

Abstract
Integrated retroviral DNA is subject to epigenetic gene silencing, but the viral and host cell properties that influence initiation, maintenance, and reactivation are not fully understood. Here we describe rapid and high-frequency epigenetic repression and silencing of integrated avian sarcoma virus (ASV)-based vector DNAs in human HeLa cells. Initial studies utilized a vector carrying the strong human cytomegalovirus (hCMV) immediate-early (IE) promoter to drive expression of a green fluorescent protein (GFP) reporter gene, and cells were sorted into two populations based on GFP expression [GFP(+) and GFP(−)]. Two potent epigenetic effects were observed: (i) a very broad distribution of GFP intensities among cells in the GFP(+) population as well as individual GFP(+) clones and (ii) high-frequency GFP reporter gene silencing in GFP(−) cells. We previously showed that histone deacetylases (HDACs) can associate with ASV DNA soon after infection and may act to repress viral transcription at the level of chromatin. Consistent with this finding, we report here that treatment with the histone deacetylase inhibitor trichostatin A (TSA) induces GFP activation in GFP(−) cells and can also increase GFP expression in GFP(+) cells. In the case of the GFP(−) populations, we found that after removal of TSA, GFP silencing was reestablished in a subset of cells. We used that finding to enrich for stable GFP(−) cell populations in which viral GFP reporter expression could be reactivated by TSA; furthermore, we found that the ability to isolate such populations was independent of the promoter driving the GFP gene. In such enriched cultures, hCMV IE-driven, but not the viral long terminal repeat-driven, silent GFP reporter expression could be reactivated by the transcriptional activator prostratin. Microscopy-based studies using synchronized cells revealed variegated reactivation in cell clones, indicating that secondary epigenetic effects can restrict reactivation from silencing. Furthermore we found that entry into S phase was not required for reactivation. We conclude that HDACs can act rapidly to initiate and maintain promoter-independent retroviral epigenetic repression and silencing but that reactivation can be restricted by additional mechanisms.
After integration, retroviral DNA becomes a segment of the host chromosome and is therefore duplicated during S phase and passed to daughter cells following mitosis (12). In addition to establishing a permanent association between viral and host DNA, integration allows for efficient retroviral gene expression. However, DNA integration does not ensure continued viral gene expression. Gene silencing is frequently observed when retroviruses are used as vectors for gene delivery; transduction of the introduced reporter gene is successful, but expression is extinguished at various times postinfection (19, 49, 57, 59). This phenomenon is prominent in embryonic or adult stem cells and has been most thoroughly studied in this context (9, 19, 24, 49, 50, 58, 63). Silencing is typically mediated by DNA methylation or chromatin modifications at the viral loci (19, 49, 57, 59). As this repressed viral state is heritable over many cell generations, retroviral silencing is by definition epigenetically controlled and may signify an active cellular mechanism to repress foreign DNA (27, 65). Epigenetic silencing is generally reversible, and retroviral gene expression can be reactivated by various stimuli.
Although retroviral DNA silencing has been well studied, the parameters that influence the initiation, maintenance, and reactivation are not fully understood. The earliest attempts to introduce murine leukemia virus into developing mouse embryos and stem cells (28) led to the discovery of a correlation between retroviral silencing and DNA methylation (59). Murine leukemia virus cis-acting sequences that mediate the repressive effects were found to reside within the viral long terminal repeat (LTR) promoter and other regions (49). However, more recent studies have implicated histone modifications in retroviral silencing (19, 22, 23, 49, 50, 59, 61, 64). A family of transcriptional activators are enzymes, histone acetyltransferases (HATs), that acetylate histone tails (29). This acetylation is thought to promote chromatin opening, as well as to serve as a biochemical mark for recruitment of other transcriptional activators. Histone deacetylases (HDACs) remove these acetyl groups and function as transcriptional repressors that maintain epigenetic silencing. Thus, HATs and HDACs can act as local antagonists to regulate gene expression. Treatment with histone deacetylase inhibitors (HDIs) results in broad histone acetylation and concomitant activation of a subset of silent cellular genes, as well as silent reporter genes (7, 8, 66). Although DNA methylation can recruit HDAC repressor complexes, there are examples of DNA methylation-independent HDAC-mediated reporter gene repression (50). Numerous studies have now indicated both independent and combinatorial roles for DNA methylation, histone modifications, and chromatin remodeling in retroviral silencing (9, 19, 24, 25, 40-42, 45, 49, 50, 57-59, 63). However, a universal model for the initiation and maintenance of retroviral silencing has not yet emerged. The apparent diversity of mechanisms may be due to differing cell- and virus-specific features. In addition, redundancies in host silencing mechanisms or temporal-specific effects may cause difficulties in correlating the various findings.
Two ways in which epigenetic modifiers (e.g., HATs, HDACs, chromatin-remodeling complexes, and DNA methyltransferases) might associate with retroviral DNA to initiate and maintain epigenetic silencing are by “spreading” from regions of host chromatin that flank the integration site (17) and/or by more direct recruitment to viral DNA (22, 49). Transcriptional activity of neighboring genes might also influence expression of the integrated viral DNA (37). Therefore, features that influence the frequency at which retroviral silencing occurs may include the location of the retroviral DNA integration site within the host chromosome (18, 30, 31, 37), the orientation of the integrated viral DNA with respect to host DNA (20), and the propensity of the viral LTR or other cis-acting sequences to recruit repressive host factors (49). The so-called “position effects” on retroviral expression are believed to be mediated by the local chromatin state; i.e., constitutive/facultative heterochromatin (closed) or euchromatin (open), corresponding to silent and potentially active regions, respectively. Features of constitutive heterochromatin include DNA methylation, presence of repressive histone code modifications, and late S-phase replication.
Initial studies revealed that the DNA of human immunodeficiency virus type 1 (HIV-1) integrates preferentially into active transcription units (protein-coding genes) (56), although the biochemical basis for this preference is not yet understood. We (47) and others (4, 44) investigated avian sarcoma virus (ASV) integration site selection in human cells, for which a modest preference for integration into genes was observed. However, transcriptional activity of these target genes at the time of integration neither stimulated nor inhibited ASV integration (47). Postintegration latency of HIV-1 is controlled in part by epigenetic mechanisms, including histone modifications (14, 23, 36, 51, 61, 64), and some studies have suggested that integration near closed chromatin may account for establishment of such rare HIV-1 latency (30, 31). Another study indicates that a wide variety of flanking genomic features can influence HIV expression (37).
While using human HeLa cells to study retroviral DNA nuclear import (32) and integration site selection (47), we found that an ASV-based green fluorescent protein (GFP) reporter vector was susceptible to rapid silencing and variegation. Other studies from our laboratory demonstrated that HDACs can physically associate with ASV DNA complexes early after infection (22). Here we report results from a series of cell population and clonal studies, which show that the epigenetically silent state of GFP reporter genes can be antagonized by HDIs, resulting in robust activation of GFP expression. We observed similar reactivation after treatment with the phorbol ester prostratin, although in this case, the response was promoter specific. By following cells microscopically during reactivation, we determined that the response to these compounds is not uniform in cell clones, and this may indicate an orderly switching between transcriptionally responsive and nonresponsive states. These studies provide further insights into the mechanisms of retroviral silencing as well as an explanation for the nonuniform response to HDIs.
MATERIALS AND METHODS
Viruses and cells.
The ASV-based vectors that utilize the LTR (ASVA-GFP) or an internal human cytomegalovirus (hCMV) immediate-early (IE) promoter (ASVA-CMVEGFP) to drive expression of the enhanced GFP gene have been described previously (33). These vectors are derivatives of an ASV-based vector that is capable of only one round infection of mammalian cells (1). A third ASV-based vector was constructed for this study, in which the hCMV IE promoter in ASVA-CMVEGFP was replaced with the elongation factor 1 (EF-1) alpha promoter by using standard cloning methods. The EF-1 alpha promoter region was PCR amplified from the pEF1 plasmid (Invitrogen). The HIV-1-based GFP vector was described previously (32). Jurkat T-cell clones containing a latent HIV-1 genome encoding a GFP reporter were kindly provided by Eric Verdin (30).
Chemicals and antibodies.
Trichostatin A (TSA), aphidicolin, nocodozole, valproic acid, sodium butyrate, and apidicin were purchased from Sigma-Aldrich. Tumor necrosis factor alpha (TNF-α), phorbol 12-myristate 13-acetate (PMA), and prostratin (12-deoxyphorbol 13-acetate) were purchased from Biomol, and the anti-GFP antibody was purchased from Abcam.
Fluorescence-activated cell sorter (FACS) analysis.
GFP expression was measured using a Becton Dickinson FACScan flow analyzer. Data were analyzed using FlowJo software.
Infections, cell sorting, and cell cloning.
HeLa cells were infected with GFP reporter vectors by incubation with several dilutions of virus stocks in the presence of 10 μg/ml DEAE-dextran. The ASV-based vectors were produced in DF-1 chicken cells, and the HIV-1-based vectors were produced using the three-plasmid transfection strategy, as described previously (32). After 48 h, cultures were examined for GFP expression by microscopy, and the percentage and intensity of GFP expression was quantitated by FACS analysis. The GFP expression patterns shown in Fig. 1A were observed reproducibly. Cultures that contained 10 to 30% GFP(+) cells were selected for further analysis. Viable cells were preparatively sorted at 7 to 10 days postinfection using a Becton Dickinson FACS-VantageSE flow cytometer. Several independent infections and sorting experiments were used to prepare GFP(+) and GFP(−) populations. The general gating strategy is outlined in Fig. 1B. For the GFP(+) fraction, cells were selected from approximately the two most intense decades of GFP signal, as these cells expressed sufficient GFP to be analyzed by microscopy. The GFP(+) cells were either sorted as a population or distributed for cloning as individual cells in 96-well dishes.
FIG. 1.

GFP intensity profiles of infected HeLa cells as analyzed by FACS. (A) Profiles of GFP expression were measured at 48 h postinfection. Shown are GFP profiles for uninfected HeLa cells (CON) (light shading) and for cells after infection with increasing amounts of the ASV-GFP vector. A parallel culture was infected with an HIV-1-based vector (HIV-GFP) (dark shading). Data were analyzed using FlowJo software. y-axis scaling was used to more readily compare the intensity profiles. (B) Gating strategy to isolate GFP(−) and GFP(+) cells. A representative profile from panel A is shown with the approximate gates used for isolation of cell populations by preparative cell sorting. In this example, the GFP(+) cells represented ca. 30% of the culture. A subfraction of brighter GFP(+) cells were selected for isolation to ensure that GFP patterns were bright enough to observe microscopically.
A multistep sorting strategy was used to purify cells in which GFP expression could be reactivated by TSA treatment. The GFP(−) cell population was treated with TSA, and the GFP-expressing cells were sorted. This population was passaged for ca. 10 days, at which time there was a significant loss of GFP expression. The residual GFP-expressing cells were then removed by cell sorting, and the resulting population was designated TI, as they were selected for TSA-induced GFP expression. On occasion, a second sorting step was required to eliminate contaminating GFP-expressing cells. An identical strategy was used to isolate TI-C, TI-L, and TI-E cells. We noted that during initial isolation of GFP(−) cells, gating of the entire symmetrical GFP(−) peak corresponding to uninfected HeLa cells (Fig. 1B) reproducibly generated cell populations in which a subset of cells responded to TSA. Higher-stringency gating to exclude cells with greater fluorescence within the GFP(−) peak appeared to reduce the fraction of cells that responded to TSA, indicating that TSA-responsive cells were not uniformly distributed within the GFP(−) peak.
Cell synchronization and microscopy.
Synchronization of HeLa cells was carried out essentially as described previously (32). Subconfluent HeLa cultures were washed with medium three times to remove weakly adherent cells and were then treated with nocodozole (16 ng/ml) for 2 h. Mitotic shakeoff was carried out, and the resultant cells were plated under dilute conditions to produce well-isolated microclones. Microscopy was carried out using a Nikon Eclipse TE800 inverted microscope fitted with a Nikon COOLPIX 950 digital camera.
qPCR.
The 5′-nuclease assays with TaqMan chemistry were used to measure the relative copy number of GFP compared to a control HeLa gene (human albumin, 4q11-q13) (39). DNA was prepared from GFP(−), GFP(+), or unsorted cells by using a DNeasy kit (QIAGEN). The Primer Express software (Applied Biosystems) was used to design primers and probes. The quantitative real-time PCR (qPCR) primer and probe sequences for albumin were 5′-CATTTATTGGTGTGTCCCCTTG-3′ (forward), 5′-ACACCAGTGAAAACAATTTAAGCC-3′ (reverse), and (6-FAM)CCCAACAGAAGAATTCAGCAGCCGTAAG(BHQ1) (probe), and those for enhanced GFP were 5′-CCCAGTCCGCCCTGAG-3′ (forward), 5′-ACGAACTCCAGCAGGACCA-3′ (reverse), and (6-FAM)CCCCAACGAGAAGCGCGATCA(BHQ1) (probe). The 5′ and 3′ ends of the probes were labeled with the reporter dye 6-FAM (6-carboxyfluorescein) (Glenn Research) and the quencher dye BHQ1 (Black Hole Quencher) (Biosearch Technologies), respectively. All primers and probes were synthesized by the Fox Chase Cancer Center Fannie Rippel Biotechnology Facility. Quantitect (QIAGEN) master mix was used for PCR. The Cepheid Smart Cycler was used for qPCR analyses. Cycling conditions were 95°C for 10 min, followed by 45 (two-step) cycles (95°C for 20 s and 62°C for 60 s). The difference in threshold cycle (CT) values (ΔCT) between GFP and albumin was used to normalize the amount of genomic DNA. A GFP standard was established by infecting HeLa cells at a very low multiplicity of infection (MOI) with the HIV-GFP vector and sorting the GFP(+) cell population (1% of the total), ensuring that a single copy of GFP was present. The ratio of GFP to genomic DNA (albumin) in this sample is normalized to 1 (one copy of GFP per cell genome), and this sample is taken as the calibrator. The differences in ΔCTs (ΔΔCT) for the samples of interest and the calibrator are used to estimate the relative quantity (RQ) of GFP (to the calibrator sample) by using the formula RQ = 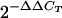 (39). The values and standard deviations in Table 1 (copy number) were obtained by averaging results from three PCRs (for each reporter) performed with inputs of 100, 20, and 4 ng of genomic DNA (based on optical density). As a negative control, DNA was prepared, using sorted GFP(−) cells from a culture that was infected with an HIV-GFP vector [50% GFP(+) cells]. As we could find no evidence for silent GFP in this GFP(−) population by conventional PCR or TSA treatment, this sample served as a rigorous negative control. Analysis of this control sample gave a CT value comparable to that of the GFP standard sample for albumin, but the CT value for GFP was lower by ca. 7 cycles (ΔΔCT = 7), indicating that the relative GFP copy number was negligible (27-fold lower).
(39). The values and standard deviations in Table 1 (copy number) were obtained by averaging results from three PCRs (for each reporter) performed with inputs of 100, 20, and 4 ng of genomic DNA (based on optical density). As a negative control, DNA was prepared, using sorted GFP(−) cells from a culture that was infected with an HIV-GFP vector [50% GFP(+) cells]. As we could find no evidence for silent GFP in this GFP(−) population by conventional PCR or TSA treatment, this sample served as a rigorous negative control. Analysis of this control sample gave a CT value comparable to that of the GFP standard sample for albumin, but the CT value for GFP was lower by ca. 7 cycles (ΔΔCT = 7), indicating that the relative GFP copy number was negligible (27-fold lower).
TABLE 1.
Copy numbers of integrated ASV-GFP reporter genes in GFP(+) and GFP(−) cellsa
| Cells | Fraction of GFP-positive cells in unsorted starting culture | GFP copy no. per cell (mean ± SD) |
|---|---|---|
| GFP standardb | NRd | 1.0 |
| GFP(+)c | 0.35 | 1.4 ± 0.2 |
| GFP(−)c | 0.35 | 0.3 ± 0.0 |
| GFP(+) | 0.20 | 1.2 ± 0.3 |
The GFP reporter gene copy number relative to the cellular albumin gene was determined by qPCR (see Materials and Methods). As a negative control, GFP(−) cells were sorted from a culture infected with an HIV-GFP vector in which ca. 50% of cells were GFP(+). Results indicated that GFP(−) cells derived from this culture did not harbor significant levels of HIV DNA (27-fold lower than the GFP standard sample).
To prepare control cells containing a single-copy GFP gene, HeLa cells were infected with an HIV-GFP vector under conditions where ca. 1% of the cells were GFP(+). The GFP(+) population was isolated by cell sorting and used to prepare the DNA standard.
The indicated GFP(+) and GFP(−) cell populations were sorted from the same starting culture.
NR, not relevant.
RESULTS
Broad range of GFP reporter expression intensities in HeLa cells after infection with an ASV-based vector.
Interspecies infection with retroviruses can be accomplished through pseudotyping, and such a strategy has been used for a variety of laboratory and gene transfer experiments. During our previous studies (32, 47), we frequently observed clonal variegation (mosaic patterns) of GFP reporter expression (by microscopy) early after infection of human HeLa cells with a pseudotyped ASV-based vector (1). Such variegated expression patterns were somewhat unexpected, as the GFP gene was driven by the strong hCMV IE promoter. As infection is limited to one round in this system, reinfection cannot contribute to this phenomenon. In addition, infection of HeLa cells with an ASV-GFP vector that is defective for DNA integration does not result in detectable GFP expression (33), and therefore the observed pattern could not be attributed to dilution of unintegrated viral DNA during cell outgrowth. We suspected that the rapid variegation was produced by epigenetic changes and might represent intermediate steps in GFP reporter gene silencing.
To examine this phenomenon in more detail, we sought to establish infection conditions whereby various GFP expression patterns could be more readily interpreted. In particular, it was important to limit the MOI, such that differences in GFP expression intensity could not be due to differences in GFP copy number. Accordingly, HeLa cells were infected with various amounts of the ASV-GFP vector to derive conditions whereby only ca. 10 to 30% of the cells expressed GFP at 7 to 10 days postinfection. In these populations we designate the resulting GFP-expressing cells GFP(+) and nonexpressing cells GFP(−). The latter cell population would be expected to include uninfected cells, as well as cells in which the viral DNA may be silenced by epigenetic processes.
After infection with the ASV-GFP vector, FACS analysis showed that the GFP intensity profile was quite broad at early times, and this pattern was generally independent of the MOI. (Fig. 1A). Such broad profiles are typically attributed to differences in reporter gene expression that result from integration at different sites within the host cell genome and/or positional variegation (mosaic patterns of reporter expression produced during outgrowth of cell clones within the population). Strikingly, introduction of an HIV-based vector that encoded a similar hCMV IE-driven GFP reporter gene (32) produced a more intense and uniform peak of GFP-expressing cells (Fig. 1A); after several additional days of culture, these intense cells became the predominant form (not shown) (16). The resulting characteristic GFP intensity profiles produced by these ASV- and HIV-based vectors remained fairly constant during long-term passage of these cell populations (not shown). The distinct GFP profiles observed with the ASV- and HIV-based vectors suggest that the epigenetic effects are not simply a response to the heterologous GFP gene (15).
Evidence for epigenetic effects on ASV-based vector GFP reporter gene expression.
We hypothesized that the broad range of GFP intensities observed in the population of ASV-based vector-infected cells was due to either (i) outgrowth of cell clones with characteristic GFP expression levels, (ii) positional variegation of GFP expression within cell clones, (iii) cell cycle effects on GFP expression, (iv) cells which represent intermediates in GFP silencing, or (v) a combination of these features. Furthermore, because of the broad expression range, we suspected that a fraction of the GFP(−) cells contained silent GFP reporter genes. To test these hypotheses, infected cultures containing ca. 30% GFP(+) cells were preparatively sorted at 2 weeks postinfection, and single GFP(+)-positive cells were deposited robotically in each well of a 96-well dish (Fig. 1B). Inspection of the wells immediately and at 1 day postplating revealed that ca. 30 wells received single, viable cells. Eleven single-cell-derived GFP(+) cell clones were ultimately expanded from these wells and were designated AP1 through AP11. By microscopy, five of the clones (including AP6, AP10, and AP11) contained apparent mixtures of bright and dim GFP(+) cells (designated variegated [V] clones), while six clones (including AP2 and AP4) displayed more intense and uniform GFP expression (designated uniform [U] clones).
We devised a strategy to follow development of the variegated patterns during outgrowth of single cells into colonies. Cultures from the GFP(+) cell pool were treated briefly with nocodazole to enrich for mitotic cells, and a mitotic shakeoff was performed. Mitotic cells were plated under dilute conditions, and they entered G1 with 70 to 90% synchrony, producing cell doublets (Fig. 2A). This method ensures that the majority of adjacent cells are daughters that will grow synchronously over several cell divisions; the synchronous growth also minimizes potential cell cycle effects on GFP intensity. Microclones that were derived from the GFP(+) population were analyzed at the four- and eight-cell stages. As shown in Fig. 2B, we observed two predominant GFP intensity patterns during outgrowth of these clones: uniform GFP cell-to-cell intensity within each colony, or variegated GFP intensities (both shown at the eight-cell stage). The variegated pattern was typically characterized by four bright cells and four dim cells (Fig. 2B, left panels), and the sectored pattern implies that the bright and dim cells were derived from single progenitors at the two-cell stage, respectively. When such colonies became larger, GFP expression was detected throughout the colony, and a classical variegated pattern was prominent (Fig. 2B, right panels). Typically, sectoring of adjacent cells of similar intensity could be observed. We interpret these results to indicate that these variegated patterns in cell clones were due to orderly epigenetic-based oscillations of GFP expression (positional variegation).
FIG. 2.

Variegation of GFP expression during outgrowth of GFP(+) microclones. (A) A mitotic shakeoff strategy was used to promote highly synchronized outgrowth of microclones from single cells. Shown is a single field, imaged ca. 2 h after plating mitotic cells. Typically, synchronization occurred with 70% to 90% efficiency. Boxed colonies have completed cytokinesis synchronously and entered G1 as indicated by the cell doublets. (B) GFP patterns were monitored during colony outgrowth. Left panels show eight-cell colonies, with the upper panel illustrating a colony displaying uniform GFP expression and the middle and bottom panels highlighting variegated colonies. Right panels show a representative variegated pattern in a large colony. Phase-contrast and fluorescent images are shown. (C) Analysis of GFP variegation during outgrowth of synchronous microclones derived from GFP(+) cell clones. One uniform (U) (AP2) and one variegating (V) (AP6) clone were examined. Representative images are shown. Arrows indicate cells displaying higher-intensity GFP expression.
To investigate this phenomenon further, we performed similar experiments with two GFP(+) cell clones, AP2 (U) and AP6 (V) (Fig. 2C). Here, we examined two-cell colonies for evidence of oscillation. In the case of the AP6 V clone, we observed frequent cell doublets in which one cell was very bright and one cell was dim; in contrast, the U clone typically produced cell doublets with similar GFP intensities. In the V clone, the substantial differences in GFP expression between the two daughter cells could more readily be accounted for by derepression of GFP in the bright daughter cell, as opposed to silencing, GFP dilution, and turnover in the dim cell. We conclude, therefore, that a fraction of HeLa cells contain integrated viral genomes that are prone to epigenetic variegation, and one possible interpretation is that such variegation is the result of oscillation between repressed and derepressed states.
Evidence for high-frequency epigenetic silencing of the ASV vector in HeLa cells.
We next asked if the epigenetic effects on GFP reporter gene expression could include complete silencing. If so, it would be expected that in addition to uninfected cells, the GFP(−) population would include cells that contain integrated viral DNA. Cellular DNA was prepared from GFP(+) and GFP(−) populations in which the amount of infecting virus had been adjusted to produce ca. 20 to 35% GFP-positive cells, to minimized the fraction of cells that harbor multiple copies of integrated DNA (see Fig. 1B for a representative FACS profile). GFP DNA could be readily detected in GFP(−) cells by semiquantitative PCR (not shown), indicating that these cells contained epigenetically silent viral DNA.
The average copy numbers of GFP DNA in the GFP(−) and GFP(+) populations were next measured by qPCR (Table 1). To facilitate quantitation, a single-copy GFP reporter gene standard was calibrated to the cellular albumin gene. As expected, qPCR analysis using two independent GFP(+) cell populations indicated that these sorted cells contained ca. 1 copy of GFP DNA on average. Analysis of the GFP(−) cells from a starting population containing 35% GFP(+) cells revealed an average GFP copy number of ca. 0.3, confirming that a significant fraction of cells contained silent viral genomes.
From these experiments, we can estimate the fraction of ASV integration events that result in silencing of GFP. As the initial conditions for infection produced approximately 35% GFP(+) cells, a copy number of 0.3 in the GFP(−) population indicates that the distribution in the starting culture was ca. 35% GFP(+), 20% GFP silent, and 45% uninfected.
Induction of GFP expression by trichostatin A.
Variegation of GFP expression in the ASV vector-infected population could be observed as early as 2 days postinfection, and vector-containing silent cells could be identified by 7 to 10 days postinfection. These rapid epigenetic changes were consistent with our previous findings that HDACs, key regulators of epigenetic silencing, can associate with ASV DNA soon after infection of HeLa cells (22). HDACs act on chromatin by removing acetyl groups from histone tails, thereby promoting transcriptional repression. Treatment of cells with HDIs results in increased global histone acetylation and thus derepresses the subset of cellular genes (several percent) that are normally repressed by HDACs.
To determine if HDACs have a role in maintaining silencing of the GFP reporter genes, we treated GFP(−) cell populations with a known HDI. For these experiments, GFP(−) cells were sorted from infected cultures that contained 20 to 30% GFP(+) cells. The sorted GFP(−) cells were then treated with TSA, an HDI frequently reported to activate epigenetically silenced host cell and reporter genes. TSA treatment resulted in a significant increase in total histone H4 acetylation within 3 to 4 h (data not shown). We observed that TSA treatment induced GFP expression in the GFP(−) population, in a dose-dependent manner as determined by FACS analysis (Fig. 3A). Western blot analysis (not shown) verified that GFP protein was initially undetectable in these GFP(−) cells but accumulated after TSA treatment. Quantitation by FACS analysis indicated that GFP expression was induced in ca. 10% of the GFP(−) cells. This percentage was lower than that found to contain the GFP gene by qPCR (ca. 30%). We considered that either some ASV genomes are silenced by a mechanism that is nonresponsive to TSA treatment, the treatment was not optimal, or the response was limited by other cellular processes (see below).
FIG. 3.

Reactivation of GFP reporter genes after treatment with HDIs. (A) Cells were sorted at 8 days postinfection, and the GFP(−) cells were passaged for several months. GFP(−) cells were treated with the indicated concentrations of TSA, and GFP expression was quantitated by FACS at 24 h posttreatment. (B) TI-C cultures that were passaged for several months were challenged with the indicated HDIs, and GFP expression was quantitated by FACS at 24 h posttreatment. con, not treated. Concentrations: dimethyl sulfoxide (DMSO), 0.05% and 0.2%; TSA, 0.5, 1, and 2 μM; apidicin, 0.5, 1, and 2 μg/ml; valproic acid (VPA), 2, 4, and 8 mM; sodium butyrate (NaBut), 2, 4, and 8 mM. (C) Northern blot analysis of GFP mRNA was carried out using standard methods. GFP mRNA was characterized in a pool of GFP(+) HeLa cells infected with the ASV construct in which the GFP gene is under control of the hCMV IE promoter (left panel). RNA loading was monitored by staining of 18S and 28S RNAs, and these species also served as sizing standards (5,025 and 1,868 nucleotides, respectively). A GFP transcript of ca. 1,700 nucleotides was identified, and this size is consistent with initiation within the internal hCMV viral promoter and 3′ processing at the 3′ LTR. This transcript was not detected in TI-C cells but was induced to significant levels after treatment with TSA (1 μM) (right panel).
To determine if TSA-induced GFP expression was heritable over many cell divisions, the drug was removed after 24 h and cultures were propagated and observed. Significant loss of GFP expression was observed over a period of ca. 1 week (not shown). As described below in detail, we determined by cell sorting that this loss was not due to inviability of cells in which GFP expression was induced. We conclude, therefore, that although treatment with the HDI was sufficient to activate expression, a remaining epigenetic signature could mediate resilencing.
Enrichment for cell populations in which retroviral silencing is controlled by HDACs.
The finding that withdrawal of TSA resulted in resilencing of GFP expression in a subset of cells allowed us to enrich for cells in which the ASV-GFP reporter gene silencing is regulated by HDACs, as follows. A GFP(−) cell population was sorted from a starting culture which contained ca. 20% GFP(+) cells. This GFP(−) population was treated with TSA, and the resulting subpopulation of GFP-expressing cells was isolated by cell sorting. Continued culturing of this population over ca. 10 days in the absence of TSA resulted in the progressive loss of GFP expression in a large percentage of the cells, as expected. Residual GFP-expressing cells were then removed by sorting. The resulting GFP(−) subpopulation was then passaged for various times and rechallenged with TSA. As shown in Fig. 3B, we observed significant TSA-inducible GFP reactivation even after long-term passage of these cells. The extent of the response never approached 100% (see below) and varied from experiment to experiment (ca. 30% to 60% [data not shown]). We have designated this enriched population TI-C cells (for TSA-inducible, hCMV IE-driven GFP). Using the method described in Table 1, the average GFP reporter gene copy number in the TI-C population was determined to be 0.8 ± 0.2. As shown in Fig. 3B, treatment of these cells with chemically diverse HDIs (sodium butyrate, apidicin, and valproic acid) also induced GFP expression.
To confirm that GFP protein expression was mediated by a GFP mRNA that initiated in the integrated viral vector DNA, Northern blot analysis was performed with total cell RNA isolated from untreated and TSA-treated TI-C cell populations. As shown in Fig. 3C, TSA treatment resulted in the appearance of an RNA transcript of the size expected if transcriptional initiation occurred at the internal hCMV IE promoter, and 3′-end processing was directed by the viral 3′ LTR. This analysis revealed that repression and reactivation by TSA were tightly regulated at the level of transcription. We note that an LTR-driven RNA transcript corresponding to full-length vector RNA was not readily detectable. This was not unexpected, as we have found independently using an LTR-driven GFP vector, that full-length viral RNA is difficult to detect in this system, possibly due to the weaker LTR promoter and excessive splicing of ASV RNA in mammalian cells (3).
Evidence for HDAC-mediated repression in GFP(+) cells.
As illustrated in Fig. 1 and 2, a broad range of GFP intensities as well as positional variegation of GFP expression was observed in the GFP(+) population of HeLa cells infected with the ASV-GFP vector. To determine if these effects could also be modulated by cellular HDACs, GFP(+) cell populations and clones were treated with TSA. A dramatic increase in GFP intensity in the GFP(+) cell population was observed following such treatment (data not shown). Similarly, treatment of representative GFP(+) V cell clones, in which GFP intensity was initially weak and broadly distributed, resulted in more uniform and intense GFP expression (Fig. 4, AP6, AP10, and AP11). Northern blot analysis confirmed an increase in GFP mRNA in GFP(+) cells following TSA treatment (not shown). We concluded, therefore, that TSA treatment derepresses GFP expression in GFP(+) cells. Although our qPCR analysis indicated that the average GFP copy number in GFP(+) cells was close to 1, we could not immediately exclude the possibility that the increase in GFP expression was the result of activation of secondary, silent GFP reporter genes that were present in GPF(+) cells. However, we note that TSA treatment caused a partial or complete shift in GFP intensity patterns from weak and broad to more uniform and intense. As the distribution of GFP intensity changes in response to TSA treatment, such patterns are inconsistent with the activation of secondary, silent GFP genes. To further address this issue, we used a linker-mediated PCR-based strategy to amplify host-virus junctions from one clone that displayed broad GFP expression (AP6); only a single integrated viral DNA was detected (not shown). TSA treatment of the representative GFP U clones, which display more uniform and stronger GFP expression, produced little change in GFP intensity (Fig. 4, AP2 and AP4). Furthermore, the small shifts in fluorescence intensities observed are likely due to TSA-induced changes in cell morphology, as treatment of uninfected HeLa cells with TSA produced a similar shift in the autofluorescence signal (Fig. 4).
FIG. 4.

FACS analysis of GFP(+) cell clones after treatment with TSA. Eleven GFP(+) clones were categorized as variegated (V) or uniform (U) by microscopy. Analyses of several representative clones (V clones, AP6, AP10, and AP11; U clones, AP2 and AP4) are presented. Cultures were treated with 0.5 μM TSA for 24 h and analyzed by FACS. Nonspecific effects were monitored by treatment of uninfected HeLa cells (HeLa). Data were processed with FlowJo software. Untreated, no fill; TSA treated, filled.
From the results described in Fig. 4 we conclude that in GFP(+) U clones, GFP reporter expression is fully derepressed. In contrast, in GFP(+) V clones, reporter expression is subject to HDAC-mediated repression. We note that the GFP intensity profiles in U clones are similar to the profiles obtained after treatment of V clones with TSA (using the same GFP intensity scale). Therefore, our analysis of the GFP(+) V clones reveals a role for HDACs in epigenetic repression, and analysis of the U clone identifies a situation in which the reporter gene is highly resistant to HDAC-mediated repression (see Discussion and the model in Fig. 9). Although we believe that these results are representative, we cannot rule out the presence of multiple vector DNAs in some clones; multiple integrations would produce a bias whereby high-level expression of GFP from one vector could obscure detection of a weaker, variegated GFP phenotype produced by a second integrated vector.
FIG. 9.

Models for epigenetic silencing and repression. (A) Model for a continuum of HDAC-mediated repression and silencing in GFP(−) and GFP(+) cells. The model is based on the broad GFP expression profiles and the stimulation of GFP expression in both GFP(−) and GFP(+) variegated (V) clones by TSA. Filled arrowheads indicate strong (large arrowhead) or weak (smaller arrowheads) HDAC effects that are modulated by the integration site. (B) Model showing interaction between Daxx/HDAC and integrated retroviral DNA (22). TI cells were selected for the ability of the GFP reporter gene to be reactivated by TSA, an HDI. In such selected cells, phorbol esters can also reactivate the GFP reporter gene, suggesting that the silent loci can be available to transcription factors (rectangle). Coactivator complexes containing HATs may overcome or displace HDACs. (C) The model indicates a common mechanism underlying two phenomena: variegation in some GFP(+) clones and variegated reactivation in GFP(−) TI cells. Expression could be restricted in one daughter cell due to transient inaccessibility of the integration site locus to transcriptional centers (48).
Promoter-independent silencing of the GFP reporter gene.
The ASV-based vector described above utilizes the strong hCMV IE promoter to drive GFP expression. To evaluate the contribution of the promoter to the observed silencing phenomena, we tested two other ASV vectors in which the GFP reporter gene was driven either by the native LTR promoter or a human cellular promoter derived from the EF-1 alpha gene. The latter promoter is reported to drive persistent reporter gene expression and has been exploited in vector design (21, 54). In the LTR-driven construct, the GFP gene is in the position of the viral v-src gene and expressed through a spliced mRNA, while the EF-1 alpha promoter replaces the internal hCMV IE promoter. HeLa cells were infected with these vectors under conditions that produced ca. 20% GFP(+) cells. GFP(−) cells were sorted from infected populations and treated with TSA. As observed previously with the hCMV IE-driven GFP vector, GFP expression could be activated in a subset of these GFP(−) cells. We then enriched for cells in which GFP could be reactivated by TSA, and these populations were designated TI-L and TI-E, corresponding to the LTR and EF-1 alpha promoters, respectively. Rechallenge of these cells with TSA resulted in robust GFP activation (Fig. 5), indicating that the HDAC-mediated silencing that we have described for ASV is not restricted to reporter gene expression that is initiated from the hCMV IE promoter.
FIG. 5.

Characterization of cell populations enriched for TSA-inducible hCMV IE-, ASV LTR-, and EF1 alpha-driven GFP expression. The indicated cell populations were treated with TSA (1 μM) for 24 h, and GFP expression was quantitated by FACS analysis. TI-C, hCMV promoter; TI-L, ASV LTR promoter; TI-E, EF-1α promoter.
Reactivation of the HDAC-repressed GFP reporter gene by prostratin is promoter specific.
HIV-1 postintegration latency is an epigenetic phenomenon. It has been demonstrated that HDAC inhibitors can activate silent HIV, and such treatment may be useful as part of a combined therapy to eliminate latently infected cells (34, 36, 64). Prostratin is a phorbol ester compound that has similar potential (35) and is able to activate latent HIV-1 in cultured cells through stimulation of the NF-κB pathway (Fig. 6A) (62). To identify factors or pathways that might cooperate with HDACs to maintain silencing, we tested a variety of compounds for their abilities to reactivate GFP expression in the TI-C and TI-L cultures. We found that treatment with prostratin, as well as another phorbol ester, PMA, resulted in robust GFP reactivation in the TI-C population (Fig. 6B). Therefore, although these cells were selected for HDAC-mediated silencing, direct inhibition of HDAC activity per se is not necessary for reactivation; rather, stimulation of the hCMV IE promoter with phorbol esters is apparently sufficient (11). As prostratin stimulates the NF-κB pathway (62), we hypothesized that the ability of this compound to reactivate the silent GFP reporter gene was mediated by NF-κB sites in the hCMV IE promoter. The ASV LTR does not contain known NF-κB sites, and, consistent with the hypothesis, treatment of TI-L cells with prostratin did not result in reactivation of GFP (Fig. 6C). However, treatment of TI-C cells with TNF-α, which stimulates the NF-κB pathway in HeLa cells, failed to reactivate GFP expression (Fig. 6D). As a positive control, we confirmed the ability of TNF-α to induce HIV expression in the model cell system described for Fig. 6A, as well as to trigger apoptosis in the TI-C culture in the presence of cycloheximide (data not shown). We conclude that the abilities of prostratin and PMA to activate hCMV IE-driven GFP expression in this system may be dependent on several response elements in the promoter/enhancer (6, 11). These results suggest that silent retroviral DNAs are accessible to the general transcription machinery and that reactivation can be mediated by local HAT recruitment and/or displacement of HDACs.
FIG. 6.

Phorbol esters can reactivate silent GFP reporter gene expression in TSA-inducible TI-C but not TI-L populations. GFP expression in treated and untreated (con) cells was quantitated by FACS analysis at 24 h posttreatment. (A) A Jurkat cell clone (10.6) harboring latent HIV encoding a GFP reporter (30, 62) was treated with prostratin (2 μM). (B) TI-C cells were treated with prostratin (0.1, 0.3, 1.0, and 1.5 μM) or TSA (2 μM). (C) TI-L cells were treated with prostratin (0.3, 1.0, and 1.5 μM) or TSA (2 μM). (D) TI-C cells were treated with prostratin (2.0, 5.0 μM), PMA (100 and 200 nM), and TNF-α (10, 20 ng/ml).
Reactivation of an HDAC-repressed viral GFP reporter gene may be dictated by switching between responsive and nonresponsive states.
Although the TI-C populations were prepared by sorting cells in which GFP was activated in response to TSA treatment, we noted routinely that challenge with HDAC inhibitors or prostratin/PMA failed to reactivate GFP expression in all cells in the population. Several microscopy-based experiments were designed to determine the basis for this incomplete response. Single cells from the TI-C population were first plated at a high dilution, allowed to grow into large colonies, and then challenged with prostratin or TSA. We found that some cells within all of the colonies failed to reactivate, leading to variegated patterns of GFP expression (not shown). We hypothesized that this nonuniform response could be due to differences in the metabolic state of cells in the colony, nonclonal outgrowth (i.e., cross contamination of colonies), or switching between responsive and nonresponsive states.
To distinguish between these possibilities, we used an approach similar to that described above to follow variegation of GFP(+) cells (Fig. 2). Mitotic shakeoff was performed on TI-C cell cultures, and the cells were diluted and plated. After 1 to 2 h, the mitotic cells enter G1 in a highly synchronous manner (70 to 90%), with “colony birth” indicated by formation of cell doublets (Fig. 2A). Synchronous growth could frequently be followed to the four- and eight-cell stages. Cultures were then treated with TSA (or prostratin) at various times postplating. In this way, the reactivation patterns of individual cells in synchronous, clonal populations could be mapped. As TSA treatment causes HeLa cells to assume a spindle-like morphology and detach from the plate more readily than untreated cells, prostratin treatment was initially used to monitor patterns of reactivation. TI-C colonies were treated with prostratin ca. 16 h after colony birth, and GFP expression was then monitored in microclones after ca. 48 h, when eight-cell colonies were present (Fig. 7A). We observed a subset of GFP-expressing colonies in which the GFP signal was present in four out of the eight cells (variegated). To investigate this phenomenon further, TI-C colonies were treated with prostratin several hours after colony birth, and GFP expression was then monitored in microclones after ca. 24 h, when the majority had reached the four-cell stage (Fig. 7B). A significant percentage of colonies showed no response by microscopy, and this is likely a reflection of the fact that this method of scoring GFP expressing cells is less sensitive than FACS analysis. However, among 38 four-cell colonies that contained GFP-expressing cells, 12 colonies included four GFP-expressing cells (4/4, uniform), while 26 included only two GFP-expressing cells (2/4, variegated). Representative images are shown in Fig. 7B. Strikingly, none of the four-cell colonies contained either one or three GFP-expressing cells. Furthermore, the intensities of the two or four GFP-expressing cells within each colony were uniform and characteristic for each clone (Fig. 7B). As these cells were obtained from a cell population, this pattern would be consistent with a unique integration site in each microclone, with the differences in GFP intensity between clones being due to integration site position effects on expression. Overall, this variegated reactivation pattern suggested a binary switch, whereby cells could cycle between responsive and nonresponsive states (see Discussion).
FIG. 7.

Analysis of GFP reactivation patterns during clonal outgrowth of TI-C microclones reveals a variegated response. TI-C cell populations were synchronized by mitotic shakeoff and plated under dilute conditions. Colonies were treated at various times postplating with prostratin, and fluorescence (right) and phase-contrast (left) images were acquired as indicated in the diagrams: black fill, nonexpressing cells; gray fill, GFP-expressing cells. (A) Treatment at the two-cell stage resulted in variegated (V) as well as uniform (U) GFP expression at the eight-cell stage. Shown is a representative colony displaying variegated expression. (B) The two-cell colonies were treated with prostratin at 2 h postplating, and colonies were imaged at the four-cell stage (boxed). Variegated and uniform expression patterns were observed. The top panel shows neighboring V- and U-type colonies, and the bottom panel shows a V-type colony. Cell-to-cell intensity of GFP expression is constant within each clone (boxed). (C) Treatment was similar to that for panel B, except that aphidicolin was included to prevent cells from progressing into S phase. Colonies were arrested at the two-cell stage and could be quantitated according to V (1/2) or U (2/2) patterns (see Fig. 8). Representative images are shown.
To test this idea further and in a more quantitative manner, we asked what was the probability of a single cell giving rise to one versus two responsive cells. We incorporated a cell cycle arrest step, in which the G1 cell doublets were treated with prostratin plus aphidicolin to prevent entry into S phase and further cell divisions. This protocol resulted in persistence of G1 cell doublets that could be scored for one out of two (1/2, variegated) or two out of two (2/2, uniform) cells in which GFP expression was reactivated (Fig. 7C). The results showed a distribution of 1/2 (variegated) and 2/2 (uniform) GFP(+) cell doublets among those that expressed GFP (Fig. 8A). These patterns were maintained at 24 and 48 h postplating, indicating that GFP reactivation was not simply lagging in one cell in the variegated doublets. This general pattern could be reproduced using TSA with both the TI-C and TI-L cells, indicating that this switching effect is not dependent on the CMV promoter or the chemical inducer (Fig. 8B). The frequency of this asymmetric response is much higher that than that expected for chromosome loss; furthermore, loss of the GFP gene at the observed rate would result in a severe and cumulative loss of response to inducers in the population, which we have not observed. To directly distinguish between loss of the GFP gene and epigenetic effects, we allowed colonies to grow to a large size (30 to 50 cells) before treatment with TSA or prostratin. We typically observed several sectors of GFP reactivation in these colonies, suggesting that the inability to respond to inducers was mediated by reversible epigenetic restrictions.
FIG. 8.

Quantitation of variegated (V) and uniform (U) GFP expression in two-cell colonies after aphidicolin arrest and reactivation with prostratin (1 μM) or TSA (0.5 μM). The experimental design is shown in Fig. 7C, and representative results are shown. (A) TI-C 2-cell colonies were quantitated for V or U patterns at 24 and 48 h after plating and prostratin treatment. (B) Comparison of TI-C and TI-L colonies induced with TSA. The sample numbers are lower with TI-L cells, as LTR-driven GFP expression is generally weaker and thus more difficult to detect by microscopy.
From these experiments, we conclude that reactivation can be restricted by additional epigenetic mechanisms. Furthermore, as cells were plated in M phase and aphidicolin treatment blocked entry into S phase, these microclone experiments established that reactivation could occur entirely within G1.
DISCUSSION
Integration of retroviral DNA establishes a stable association with the host genome but does not ensure viral gene expression. Over the last two decades, numerous studies have addressed the role of viral and host determinants in retroviral epigenetic silencing, with major focus on the behavior of retrovirus-based vectors in stem cells. Here we describe high-frequency HDAC-mediated epigenetic silencing and repression of an ASV-based vector in HeLa cells. We believe that this behavior reflects specific host-virus interactions, as we found that the hCMV IE GFP cassette is highly resistant to silencing and repression when introduced into HeLa cells with an HIV-1-based vector (Fig. 1 and data not shown).
Based on our current and previous findings, we can outline several ways in which host-virus interactions could influence silencing initiation, maintenance, and reactivation (Fig. 9). Although we have observed variations in GFP reporter gene expression that could be attributed to position affects and positional variegation, we also found that a large fraction of integrated viral DNAs are subject to epigenetic repression or silencing, as indicated by the response to HDIs (Fig. 9A). This feature is illustrated by the finding that GFP intensities in some GFP(+) cell clones increased and became more uniform after TSA treatment and that a large fraction of GFP(−) cells harbor silent, TSA-inducible viral GFP reporter genes. In contrast, some GFP(+) clones displayed more intense and uniform GFP expression which was unaffected by TSA treatment. These findings suggest the existence of a broad continuum of HDAC-mediated epigenetic effects of varying severity (Fig. 9A). The pervasiveness of the HDAC-mediated repression and silencing in this ASV system suggests that a particular cue (cis-acting viral signal or cell-virus interaction) may trigger this response.
We and others have previously characterized the integration site selection preferences for ASV DNA in HeLa cells and have found that protein-coding genes are preferred modestly as integration targets (4, 47). As a large fraction of integrated genomes are subject to either epigenetic repression or silencing, it is unlikely that the integration site locus is the sole determinant of the epigenetic fate of the integrated DNA. Furthermore, we have found that integration site selection patterns in GFP(+) and GFP silent TI cell populations are similar (not shown). These findings disfavor a model in which repression and silencing are due to integration in or near constitutive heterochromatin. It will be important to characterize integration sites and GFP expression in individual cell clones to substantiate this interpretation. Overall, the high-frequency and TSA-sensitive repression and silencing described here are consistent with our previous findings using chromatin immunoprecipitation, which showed that ASV DNA complexes associate with the transcriptional repressor Daxx and HDACs soon after infection (22) (Fig. 9B). Daxx is known to interact with HDACs (26) and is therefore an attractive candidate for initiating these events.
Based on our collective findings and the interpretations discussed above, our current model is that HDACs associate with ASV DNA as part of an antiviral response (22). HDACs have also been implicated as part of an intrinsic cellular response that inhibits hCMV infection (52, 55), and HDIs have been shown to stimulate expression of a variety of viral genomes (38, 43, 46, 67). Interestingly, the avian adenovirus protein Gam1 binds to and inhibits HDAC1 and may represent a virus-encoded HDAC inhibitor that serves to counter the antiviral effects of HDACs (10, 13, 38). Similarly, the hCMV pp71 protein apparently acts to relieve Daxx-mediated transcriptional repression of hCMV gene expression (5, 52, 55).
A correlation between DNA methylation and retroviral silencing is well established, and some studies have provided strong evidence for a causal relationship (58, 59). We found that in our experimental system, HDAC inhibitors are sufficient to reactivate silent viral DNA in a promoter-independent fashion, and the DNA methyltransferase inhibitor 5-azacytidine has only marginal and temporal-specific effects (data not shown). Thus, the phenomenon that we have described appears to be an example of DNA methylation-independent, HDAC-mediated silencing (50). We hypothesize that the repressive action of HDACs is mediated through removal of acetyl groups of histone tails, minimally within the silent promoter regions, as has been demonstrated previously for the hCMV promoter (8). However, we cannot rule out that reactivation by HDIs is mediated through an indirect mechanism.
Our studies have also shown that the phorbol esters prostratin and PMA can reactivate the silenced GFP reporter gene and that this effect is promoter specific (i.e., occurs in TI-C but not TI-L cells). The hCMV IE promoter can be activated by phorbol esters, likely through stimulation of several transcription factors that target this promoter (6). These transcription factors may cause dissociation of HDACs and/or promote recruitment of HAT coactivator complexes (Fig. 9B). Our results suggest that reactivation does not require global inhibition of HDAC activities but rather that local, promoter-specific antagonism of HDACs is sufficient (Fig. 9B) and that the integrated vector DNAs can be accessible to transcription factors. These results are incompatible with a model in which silencing is driven by integration into inaccessible heterochromatin.
A major and novel finding of this study relates to the mechanism of reactivation from silencing. We used a sorting strategy to enrich for a cell population in which the GFP reporter gene was silent but could be reactivated by TSA (TI cells). Despite this selection, we found that rechallenge of asynchronous TI cell populations with TSA induced GFP expression in only a subset of cells. By following reactivation in synchronous microclones, we found that variegated patterns of GFP reactivation, where detected, appeared to be due to orderly “switching” between HDI-responsive and nonresponsive states (Fig. 9C). The time scale of our experiments (e.g., four-cell colonies) apparently favors the detection of a single switching event, manifested as 2/4 GFP-expressing cells. Analysis of eight-cell colonies occasionally revealed a 2/8 GFP(+) sectored pattern, suggesting that “switching” occurred during the second synchronous division. Furthermore, treatment of larger colonies resulted in a sectored pattern featuring clusters of GFP(+) cells (not shown).
One interpretation of these results is that a dominant, epigenetic phenomenon, which imparts HDI resistance, mediates this variegated response. We speculate that this switching is a manifestation of temporal variations in transcriptional competence of the integrated DNA, as has been observed for cellular genes and introduced reporter genes (2, 48, 53), and such effects may be more readily observed in our system due to the fact that the viral locus is haploid. In support of this interpretation, we note that the characteristic variegated patterns of drug-induced GFP expression in microclones are similar to the patterns of GFP expression observed during outgrowth of some GFP(+) microclones (e.g., compare Fig. 2B left middle panel and 7A; Fig. 9C). Taken together, the patterns suggest that depending on the integration site, the viral loci may be only transiently accessible for transcription (2, 48, 53), resulting in variegated expression. In some GFP(−) TI cell microclones, interrogation with the HDIs or activators reveals this variegated response, which we interpret as being the result of underlying “on-off switching” of transcriptional competence; GFP sectoring after challenge would be expected if such states are epigenetically heritable over several cell divisions. We suggest, therefore, that transient, locus-specific transcriptional inaccessibility accounts for both the variegated GFP reactivation observed after HDI or phorbol ester treatment and the variegated expression in GFP(+) colonies. Overall, these studies have apparently uncovered an important variable that can contribute to the effectiveness of HDAC inhibitors. Furthermore, our results may explain the transcriptional oscillations of some viral reporter genes, as well as the nonuniform reactivation of silent retroviral DNA observed in other systems (20, 62). In the course of these microscopy-based studies, we found that reactivation could occur in synchronized cells that were arrested at the G1-S border of the cell cycle (Fig. 7C). We have confirmed this result using FACS analysis (data not shown). This finding suggests that reactivation can occur through local HAT-mediated acetylation of preexisting nucleosomes, rather than through DNA replication-coupled deposition of acetylated histones (60).
In summary, we report high-frequency epigenetic silencing and repression of retroviral DNA expression. Our studies reveal that such silencing is rapid and is likely mediated by HDACs that associate with viral DNA early after infection. The interpretation of responses to chemical treatments suggests that reactivation can occur either through inhibition of HDACs or by local recruitment of transcriptional activators. Overall, our results suggest that such silencing and repression can occur independently of the integration site but that the location can influence the degree of repressive effects as well as the ability to respond to reactivating stimuli.
Acknowledgments
This work was supported by National Institutes of Health grants CA71515 and CA06927 and also by an appropriation from the Commonwealth of Pennsylvania. Support for R.A.K. was funded in part under a grant with the Pennsylvania Department of Health.
The following Fox Chase Cancer Center Shared Facilities were used in the course of this work: the Automated DNA Sequencing Facility, the Flow Cytometry and Cell Sorting Facility, and The Fannie E. Rippel Biochemistry and Biotechnology Facility.
We thank Joe Ramcharan for providing the ASV-EF-1 alpha GFP vector and Yanacha Toporovskaya, Elizabeth Farley, Kim Boland, and Paul Garr for technical assistance. We also thank Konstantin Taganov for analyses of integrated ASV-GFP vectors and helpful discussions. We are also grateful to Eric Verdin for providing HIV-GFP-infected Jurkat cell clones. We thank Severin Gudima and Jared Evans for critical comments on the manuscript and Marie Estes for assistance in preparing the document.
The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or any other sponsoring organization. The Pennsylvania Department of Health specifically disclaims responsibility for any analyses, interpretations, or conclusions.
Footnotes
Published ahead of print on 3 January 2007.
REFERENCES
- 1.Barsov, E. V., and S. H. Hughes. 1996. Gene transfer into mammalian cells by a Rous sarcoma virus-based retroviral vector with the host range of the amphotropic murine leukemia virus. J. Virol. 70:3922-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baur, J. A., J. W. Shay, and W. E. Wright. 2004. Spontaneous reactivation of a silent telomeric transgene in a human cell line. Chromosoma 112:240-246. [DOI] [PubMed] [Google Scholar]
- 3.Berberich, S. L., M. Macias, L. Zhang, L. P. Turek, and C. M. Stoltzfus. 1990. Comparison of Rous sarcoma virus RNA processing in chicken and mouse fibroblasts: evidence for double-spliced RNA in nonpermissive mouse cells. J. Virol. 64:4313-4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bushman, F., M. Lewinski, A. Ciuffi, S. Barr, J. Leipzig, S. Hannenhalli, and C. Hoffmann. 2005. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 3:848-858. [DOI] [PubMed] [Google Scholar]
- 5.Cantrell, S. R., and W. A. Bresnahan. 2006. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80:6188-6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan, Y. J., C. J. Chiou, Q. Huang, and G. S. Hayward. 1996. Synergistic interactions between overlapping binding sites for the serum response factor and ELK-1 proteins mediate both basal enhancement and phorbol ester responsiveness of primate cytomegalovirus major immediate-early promoters in monocyte and T-lymphocyte cell types. J. Virol. 70:8590-8605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, W. Y., E. C. Bailey, S. L. McCune, J. Y. Dong, and T. M. Townes. 1997. Reactivation of silenced, virally transduced genes by inhibitors of histone deacetylase. Proc. Natl. Acad. Sci. USA 94:5798-5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, W. Y., and T. M. Townes. 2000. Molecular mechanism for silencing virally transduced genes involves histone deacetylation and chromatin condensation. Proc. Natl. Acad. Sci. USA 97:377-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherry, S. R., D. Biniszkiewicz, L. van Parijs, D. Baltimore, and R. Jaenisch. 2000. Retroviral expression in embryonic stem cells and hematopoietic stem cells. Mol. Cell. Biol. 20:7419-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiocca, S., V. Kurtev, R. Colombo, R. Boggio, M. T. Sciurpi, G. Brosch, C. Seiser, G. F. Draetta, and M. Cotten. 2002. Histone deacetylase 1 inactivation by an adenovirus early gene product. Curr. Biol. 12:594-598. [DOI] [PubMed] [Google Scholar]
- 11.Clesham, G. J., H. Browne, S. Efstathiou, and P. L. Weissberg. 1996. Enhancer stimulation unmasks latent gene transfer after adenovirus-mediated gene delivery into human vascular smooth muscle cells. Circ. Res. 79:1188-1195. [DOI] [PubMed] [Google Scholar]
- 12.Coffin, J. M., S. H. Hughes, and H. Varmus. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY. [PubMed]
- 13.Colombo, R., R. Boggio, C. Seiser, G. F. Draetta, and S. Chiocca. 2002. The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep. 3:1062-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coull, J. J., F. Romerio, J. M. Sun, J. L. Volker, K. M. Galvin, J. R. Davie, Y. Shi, U. Hansen, and D. M. Margolis. 2000. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 74:6790-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalle, B., J. E. Rubin, O. Alkan, T. Sukonnik, P. Pasceri, S. Yao, R. Pawliuk, P. Leboulch, and J. Ellis. 2005. eGFP reporter genes silence LCRbeta-globin transgene expression via CpG dinucleotides. Mol. Ther. 11:591-599. [DOI] [PubMed] [Google Scholar]
- 16.Daniel, R., J. G. Greger, R. A. Katz, K. D. Taganov, X. Wu, J. C. Kappes, and A. M. Skalka. 2004. Evidence that stable retroviral transduction and cell survival following DNA integration depend on components of the nonhomologous end joining repair pathway. J. Virol. 78:8573-8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eissenberg, J. C., and L. L. Wallrath. 2003. Heterochromatin, position effects, and the genetic dissection of chromatin. Prog. Nucleic Acid Res. Mol. Biol. 74:275-299. [DOI] [PubMed] [Google Scholar]
- 18.Ellis, J. 2005. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 16:1241-1246. [DOI] [PubMed] [Google Scholar]
- 19.Ellis, J., and S. Yao. 2005. Retrovirus silencing and vector design: relevance to normal and cancer stem cells? Curr. Gene Ther. 5:367-373. [DOI] [PubMed] [Google Scholar]
- 20.Feng, Y. Q., M. C. Lorincz, S. Fiering, J. M. Greally, and E. E. Bouhassira. 2001. Position effects are influenced by the orientation of a transgene with respect to flanking chromatin. Mol. Cell. Biol. 21:298-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gill, D. R., S. E. Smyth, C. A. Goddard, I. A. Pringle, C. F. Higgins, W. H. Colledge, and S. C. Hyde. 2001. Increased persistence of lung gene expression using plasmids containing the ubiquitin C or elongation factor 1 alpha promoter. Gene Ther. 8:1539-1546. [DOI] [PubMed] [Google Scholar]
- 22.Greger, J. G., R. A. Katz, A. M. Ishov, G. G. Maul, and A. M. Skalka. 2005. The cellular protein daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J. Virol. 79:4610-4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He, G., and D. M. Margolis. 2002. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell. Biol. 22:2965-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He, J., Q. Yang, and L. J. Chang. 2005. Dynamic DNA methylation and histone modifications contribute to lentiviral transgene silencing in murine embryonic carcinoma cells. J. Virol. 79:13497-13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hejnar, J., P. Hajkova, J. Plachy, D. Elleder, V. Stepanets, and J. Svoboda. 2001. CpG island protects Rous sarcoma virus-derived vectors integrated into nonpermissive cells from DNA methylation and transcriptional suppression. Proc. Natl. Acad. Sci. USA 98:565-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hollenbach, A. D., C. J. McPherson, E. J. Mientjes, R. Iyengar, and G. Grosveld. 2002. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 115:3319-3330. [DOI] [PubMed] [Google Scholar]
- 27.Jaenisch, R., and A. Bird. 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33(Suppl.):245-254. [DOI] [PubMed] [Google Scholar]
- 28.Jahner, D., H. Stuhlmann, C. L. Stewart, K. Harbers, J. Lohler, I. Simon, and R. Jaenisch. 1982. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature 298:623-628. [DOI] [PubMed] [Google Scholar]
- 29.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science 293:1074-1080. [DOI] [PubMed] [Google Scholar]
- 30.Jordan, A., D. Bisgrove, and E. Verdin. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22:1868-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan, A., P. Defechereux, and E. Verdin. 2001. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 20:1726-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz, R. A., J. G. Greger, P. Boimel, and A. M. Skalka. 2003. Human immunodeficiency virus type 1 DNA nuclear import and integration are mitosis independent in cycling cells. J. Virol. 77:13412-13417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katz, R. A., J. G. Greger, K. Darby, P. Boimel, G. F. Rall, and A. M. Skalka. 2002. Transduction of interphase cells by avian sarcoma virus. J. Virol. 76:5422-54234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korin, Y. D., D. G. Brooks, S. Brown, A. Korotzer, and J. A. Zack. 2002. Effects of prostratin on T-cell activation and human immunodeficiency virus latency. J. Virol. 76:8118-8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulkosky, J., J. Sullivan, Y. Xu, E. Souder, D. H. Hamer, and R. J. Pomerantz. 2004. Expression of latent HAART-persistent HIV type 1 induced by novel cellular activating agents. AIDS Res. Hum. Retroviruses 20:497-505. [DOI] [PubMed] [Google Scholar]
- 36.Lehrman, G., I. B. Hogue, S. Palmer, C. Jennings, C. A. Spina, A. Wiegand, A. L. Landay, R. W. Coombs, D. D. Richman, J. W. Mellors, J. M. Coffin, R. J. Bosch, and D. M. Margolis. 2005. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet 366:549-555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewinski, M. K., D. Bisgrove, P. Shinn, H. Chen, C. Hoffmann, S. Hannenhalli, E. Verdin, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2005. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 79:6610-6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lieberman, P. M. 2006. Chromatin regulation of virus infection. Trends Microbiol. 14:132-140. [DOI] [PubMed] [Google Scholar]
- 39.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 40.Lorincz, M. C., D. Schubeler, S. C. Goeke, M. Walters, M. Groudine, and D. I. Martin. 2000. Dynamic analysis of proviral induction and de novo methylation: implications for a histone deacetylase-independent, methylation density-dependent mechanism of transcriptional repression. Mol. Cell. Biol. 20:842-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorincz, M. C., D. Schubeler, and M. Groudine. 2001. Methylation-mediated proviral silencing is associated with MeCP2 recruitment and localized histone H3 deacetylation. Mol. Cell. Biol. 21:7913-7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lorincz, M. C., D. Schubeler, S. R. Hutchinson, D. R. Dickerson, and M. Groudine. 2002. DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol. Cell. Biol. 22:7572-7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meier, J. L. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell, R. S., B. F. Beitzel, A. R. Schroder, P. Shinn, H. Chen, C. C. Berry, J. R. Ecker, and F. D. Bushman. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2:E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizutani, T., T. Ito, M. Nishina, N. Yamamichi, A. Watanabe, and H. Iba. 2002. Maintenance of integrated proviral gene expression requires Brm, a catalytic subunit of SWI/SNF complex. J. Biol. Chem. 277:15859-15864. [DOI] [PubMed] [Google Scholar]
- 46.Murphy, J. C., W. Fischle, E. Verdin, and J. H. Sinclair. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narezkina, A., K. D. Taganov, S. Litwin, R. Stoyanova, J. Hayashi, C. Seeger, A. M. Skalka, and R. A. Katz. 2004. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 78:11656-11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Osborne, C. S., L. Chakalova, K. E. Brown, D. Carter, A. Horton, E. Debrand, B. Goyenechea, J. A. Mitchell, S. Lopes, W. Reik, and P. Fraser. 2004. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36:1065-1071. [DOI] [PubMed] [Google Scholar]
- 49.Pannell, D., and J. Ellis. 2001. Silencing of gene expression: implications for design of retrovirus vectors. Rev. Med. Virol. 11:205-217. [DOI] [PubMed] [Google Scholar]
- 50.Pannell, D., C. S. Osborne, S. Yao, T. Sukonnik, P. Pasceri, A. Karaiskakis, M. Okano, E. Li, H. D. Lipshitz, and J. Ellis. 2000. Retrovirus vector silencing is de novo methylase independent and marked by a repressive histone code. EMBO J. 19:5884-5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pion, M., A. Jordan, A. Biancotto, F. Dequiedt, F. Gondois-Rey, S. Rondeau, R. Vigne, J. Hejnar, E. Verdin, and I. Hirsch. 2003. Transcriptional suppression of in vitro-integrated human immunodeficiency virus type 1 does not correlate with proviral DNA methylation. J. Virol. 77:4025-4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Preston, C. M., and M. J. Nicholl. 2006. Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87:1113-1121. [DOI] [PubMed] [Google Scholar]
- 53.Raj, A., C. S. Peskin, D. Tranchina, D. Y. Vargas, and S. Tyagi. 2006. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 4:e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramezani, A., T. S. Hawley, and R. G. Hawley. 2000. Lentiviral vectors for enhanced gene expression in human hematopoietic cells. Mol. Ther. 2:458-469. [DOI] [PubMed] [Google Scholar]
- 55.Saffert, R. T., and R. F. Kalejta. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863-3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schroder, A. R., P. Shinn, H. Chen, C. Berry, J. R. Ecker, and F. Bushman. 2002. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110:521-529. [DOI] [PubMed] [Google Scholar]
- 57.Svoboda, J., J. Hejnar, J. Geryk, D. Elleder, and Z. Vernerova. 2000. Retroviruses in foreign species and the problem of provirus silencing. Gene 261:181-188. [DOI] [PubMed] [Google Scholar]
- 58.Swindle, C. S., H. G. Kim, and C. A. Klug. 2004. Mutation of CpGs in the murine stem cell virus retroviral vector long terminal repeat represses silencing in embryonic stem cells. J. Biol. Chem. 279:34-41. [DOI] [PubMed] [Google Scholar]
- 59.Swindle, C. S., and C. A. Klug. 2002. Mechanisms that regulate silencing of gene expression from retroviral vectors. J. Hematother. Stem Cell Res. 11:449-456. [DOI] [PubMed] [Google Scholar]
- 60.Taddei, A., D. Roche, W. A. Bickmore, and G. Almouzni. 2005. The effects of histone deacetylase inhibitors on heterochromatin: implications for anticancer therapy? EMBO Rep. 6:520-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Lint, C., V. Quivy, D. Demonte, A. Chariot, C. Vanhulle, S. de Walque, G. Gaudray, E. Veithen, V. Bours, J. Piette, and A. Burny. 2004. Molecular mechanisms involved in HIV-1 transcriptional latency and reactivation: implications for the development of therapeutic strategies. Bull. Mem. Acad. R Med. Belg. 159:176-189. [PubMed] [Google Scholar]
- 62.Williams, S. A., L. F. Chen, H. Kwon, D. Fenard, D. Bisgrove, E. Verdin, and W. C. Greene. 2004. Prostratin antagonizes HIV latency by activating NF-kappaB. J. Biol. Chem. 279:42008-42017. [DOI] [PubMed] [Google Scholar]
- 63.Yao, S., T. Sukonnik, T. Kean, R. R. Bharadwaj, P. Pasceri, and J. Ellis. 2004. Retrovirus silencing, variegation, extinction, and memory are controlled by a dynamic interplay of multiple epigenetic modifications. Mol. Ther. 10:27-36. [DOI] [PubMed] [Google Scholar]
- 64.Ylisastigui, L., N. M. Archin, G. Lehrman, R. J. Bosch, and D. M. Margolis. 2004. Coaxing HIV-1 from resting CD4 T cells: histone deacetylase inhibition allows latent viral expression. AIDS 18:1101-1108. [DOI] [PubMed] [Google Scholar]
- 65.Yoder, J. A., C. P. Walsh, and T. H. Bestor. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13:335-340. [DOI] [PubMed] [Google Scholar]
- 66.Yoshida, M., A. Matsuyama, Y. Komatsu, and N. Nishino. 2003. From discovery to the coming generation of histone deacetylase inhibitors. Curr. Med. Chem. 10:2351-2358. [DOI] [PubMed] [Google Scholar]
- 67.Zhang, Y., and C. Jones. 2001. The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J. Virol. 75:9571-9578. [DOI] [PMC free article] [PubMed] [Google Scholar]