocytes is dependent on the expression and phosphorylation of caveolin-2. J. Biol. Chem. 280:4864-4872. [DOI] [PubMed] [Google Scholar]
- 57.Zaidi, T. S., S. M. Fleiszig, M. J. Preston, J. B. Goldberg, and G. B. Pier. 1996. Lipopolysaccharide outer core is a ligand for corneal cell binding and ingestion of Pseudomonas aeruginosa. Investig. Ophthalmol. Vis. Sci. 37:976-986. [PubMed] [Google Scholar]
- 58.Zhang, Z., J. P. Louboutin, D. J. Weiner, J. B. Goldberg, and J. M. Wilson. 2005. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect. Immun. 73:7151-7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.