

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) persists as episomes in infected cells by circularizing at the terminal repeats (TRs). The KSHV episome carries multiple reiterated copies of the terminal repeat, and each copy is capable of supporting replication. Expression of the latency-associated nuclear antigen (LANA) is critical for the replication of TR-containing plasmids. A 32-bp sequence upstream of LANA binding site 1 (LBS1), referred to as RE (replication element), along with LANA binding sites 1 and 2 (RE-LBS1/2), is sufficient to support replication (J. Hu and R. Renne, J. Virol. 79:2637-2642, 2005). In this report we demonstrate that the minimal replicator element (RE-LBS1/2) replicates in synchrony with the host cellular DNA, and only once, in a cell-cycle-dependent manner. Overexpression of the mammalian replication inhibitor geminin blocked replication of the plasmid containing the minimal replicator element, confirming the involvement of the host cellular replication control mechanism, and prevented rereplication of the plasmid in the same cell cycle. Overexpression of Cdt1 also rescued the replicative ability of the RE-LBS1/2-containing plasmids. A chromatin immunoprecipitation assay performed using anti-origin recognition complex 2 (α-ORC2) and α-LANA antibodies from cells transfected with RE-LBS1/2, RE-LBS1, LBS1, or RE showed the association of ORC2 with the RE region. Expression of LANA increased the number of copies of chromatin-bound DNA of replication elements, suggesting that LANA is important for the recruitment of ORCs and may contribute to the stabilization of the replication protein complexes at the RE site.
Kaposi's sarcoma-associated herpesvirus (KSHV), discovered using a subtractive hybridization technique from the Kaposi sarcoma lesions, is also associated with at least two lymphoproliferative diseases, primary effusion lymphoma (PEL) and multicentric Castleman's disease (9-11, 17, 54, 59). KSHV persists as multicopy episomal DNA in infected cells with the expression of a small subset of genes (18, 34, 55, 65). Expression of the latency-associated nuclear antigen (LANA) is considered one of the crucial signatures of KSHV infection. Serum from KSHV-positive patients was initially used for the detection of LANA in infected cells (16, 32). LANA is a large nuclear protein detected in a punctate pattern in KSHV-infected cells (37, 48). LANA dots, which roughly correspond to the number of KSHV episomal copies, range from 15 to 120 per cell (13, 23). Detection of LANA and KSHV genomic DNA in an immunofluorescent in situ hybridization assay on chromosome spreads of KSHV-infected cells showed perfect colocalization of genomic DNA with LANA dots, suggesting the involvement of LANA in episomal tethering (13).
The role of LANA in the persistence of the KSHV genome was evaluated using the 33-kb left-end Z6 cosmid of KSHV in BJAB cells expressing LANA under G418 selection (4). Z6 cosmid DNA efficiently persisted in LANA-expressing cells, yielding outgrowth in 99% of the microtiter wells, whereas LANA-negative cells had significantly lower outgrowth (7% of the microtiter wells) (4). This suggested the presence of a replication element (RE) in the Z6 fragment of the KSHV genome. The binding of LANA to the terminal repeat (TR) was determined by using in vitro binding of the end-labeled KSHV genome fragment as well as gel shift assays (5, 14). The persistence of plasmids containing Z6 and its derivatives, including single copies and three copies of the TR, was evaluated by in situ lysis gel analysis, which showed maintenance of plasmids containing the TR element (5). LANA binding sites within the TR were mapped by coimmunoprecipitation of a small DNA fragment library as well as TR fragments with LANA and by a gel shift assay (5, 14). These results demonstrated that a minimum of 13 bp of TR sequence is required for binding to LANA in vitro (14). In a subsequent study, another LANA binding site, with lower affinity, referred to as LANA binding site 2 (LBS2), was detected in the terminal repeats (24). The previously identified LANA binding site, which lies between positions 571 and 589, is termed LANA binding sequence 1 (LBS1) (24, 36).
LANA binds to both the LANA binding sequences (LBS1/2) of the terminal repeats and suppresses transcriptional activity when fused to a reporter plasmid (24). The first open reading frame of KSHV, K1, which lies immediately after the terminal repeat and has a large portion of the promoter within the TR, was down-regulated with LANA expression (58). However, binding of LANA to its cognate sequence is essential for tethering the viral genome to the host chromosomes (19, 43, 59). LANA binds to DNA through its C-terminal domain, mapped to amino acids 996 to 1139 (33). Scanning deletion mutagenesis of this region concluded that amino acids 1007 to 10021 may be the DNA contact domain, since deletion of this region abolished LANA's ability to bind DNA and to support replication and episome persistence (33). The N terminus of LANA binds to the host chromatin and thus tethers the viral genome to the host chromosomes, bound to the C terminus of LANA (6, 13, 35, 53, 59).
Previous studies have demonstrated that LANA modulates the activities of various cellular pathways to favor cell growth by interacting with the cellular proteins (3, 20, 21, 47). LANA does not posses any enzymatic activity but is critical for the replication of TR-containing plasmids (26, 29, 33, 57). LANA supports DNA replication, most probably by recruiting host cellular replication machinery at the TR (56, 57). The detailed mechanism of replication and the dynamics of the recruitment of cellular proteins involved in the replication of TR-containing plasmids have yet to be determined.
Latent replication of Epstein-Barr virus (EBV), another member of the gammaherpesvirus family that infects humans, has been studied in more detail and has significantly helped in understanding the mechanism of replication of KSHV DNA (30, 38). EBV-encoded latent nuclear antigen 1 (EBNA1) is the functional analog of LANA (26, 29). Both of these proteins help in tethering of the respective episomes to the host chromosomes (4, 13, 31). Like EBNA1, LANA has been shown to recruit origin recognition complexes (ORCs) at the replication origins, suggesting a similar mechanism of replication within these two viruses (12, 15, 50, 52, 57). However, the organizations of the DNA elements at the replication origins in KSHV and EBV are highly divergent (30). EBV oriP is a 1.8-kbp cis element consisting of the family-of-repeats (FR) and dyad symmetry (DS) elements (49). The FR element is a cluster of 20 EBNA1 binding sites that mediate the maintenance of oriP-dependent episomes (2, 49, 62). The DS element contains four EBNA1 binding sites and also the DNA replication initiation site within or in close proximity to DS (62). Deletion of the DS element abolished the binding of ORCs and reduced replication initiation to basal levels (12, 46, 52). In contrast, KSHV does not have corresponding DS and FR elements (30). However, the presence of multiple copies of the TR has been proposed to function like the FR element, because multiple copies of the terminal repeat are required for efficient maintenance of the plasmid during long-term persistence, a requirement similar to that for FR in EBV persistence (30). Comparison of the organization of the LANA binding sequences in the TR unit and oriP of EBV has shown some conservation (30). The KSHV TR unit has both LANA binding sequences (LBS1/2) separated by 22 bp, compared to the 21 bp in the EBNA1 binding sequence within the DS element, but has half as many binding sites as the DS element (24, 28). EBV has replication initiation sites within the DS element, unlike KSHV, which requires a 32-bp GC-rich DNA element for replication, referred to as the replication element (30).
Previous reports published by our lab as well as other have shown the involvement of host cellular replication machinery in the replication of KSHV episomal DNA (56, 57). The host genome replicates in a very precise manner to maintain genetic integrity by ensuring that no segment of DNA replicates more than once per cell cycle (8, 44). Replication is divided into two major steps. The first step, which occurs during late G1 and early S phase, is licensing of the replication origin sites for use in S phase by sequential loading of ORCs, cdc6, Cdt1, and the heterohexameric complex MCM2-7 (minichromosome maintenance proteins 2 through 7) to the origins (7). Upon loading of these proteins, origins are licensed to form a prereplicative complex (pre-RC), followed by replication of DNA (7). The second step involves the prevention of rereplication by the prevention of relicensing (reloading of MCMs) of the origins before the cell enters into S phase (7). High activity of cyclin-dependent kinases from late G1 until the end of mitosis negatively regulates rereplication (7). Metazoans have another replication inhibitor, geminin, which specifically binds to Cdt1 and inhibits loading onto the pre-RC (60). Like cyclin-dependent kinases, geminin is active throughout the S phase, G2, and mitosis, followed by polyubiquitination by the anaphase-promoting complex for proteasome-mediated degradation (40, 60).
In this report we demonstrate that the minimal replicator element (MRE) of the KSHV genome replicates once in a cell-cycle-dependent manner. The plasmid containing the MRE undergoes a few rounds of replication even in the absence of LANA, but the replicated DNA disappears after 48 h without LANA. Geminin, which is the inhibitor of rereplication, blocked replication of the KSHV replicator element. Expression of Cdt1, an essential component of the pre-RC, rescued replication of the MRE-containing plasmid, suggesting the involvement of the host cellular replication control mechanism in the replication of the KSHV genome. Short-term replication assays of the MRE and its derivatives showed that LBS2 is dispensable for replication. Additionally, chromatin immunoprecipitation (ChIP) assays demonstrated that the binding of ORC2 with the associated chromatin of the MREs is increased in the presence of LANA.
MATERIALS AND METHODS
Cells, plasmids, and antibodies.
Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin (5 U/ml and 5 μg/ml, respectively).
The MRE and its deletion mutants, containing single standard oligonucleotides and their complementary strands, were synthesized at IDT (Coralville, IA). Both the strands were annealed, generating overhang of a BamHI site. RE-LBS1/2, RE-LBS1, RE, and LBS1 were ligated at the BamHI site of pBSpuro, generating pBSpuroRE-LBS1/2, pBSpuroRE-LBS1, pBSpuroLBS1, and pBSpuroRE. Clones containing these fragments were confirmed by sequencing. Generation of pBSpuro and the myc-tagged LANA expression vector pA3M have been described previously (57). A hemagglutinin (HA)-tagged human geminin expression vector was generated after PCR amplification using KS43 as a template (a generous gift from Bruce Stillman, Cold Spring Harbor Laboratory, NY) (41) and was cloned into the pCDNA3.1HA vector. HA-tagged human Cdt1 (hCdt1) was kindly provided by Hideko Nishitani (Kyushu University, Japan) (45). The BamC fragment of the EBV genome was digested with ApaLI, followed by end blunting with Klenow fragment. The 1.8-kb oriP region was excised from the ApaLI-linearized and blunted BamC fragment by digestion with EcoRI and was cloned into the BamHI (blunted) and EcoRI sites of pBSpuro, generating pBSpuro-oriP.
Myc-tagged LANA was detected using a supernatant from the 9E10 hybridoma (57). HA-tagged proteins (geminin and Cdt1) were detected using a supernatant from the 12CA5 hybridoma, described previously (58). The monoclonal anti-ORC2 (α-ORC2) antibody used for chromatin immunoprecipitation and Western blotting was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Immunoreactive signals were detected using infrared antibodies with an Odyssey infrared scanner (Li-COR, Lincoln, NE).
BrdU labeling of replicating plasmid and separation of BrdU-substituted DNA in CsCl density equilibrium.
The MRE-containing plasmid pBSpuroLBS1/2RE and the control plasmid pBSpuro were cotransfected with pA3M-LANA into 2 × 107 293 cells and labeled with bromodeoxyuridine (BrdU) as described previously with slight modifications (2, 51). Briefly, at 24 h posttransfection, cells were pulsed with BrdU (30 μg/ml) under puromycin selection for different times, allowing one or two rounds of replication. HEK293 cells divide in approximately 24 h in the presence of puromycin; therefore, cells at 0 h, 24 h, and 48 h were harvested, allowing no replication, a single round, and two rounds of replication, respectively. Total DNA isolated from the cells harvested at these time points were sonicated to an average length of 600 bp and loaded onto a CsCl density gradient with a refractive index of 1.4038. Tubes were spun in a Beckman SW41 rotor for 24 h at 40,000 rpm, followed by another 24 h at 30,000 rpm. Gradient-separated DNAs were collected as 250-μl fractions. The refractive index of each fraction was determined using a refractometer, and the density of each fraction was calculated based on a set standard. Each fraction was dialyzed by a drop dialysis technique using a 0.025 μm-pore-size Millipore membrane filter according to the manufacturer's recommendation (Millipore Inc., Bedford, MA). DNA in each fraction was precipitated with sodium acetate and ethanol. Total DNA in each fraction was quantified using SYBR green in a real-time PCR machine. The amount of DNA in each fraction was plotted against the respective fraction. Plasmid copies in each fraction were detected by using the following primer pair to amplify a region of the puromycin gene: 5′-CCGCGCAGCAACAGATGGAA-3′(sense) and 5′-AAGCCGAGCCGCTCGTAGAA-3′(antisense). Cellular DNA, based on copies of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene, was detected using the following primer pair: 5′-TGCACCACCAACTGCTTAG-3′(sense) and 5′-GATGCAGGGATGATGTTC-3′(antisense).
293 cells transfected with pBSpuro MRE and pBSpuro-oriP along with their respective trans-acting proteins were blocked in G1 phase by using mimosine (0.5 mM) for 18 h. G1-arrested cells were transferred to Dulbecco's modified Eagle medium containing 30 μg/ml BrdU for labeling of the replicating DNA. Cells were harvested at 0, 4, 8, 12, 24, and 48 h post-BrdU pulsing, and total DNA was extracted. Sonicated DNA was spun for the separation of density-labeled DNA on a CsCl density gradient as described above. The density of each fraction (250 μl each) collected from the top was determined, and each fraction was diluted three times with water to reduce the CsCl content for the precipitation of DNA. Plasmids (pBSpuro MRE and pBSpuro-oriP) and the cellular DNA in each fraction were detected by real-time quantitative PCR using the primers given above.
Short-term replication assay.
pBSpuro containing the minimal replicator element LBS1/2RE (MRE) and its deletion mutants were transfected into HEK293 cells using a Gene Pulser at 210 V and 975 μF (Bio-Rad Inc., Hercules, CA). Twelve million cells were transfected with 30 μg pBSpuroLBS1/2RE and its derivatives, pBSpuro LBS1RE, pBSpuroLBS1, pBSpuroRE, and vector control pBSpuro with 10 μg of the LANA expression vector. HA-tagged human geminin was transfected at 5 μg, and human Cdt1 (HA tagged) was expressed by transfecting 5 and 15 μg of pCDNA3.1HA hCdt1. Puromycin (1 μg/ml) was added to the medium for the selection of transfected cells. Forty-eight hours posttransfection, cells were replated with fresh puromycin and a fraction of the cells was analyzed for Western blot detection of the proteins. Ninety-six hours posttransfection, plasmid DNA was extracted using a modified Hirt procedure (27). Briefly, the medium from the 100-mm plates was removed, and cells were washed with phosphate-buffered saline (PBS), followed by lysing of the cells in plates with a 2.4-ml 1:2 mixture of solutions I and II (solution I, Tris, glucose, and EDTA; solution II, sodium dodecyl sulfate [SDS] and NaOH). Lysed cells were transferred to a tube, and 1.2 ml potassium acetate was added (solution III). The lysate was incubated on ice for 10 min, followed by centrifugation at 8,000 rpm for 10 min. The supernatant was further extracted with phenol and phenol-chloroform-isoamyl alcohol, and DNA was precipitated using 0.6 volume of 2-propanol. The pellet was dried, dissolved in Tris-EDTA with RNase, and incubated at 37°C for 30 min, followed by proteinase K treatment. The samples were subjected to a second phenol extraction, followed by precipitation with sodium acetate and ethanol. DNA was pelleted, washed with 70% ethanol, dried, and dissolved in water. DNA purified by the Hirt procedure was digested either with BamHI alone (to linearize) or with BamHI plus DpnI for at least 24 h. The digested DNA samples were electrophoresed on a 0.8% agarose gel, transferred to a GeneScreen membrane (Perkin-Elmer, Wellesley, MA), and hybridized with a 32P-labeled puromycin cassette probe. Hybridization signals were detected using a PhosphorImager plate (Molecular Dynamics Inc.). Signals were quantified using ImageQuant software (Molecular Dynamics Inc.).
Measurement of replicated DNA by quantitative PCR.
The plasmid containing MRE (pBSpuro MRE) and the vector control (pBSpuro) were cotransfected with either pA3M-LANA or an empty vector into 293 cells. Plasmid DNA was extracted at 24, 48, 72, and 96 h posttransfection using a modified Hirt procedure (27). Ninety percent of the extracted DNA was digested for 24 h with DpnI, and the remaining 10% was digested with BamHI for linearization of the plasmid. Fifty percent of each digested DNA was resolved on a 0.8% agarose gel for Southern blot detection of replicated copies using a 32P-labeled puromycin cassette as a probe. Hybridization signals were detected using a PhosphorImager plate and quantified using ImageQuant software (Molecular Dynamics Inc.). The remaining 50% of the digested DNA was precipitated following phenol extraction. DpnI-resistant copies of the MRE and vector backbone plasmids were quantified by real-time quantitative PCR using a primer pair flanking the DpnI restriction site. The sequences of the primers used for the quantitation are 5′-GGGTCACCGAGCTGCAAGAA-3′(sense) and 5′-GCCTTCCATCTGTTGCTGCG-3′(antisense). The number of replicated copies per cell was calculated by normalizing the percentage of transfected cells by transfecting the green fluorescent protein-expressing plasmid, pEGFP-C1.
ChIPassay.
HEK293cells transfected with pBSpuroLBS1/2RE, pBSpuroLBS1RE, or pBSpuroRE with or without the LANA expression vector were selected with puromycin for 48 h before chromatin immunoprecipitation. Puromycin-selected cells were cross-linked with 1% formaldehyde by rocking for 10 min at room temperature, followed by addition of 125 mM glycine to stop the cross-linking reaction. Cells were washed twice with cold PBS and collected in PBS containing protease inhibitors (1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin, and 1 mM phenylmethylsulfonyl fluoride). Cells were resuspended in cell lysis buffer [5 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES)-KOH (pH 8.0)-85 mM KCl-0.5% NP-40] containing protease inhibitors and were incubated on ice for 10 min. Cells were subjected to Dounce homogenization for efficient lysis, followed by centrifugation at 5,000 rpm for 5 min at 4°C. Nuclei were resuspended in nuclear lysis buffer (50 mM Tris [pH 8.0]-10 mM EDTA-1% SDS containing protease inhibitors), followed by incubation on ice for 10 min. Chromatin was sonicated to an average length of 700 bp, and cell debris was removed by centrifugation at high speed for 15 min at 4°C. The supernatant containing sonicated chromatin was diluted fivefold with ChIP dilution buffer (0.01% SDS-1.0% Triton X-100-1.2 mM EDTA-16.7 mM Tris [pH 8.1]-167 mM NaCl including protease inhibitor). Samples were precleared with a salmon sperm DNA-protein A-protein G Sepharose slurryfor 30 min at 4°C with constant rotation. The supernatant was collected after a brief centrifugation at 4°C. Ten percent of the total supernatant was saved for input in Western blotting, and 15% was saved for input of chromatin. The remaining 75% was divided into three fractions, to which was added 5 μg of (i) a control antibody, (ii) anti-ORC2 (Santa Cruz, CA), or (iii) anti-LANA antibodies, respectively. Reaction complexes were rotated overnight at 4°C, followed by precipitation of the immune complex by using a salmon sperm DNA-protein A-protein G slurry. Beads were then washed sequentially with a low-salt buffer (0.1% SDS-1.0% Triton X-100-2 mM EDTA-20 mM Tris [pH 8.1]-150 mM NaCl), a high-salt buffer (0.1% SDS-1.0% Triton X-100-2 mM EDTA-20 mM Tris [pH 8.1]-500 mM NaCl), and a LiCl wash buffer (0.25 M LiCl-1.0% NP-40-1% deoxycholate-1 mM EDTA-10 mM Tris [pH 8.0]) and twice in Tris-EDTA. Forty percent of the immunoprecipitated chromatin was taken for Western blot detection. Chromatin was eluted using an elution buffer (1% SDS-0.1 M NaHCO3) and reverse cross-linked by adding 0.3 M NaCl at 65°C for 4 to 5 h. Eluted DNA was precipitated, treated with proteinase K at 45°C for 2 h, and purified. Purified DNA was subjected to amplification of the puromycin target gene with the primers described previously, using eluted DNA as the template.
RESULTS
Plasmids containing the KSHV minimal replicator element (MRE) replicates in synchrony with cellular DNA.
Replication mediated by the KSHV terminal repeat element has been reproducibly shown by various laboratories (26, 29, 56, 57). KSHV has multiple copies of identical TR units, and a single copy of TR is capable of supporting replication (57). The minimal replicator element (Fig. 1A), which consists of 32-bp RE and LANA binding sequences (LBS1/2), replicates in a LANA-dependent manner (30). In order to determine if the MRE-containing plasmid replicates in synchrony with cellular DNA, we used the Meselson-Stahl experiment, which involves labeling of the replicating DNA with heavy nucleotides (42). We used BrdU, an analog of thymidine but with a greater density, to label the newly synthesized copies of the plasmid. In a semiconservative mode of replication, the newly synthesized plasmid incorporates the density label (BrdU) in one strand after a single round of replication, thus increasing the density of the DNA; this molecule is called heavy:light (H:L). The DNA molecule that replicates more than once incorporates BrdU in both strands and forms a heavy:heavy (H:H) molecule. The molecules that do not replicate fail to incorporate BrdU and thus have the lowest density; they are referred to as light:light (L:L). These species of DNA molecules have sufficiently different densities to be separated using the CsCl density gradient fractionation technique (Fig. 1B).
FIG. 1.

(A) Schematic showing single units of the KSHV TR and the sequence of the minimal replicator element (RE-LBS1/2). (B) Theoretical models of DNA replication. Solid lines, parental (original) DNA; shaded lines, newly synthesized DNA. In a conservative mode of replication, the original DNA molecule remains intact and generates a completely new molecule with H:H density in the presence of a density label. In a dispersive DNA replication mode, the two DNA molecules are generated with sections of both old and new DNA interspersed along each strand. In semiconservative DNA replication, the replicated DNA molecule is composed of one old and one new strand, thus yielding a molecule with intermediate density (H:L) after one round, which becomes H:H and H:L after the second round of replication.
The refractive indices of fractions 1 (top) though 16 (bottom) ranged from 1.388 to 1.411. Densities calculated based on the refractive indices of these samples ranged from 1.5765 to 1.8261 g/cm3 for fractions 1 to 16, respectively. Relative amounts of total DNA in fractions 3 to 15 showed a peak predominantly at a density of 1.7 g/cm3 in 0-h BrdU-treated 293 cells. DNA from cells exposed to BrdU for 24 h and 48 h showed peaks at 1.75 and 1.8 g/cm3, respectively. Previous studies have shown that nonsubstituted DNA (L:L) peaks at a buoyant density of 1.7 g/cm3, whereas the hemisubstituted and fully substituted DNAs peak at 1.75 and 1.8 g/cm3, respectively (2, 51). Indeed, the fractions from both pBSpuro MRE and the pBSpuro vector showed peaks at the densities corresponding to the L:L, H:L, and H:H species of the DNA (Fig. 2A and B). This showed that pBSpuro MRE- and pBSpuro-transfected cells were growing at comparable rates as the cells incorporated the density label, BrdU, and the DNA content moved to the higher buoyant densities (Fig. 2A and B).
FIG. 2.

The plasmid containing the KSHV minimal replicator element replicates in synchrony with the cellular DNA. 293 cells cotransfected with pBSpuro MRE or pBSpuro and the LANA expression vector were labeled with 30 μg/ml BrdU for 0 h, 24 h, and 48 h, allowing no replication, one round, and two rounds of replication in the presence of the density label, respectively. DNA extracted from these cells was sonicated and separated on a CsCl density gradient. Fractions of 250 μl were collected from the top and subjected to density determination of each fraction and DNA extraction. Relative amounts of DNA in each fraction (fractions 3 to 15) are plotted against the densities of the fractions. (A and B) The distribution of total DNA from 293 cells transfected with pBSpuro MRE (A) or pBSpuro (B) in these fractions at 0 h (triangles), 24 h (circles), and 48 h (squares) is shown. Distribution of DNA was detected at three peaks with densities of 1.7, 1.75, and 1.8 g/cm3, which correspond to the unreplicated L:L, semireplicated H:L, and fully replicated H:H DNA molecules, respectively. Proportions of the transfected plasmid in these fractions were detected by PCR amplification using a specific primer targeted to the puromycin gene. Relative amounts of amplicons were quantified in these fractions. (C) Levels of pBSpuro MRE copies in these fractions (fractions 3 to 15) at 0 h (triangles), 24 h (circles), and 48 h (squares) after BrdU pulsing are presented based on the relative densities of the amplicons. Relative amounts of cellular DNA from these fractions, amplified using the GAPDH gene, were quantified, and the relative densities of the bands are presented as a graph. (D) Quantitation of the pBSpuro vector in different fractions of the gradient. Amplification of the puromycin target showed a peak of the plasmid template at only L:L density, suggesting no replication. Distribution of cellular DNA was detected as L:L, H:L, and H:H densities after 0, 24, and 48 h of BrdU pulsing, suggesting semiconservative replication of the cellular gene.
PCR amplification of the puromycin gene of the plasmid backbone in fractions 3 to 15 detected amplification in fractions 8 and 9, which correspond to the L:L density (nonsubstituted DNA) (Fig. 2C). DNA collected from the 24-h BrdU-pulsed cells showed amplification of the puromycin target in fraction 11 along with fraction 8, demonstrating a shift in buoyant density due to the incorporation of BrdU. Since fraction 11 corresponds to the H:L density, this indicated the substitution of BrdU in only one strand of the plasmid, yielding a hybrid of substituted and nonsubstituted double-stranded DNA (H:L) (Fig. 2C). DNA from the cells pulsed for a longer period (48 h, allowing two rounds of replication) showed amplification of the puromycin target in fractions 11 and 14 along with fraction 8. Fraction 14 corresponds to the density of H:H (fully substituted) DNA molecules, therefore suggesting that some copies of the plasmid had undergone two rounds of replication, yielding BrdU incorporation in both the strands of the plasmid DNA.
Detection of cellular DNA in these fractions (fractions 3 to 15) using GAPDH as a target showed peaks at fractions 7 to 9 in 0-h BrdU-pulsed cells; the peak was shifted toward a higher density (fraction 11), corresponding to the H:L hybrid, after 24 h of BrdU pulsing, due to the substitution of one strand of the plasmid after a single round of replication. The fraction from 48-h BrdU-pulsed cells showed peaks at the H:H density (fraction 14), suggesting that both the strands of the plasmid DNA were substituted after two rounds of replication. Since we know that HEK293 cells replicate once in approximately 24 h, it is expected that cellular DNA would show substitution and peaks at H:L after 24 h and H:H after 48 h. Importantly, plasmid DNA carrying the minimal replicator element (pBSpuro MRE) also showed a similar pattern of BrdU incorporation, suggesting that the minimal replicator element replicates in synchrony with the cellular DNA.
In contrast, the fraction from 293 cells transfected with the empty vector (pBSpuro) and labeled for 0, 24, and 48 h with BrdU showed amplification of the puromycin target gene only in fractions corresponding to the L:L density (Fig. 2D). This suggested that the empty vector was unable to replicate and thus did not substitute its strand; hence, it showed peaks at the L:L fraction at all three time points (Fig. 2D). Some reduced signals were seen in the H:L peak, indicating some level of BrdU incorporation. However, this is likely to be due to an incorporation/repair process, since this signal was not seen in the H:H fraction. Amplification of cellular DNA using GAPDH as a target detected strong amplification in fractions 8 to 9 at 0 h, in fraction 11 at 24 h, and in fraction 14 at 48 h of the BrdU pulse period. This indicated that cellular DNA replicated and incorporated BrdU following replication. Quantitative detection of the band in each fraction is plotted and was also confirmed by real-time semiquantitative PCR analysis.
The MRE replicates once per cell cycle.
Previous experiments suggested that the MRE-containing plasmid replicates along with the cellular DNA, since both showed similar peaks at H:L and H:H densities. However, that did not answer the question of whether the plasmid replicated with the same kinetics in the same S phase. This was determined by analyzing replication mediated by the MRE in a time shorter than the doubling time of the cells, which is approximately 24 h. To make sure that the cells were in the same cell cycle phase, we synchronized the 293 cells cotransfected with pBSpuro MRE and pA3M-LANA in G1 phase by using mimosine (Fig. 3A). To ensure the labeling of replicating DNA, we used an oriP-containing plasmid (pBSpuro oriP) along with the EBNA1 expression vector as a control. G1-arrested 293 cells containing MRE and oriP along with their respective trans-acting proteins were released into normal medium, which, however, contained BrdU as a density label. In order to determine the replication time, we harvested cells pulsed with BrdU for different times (0, 4, 8, and 12 h). According to fluorescence-activated cell sorter analysis, a fraction of cells showed duplicated DNA content at approximately 8 h after release from the G1 block (Fig. 3A to C). Total DNA was extracted from the cells containing MRE and oriP at these time points and analyzed on a CsCl density gradient. DNA in each fraction of the gradient was purified for the quantitation of plasmid (pBSpuro MRE and pBSpuro oriP) copies by real-time PCR analysis. The relative number of plasmid copies, which was calculated based on amplification of the puromycin gene, in each fraction of the gradient suggested that pBSpuro MRE and pBSpuro oriP incorporated the density label and shifted to the H:L density at 8 h (Fig. 3C). Cellular DNA, which was detected by the relative amplification of the GAPDH gene, also showed peaks at the H:L density at 8 h of BrdU labeling (Fig. 3C). This suggests that both the viral origins (MRE and oriP) replicated in a manner similar to that of the cellular DNA. The relative amounts of plasmid and cellular DNA at 12 h post-BrdU pulsing showed peaks at the H:L density (Fig. 3D), suggesting that both follow the same pattern of replication. The plasmid DNA content detected in the fractions from the cells labeled for 24 h (the doubling time of the 293 cells) showed peaks at H:L at 24 h (Fig. 3E). The similarity of the patterns between cellular DNA and viral DNA is very distinct throughout one cell division, suggesting that the plasmids (pBSpuro MRE and pBSpuro oriP) replicated at a rate similar to that of cellular DNA and only once per cell cycle. Cells that underwent two complete rounds of replication showed peaks at H:H in the case of both plasmids as well as cellular DNA. This strongly supports the previous results, which show efficient replication of MRE of the KSHV TR (Fig. 3F). Based on these data, we conclude that the plasmids containing viral origins MRE and oriP replicate once and only once during the S phase of the cell cycle in a fashion similar to that of cellular DNA.
FIG. 3.

The KSHV MRE-containing plasmid replicates only once, in a cell-cycle-dependent manner. (A) 293 cells cotransfected either with pBSpuro MRE and pA3M-LANA or with pBSpuro oriP and the EBNA1 expression vector were synchronized in G1 phase by treatment with mimosine (0.5 mM) for 18 h. Synchronized cells were released into a normal medium containing 30 μg/ml BrdU. Cells were harvested at the indicated time points (0, 4, 8, 12, 24, and 48 h), and a fraction of cells was subjected to fluorescence-activated cell sorter analysis. Total DNAs from the remaining cells were extracted and sonicated to an average length of 600 bp, followed by CsCl density gradient centrifugation. Fractions of 250 μl were collected from the top, and DNAs from these fractions were then purified. Plasmid DNAs in these fractions were detected by real-time quantitative PCR using a primer targeted to the amplification of the puromycin gene on the plasmid backbone. Solid and open bars, relative amplification of the plasmid DNAs containing MRE and oriP, respectively. Quantification of cellular DNA based on the amplification of the GAPDH gene was also performed for these fractions, and the results were plotted (shaded bars) along with the plasmid DNA content in the respective fraction. The density of each fraction was detected. Dotted lines indicate fractions with L:L, H:L, and H:H densities. Only fractions 6 to 13 are shown for the 0-h (A), 4-h (B), 8-h (C), and 12-h (D) time points. Fraction 14, which is mainly the H:H fraction, is shown in the DNA from cells labeled with BrdU for 24 h (E) and 48 h (F).
MRE-containing plasmids replicate at least two rounds without LANA and require LANA for efficient replication.
oriP of EBV has been shown to undergo at least two rounds of replication without the trans-acting protein EBNA1; therefore, we wanted to determine whether MRE can initiate replication and how long the newly synthesized DNA can persist in cells without LANA expression. In order to address this question, we cotransfected pBSpuro MRE with equal amounts of either pA3M-LANA or the pA3M vector into 293 cells. Transfected cells were pooled and plated in 100-mm plates. Cells were harvested at the indicated time points posttransfection, and the DNA extracted by the Hirt procedure was subjected to quantitation of replicated copies by Southern blotting (Fig. 4A) and real-time PCR analysis (Fig. 4B). At 48 h posttransfection, there was a faint Dpn- resistant band even in the absence of LANA (Fig. 4A, lane 2). However, in the presence of LANA, the ratio of replicated copies were significantly higher than the no LANA cells (Fig. 4A, lane 4). Quantitation of DpnI-resistant copies by real-time PCR showed that there were approximately 2 copies of the replicated plasmid per cell (Fig. 4B). In contrast, LANA-expressing cells had approximately 4.5 copies of the replicated DNA (Fig. 4B). This suggests that MRE was able to initiate and support replication during the first 24 to 48 h but requires LANA for efficient replication of the plasmid.
FIG. 4.

The plasmid containing the KSHV MRE replicates at lower levels in the absence of LANA, and the newly synthesized DNA disappears after 48 posttransfection. The pBSpuro MRE plasmid was transfected into 293 cells with or without the LANA expression vector. Transfected cells were harvested at 48 h, 72 h, and 96 h posttransfection, followed by Hirt extraction of the episomal DNA. Extracted DNA was digested with BamHI (B) (to linearize) and with BamHI plus DpnI (B+D). (A) Fifty percent of the total digested DNA was subjected to Southern blot detection of the replicated copies after resolution of the digested DNA on 0.8% agarose and transfer to a GeneScreen membrane. The signals were detected using a 32P-labeled puromycin cassette after hybridization. The intensities of the bands were quantified and presented as relative densities (RD) using ImageQuant software (Molecular Dynamics). (B) The remaining 50% of the BamHI- and DpnI-digested DNA was phenol extracted and precipitated for detection of DpnI-resistant copies. A set of primers (flanking the DpnI restriction site) targeted to the amplification of the puromycin gene was used in a real-time quantitative PCR for detection of the DpnI-resistant copies. The copies of the replicated MRE-containing plasmid in the presence and absence of LANA were calculated based on the copy number from a known amount of the pBSpuro MRE plasmid.
At 72 and 96 h posttransfection, the number of copies of the MRE-containing plasmid was further decreased to less than 1 copy per cell, whereas in LANA-expressing cells, the number of replicated copies remained almost constant. Interestingly, we detected slightly fewer copies per cell (∼3.5 copies/cell) in LANA-expressing cells at 96 h, which may be due to the absence of any selection pressure. However, the ratio of plasmid DNA in LANA-positive cells was approximately 10-fold greater than that in LANA-negative cells (Fig. 4B).
The replication inhibitor geminin blocks the replication of plasmids containing the KSHV minimal replicator element.
In order to determine whether the replication inhibitor geminin can block TR-mediated replication, we used a plasmid with the MRE, pBSpuro MRE, in a short-term replication assay in the presence of geminin expression. Geminin, an inhibitor of replication, interacts with one of the components of the prereplication complex, Cdt1, to block the loading of MCMs (7). Comparison of DpnI-resistant bands (replicated copies of the plasmid) in Fig. 5B, lanes 2 and 4, clearly shows that the expression of geminin blocked the replication of a plasmid containing the LBS1/2RE element of the TR. This confirms the involvement of the host cellular replication machinery in the replication and the rereplication control mechanism.
FIG. 5.

Human geminin blocks replication of a plasmid mediated by the MRE. (A) The plasmid containing the minimal replicator element was cotransfected with the LANA expression vector as well as human geminin and Cdt1 in the indicated lanes. Western blotting using an anti-myc antibody shows expression of LANA in all four transfections. HA-tagged hCdt1 and human geminin were detected by Western blotting using an anti-HA antibody. (B) Hirt-extracted DNAs from all four samples were digested with BamHI (B) (to linearize) and with BamHI plus DpnI (B+D) overnight. Digested DNA was resolved and transferred to a GeneScreen membrane, followed by hybridization with a 32P-labeled puromycin gene probe. DpnI-resistant bands were quantified using ImageQuant software, and relative densities (RD) of DpnI-resistant bands are shown after normalization with the density of the input lane as 10%.
Expression of Cdt1 has been shown to reverse the repressive effect of geminin (15); therefore, we transfected the Cdt1 expression vector in amounts equivalent to that of geminin as well as in a threefold molar excess. As expected, Cdt1 rescued replication of the plasmid mediated by the minimal replicator element (Fig. 5B, lane 6). Expression of Cdt1 in a threefold excess compared to geminin resulted in increased replication (Fig. 5B, lane 8), suggesting that Cdt1 is an important component of TR replication and part of the pre-RC complex at the terminal repeat. Linearized plasmids are in odd-numbered lanes (Fig. 5B, lanes 1, 3, 5, and 7). Quantitation of DpnI-resistant bands relative to linearized bands (10% input) is shown below (Fig. 5B). Expression of LANA in all the samples is shown by Western blot analysis with an anti-myc antibody (Fig. 5A). Geminin and Cdt1 expression were detected with an anti-HA antibody (Fig. 5A).
LBS2 of the MRE is dispensable for replication.
Our previous study has shown that ORC2 associates with LANA when LANA is bound to its cognate sequence, LBS1 of the TR (57). Another study has shown that the 32-bp element upstream of LBS1 is critical for replication (30). Therefore, we wanted to determine whether plasmids lacking the low-affinity LANA binding sequences (LBS2) or both the LANA binding sites are able to replicate. We transfected the minimal replicator element-containing plasmid and its deletion mutants, pBSpuro RE-LBS1/2, pBSpuroRE-LBS1, pBSpuroLBS1, and pBSpuroRE, as well as the vector backbone pBSpuro, along with the LANA expression vector into 293 cells to determine their replicative ability. A schematic of the minimal replicator element and its derivatives is shown in Fig. 6A. The trans-acting protein LANA was detected in all the samples with an anti-myc antibody (Fig. 6B).
FIG. 6.

LBS2 is dispensable for replication. (A) An empty vector and pBSpuro containing the MRE (RE-LBS1/2) with its deletion mutants (RE-LBS1, LBS1, and RE) were transfected into 293 cells along with the LANA expression vector. (B) Expression of LANA was detected by Western blotting (WB) with an anti-myc antibody. (C) DNAs extracted from these cells by the Hirt procedure 96 h posttransfection were digested with BamHI (B) (to linearize) and BamHI plus DpnI (B+D) overnight. Digested DNA was resolved and transferred to a GeneScreen membrane (Perkin-Elmer). DpnI-resistant bands were detected after hybridization with a 32P-labeled puromycin gene probe. Relative densities (RD) of the DpnI-resistant bands were quantified using ImageQuant software (Molecular Dynamics).
DNAs were extracted by the Hirt procedure at 96 h posttransfection from the transfected plasmids listed above, digested with either BamHI alone (to linearize) or BamHI plus DpnI, and subjected to Southern blot detection of the DpnI-resistant band. The result of a representative experiment showed that the minimal replicator element-containing plasmid was able to replicate, a finding consistent with previous reports (Fig. 6C, lane 2) (30). Additionally, the minimal replicator element lacking the low-affinity LANA binding sequence (LBS2) also showed replication of the plasmid under similar conditions, although it was slightly less efficient, with an approximately 40% drop in efficiency compared to the wild type (Fig. 6C, lane 4). Plasmids lacking the RE (having only LBS1) did not show any detectable replication, suggesting that the RE is critical for replication initiation (Fig. 4C, lane 6). Additionally, the plasmid containing only the RE was unable to replicate (Fig. 6C, lane 8). This suggests that binding of LANA is important for replication mediated by the minimal replicator element. Quantitation of the DpnI-resistant bands in all these samples with the input as 10% is shown below the respective bands.
LANA enhances the binding of ORC2 to the minimal replicator element.
The short-term replication experiment suggested that LANA is important for the replication of the minimal replicator element; therefore, we wanted to determine the binding status of the ORCs at the MRE using a chromatin immunoprecipitation assay. We performed chromatin immunoprecipitation assays on cells cotransfected with pBSpuro RE-LBS1/2, RE-LBS1, LBS1, RE, or vector alone with and without the LANA expression vector. A schematic of pBSpuro RE-LBS1/2 and its deletion mutants is shown in Fig. 7A. Precipitation of chromatin with LANA- and ORC2-specific antibodies was detected by Western blotting, which showed significant amounts of chromatin precipitation with the respective antibodies. A representative Western blot is shown in Fig. 7B.
FIG. 7.

LANA enhances the association of ORCs with minimal replicator elements. (A) Schematic of the pBSpuro vector backbone showing the site of the primer used for amplification of the region on the immunoprecipitated DNA (indicated by two half-arrows).(B) Plasmids pBSpuroRE-LBS1/2, pBSpuroRE-LBS1, pBSpuroLBS1, and pBSpuro RE were transfected separately in 293 cells with either pA3M (−LANA) or pA3M LANA (+LANA).Forty-eight hours posttransfection, chromatin was prepared from the cells of all the sets. Chromatin was subjected to immunoprecipitation (IP) with a matched control antibody (CoAb), an α-ORC2 antibody, or an α-LANA antibody. The α-LANA and α-ORC2 antibodies showed significant amounts of chromatin immunoprecipitation. (C) DNA was recovered from the chromatin after reverse cross-linking of the chromatin. Precipitated DNA was purified and subjected to amplification of the puromycin target gene (adjacent to the replicator element) by PCR. Relative densities of the amplified bands were quantified, and the numbers of immunoprecipitated copies of the minimal replicator element and the deletion mutant elements were detected relative to the input lane (10%). The replication element showed very few copies of the DNA immunoprecipitated with only the α-ORC2 antibody even in the absence of LANA. Expression of LANA did not change the binding of ORC2 to the RE in the plasmid containing the RE only. Chromatin from the plasmid containing LBS1 immunoprecipitated plasmid copies with LANA as well as ORC2, but the numbers of chromatin-bound copies were slightly higher in LANA-expressing cells. Additionally, with the plasmids containing the LANA binding sequence along with the RE (RE-LBS1/2 and RE-LBS1), LANA greatly enhanced the binding of ORC2 to the plasmid, suggesting that LANA stabilizes the association of ORC2 with the replicator element.
DNA recovered from the immunoprecipitated chromatin was subjected to PCR amplification of a region on the puromycin gene in close proximity to the replicator element (Fig. 7A). Amplified bands in the DNA recovered from the chromatin are shown in Fig. 7C. As expected, LANA was able to immunoprecipitate chromatin-bound DNA of pBSpuro RE-LBS1/2, pBSpuro RE-LBS1, and pBSpuroLBS1, but not the RE-containing plasmid, due to the lack of a LANA binding site on that plasmid (Fig. 7C, lanes 7 in all panels). Amplification of the target site in the DNA recovered from the chromatin immunoprecipitated with ORC2 showed significant binding of the RE-LBS1 and RE-LBS1/2 plasmids (Fig. 7C, lane 8). Interestingly, some level of amplification was detected in the DNA from the plasmid containing just the RE site, suggesting that ORC2 may have some binding affinity to the RE region (Fig. 7C, lane 8, RE). The plasmid containing the binding site of LANA (LBS1) was also immunoprecipitated with ORC2 as well as anti-LANA antibodies, suggesting that binding of LANA brings the ORCs in close proximity to the DNA. DNA extracted from the chromatin immunoprecipitated using a control antibody showed no amplification, thus confirming the specificity of the assay (Fig. 7C, lane 6). Chromatin immunoprecipitated using an anti-ORC2 antibody from cells lacking LANA expression detected copies of the plasmids containing RE-LBS1/2, RE-LBS1, and RE (Fig. 7C, lane 4). Plasmids containing LBS1 showed significantly reduced levels of amplification in ORC2-immunoprecipitated complexes (Fig. 7C, lane 4). Chromatin immunoprecipitated with the anti-LANA antibody showed no amplification of the plasmids (Fig. 7C, lane 3), like the DNA recovered from the chromatin immunoprecipitated with the control antibody (Fig. 7C, lane 2). Lanes 1 and 5 represent the plasmid DNA in 10% of the total chromatin used for immunoprecipitation with specific antibodies (Fig. 7C). Quantitative analysis based on three independent experiments suggested a three- to sixfold increase in the association of ORC2 with the minimal replicator element in the presence of LANA.
Enhancement of the binding of ORC2 to the MRE in the presence of LANA was also seen in a DNA affinity column. RE, LBS1, RE-LBS1, RE-LBS1/2, and scrambled DNA sequences were synthesized and covalently ligated to CNBr-activated Sepharose beads. Nuclear extracts prepared from 293 cells expressing LANA as well as the vector were subjected to binding to the DNA affinity columns as described previously (57). Proteins eluted after thorough washing of the column followed by detection of ORC2 in the eluate suggested enhancement of the binding of ORC2 to the MRE in the presence of LANA (data not shown).
DISCUSSION
The KSHV genome persists as an episome in infected cells with the expression of a limited number of genes (43, 59). LANA is one of the major proteins expressed during latency and is important for the persistence of the viral genome in infected cells (4, 13, 59). KSHV BAC36, deleted for the LANA open reading frame and the PEL-expressing LANA short hairpin RNA, greatly reduced the episomal copy number, suggesting LANA's role in persistence (25, 64). Episome persistence is achieved through direct binding of LANA to the TR and by hooking up the LANA and episomal complex to the host chromatin with the help of cellular proteins (6, 35, 43, 53, 59). The number of viral episomal copies ranges from 20 to 150 per cell, and interestingly, the copy number per cell is maintained in tumors as well as in cultured cells after multiple passages (22). This led us to determine the nature of KSHV genome replication. Since a single TR unit and, more recently, the 71-bp MRE has been shown to be sufficient for the replication of a plasmid, we cloned the MRE in the pBSpuro vector, which lacks the ability to support eukaryotic replication, for the evaluation of replication potential. The MRE was able to support replication in LANA-expressing cells, a finding similar to that reported previously (30).
DNA replicates in a semiconservative manner which produces molecules with both old and new strands, and both the strands are composed of one old and one new strand. To confirm that the MRE can utilize a semiconservative mode of replication, a classic Meselson-Stahl experiment was performed. This involves labeling of the replicating DNA with heavy nucleotides for a single round and two rounds of replication and the separation of the labeled DNA on a CsCl density gradient (42).
Unlike the semiconservative mode of replication, if the DNA follows a conservative DNA replication method, then both the strands will be substituted with the heavy label (in this case BrdU) and thus will band at the H:H fraction just after the single round of replication. Similarly, if the replication is dispersive, the density label will produce DNA of intermediate density (H:L,) which will be similar to the semiconservative mode of replication. However, the semiconservatively replicating DNA will produce a DNA molecule with two distinct densities, intermediate (H:L) and heavy (H:H), after the second round of replication. In contrast, the dispersive mode of replication will again produce molecules of intermediate densities even after a second round of replication.
The plasmid containing MRE showed a peak at an intermediate density (H:L) after a single round of replication, which was further moved to the H:H density after two rounds of replication. These data confirmed that the MRE utilizes a semiconservative mode of replication. Since the cellular DNA replicates in a semiconservative manner, comparison of the density-labeled peaks at 24 and 48 h suggested that the MRE-containing plasmid also replicated semiconservatively in synchrony with the cellular DNA. The replication element (oriP) of another member of the gammaherpesvirus family, EBV, also replicates semiconservatively and once per cell cycle, confirming that the replication modes are conserved between these two viruses (1, 63).
KSHV-infected cells maintain the viral episomal copies after successive rounds of cell division by the fact that the episome replicates once in a cell cycle along with the host cells and segregates during mitosis to the dividing cells. There are two major steps during eukaryotic genome replication, which enables cells to receive the complete genomic copies. These are the S (synthesis) phase, during which the DNA of the chromosomes is replicated, and the M (mitosis) phase, which involves the segregation of replicated DNA into the two newly divided cells (7). To ensure genomic stability, the S phase of cells is tightly regulated, allowing replication of the genome only once in a cell cycle (7). The replication origin becomes competent to fire or initiate replication after a series of events termed licensing, which involve the loading of Cdc6, Cdt1, and the six MCM proteins at the already bound ORC proteins (39). In order to prevent refiring of the replication origins, geminin (expressed in S phase) associates with a licensing factor, Cdt1, and prevents rereplication (60). Geminin gets degraded at the end of metaphase in a proteasome-dependent manner (7). The accumulation of Cdt1 proteins in the nucleus and their association with the chromatin permits licensing and initiation of a new round of DNA replication (40, 60). However, expression of Cdt1 in a molar excess can block the repression mediated by geminin (15). Our short-term replication data for MRE showed inhibition of replication activity similar to that of cellular DNA replication due to geminin (7). Expression of the licensing factor Cdt1 rescued the replication ability of MRE by reversing the inhibitory effect of geminin (Fig. 5). These data suggests that the MRE uses the cellular replication machinery and its rereplication control mechanism to control overreplication of the KSHV episome, thus allowing only one round of replication per cell cycle (Fig. 8).
FIG. 8.
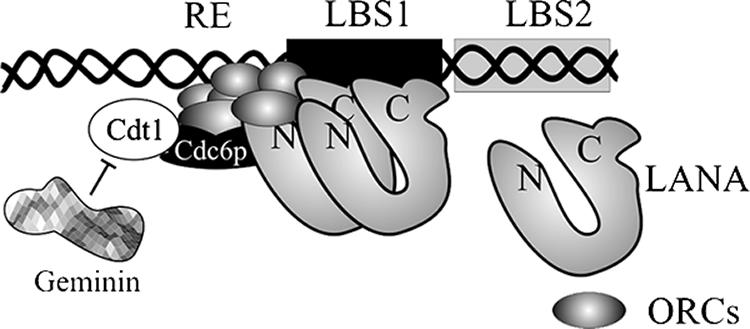
A possible model for the involvement of cellular replication proteins at the MRE. LANA binds to its cognate sequence through its carboxyl terminus, which has also been shown to bind to the ORCs. The binding of ORCs to LANA most probably allows association of ORCs with the DNA element in close proximity to the RE. LANA-bound ORCs may serve as a launching pad for the loading of other cellular replication proteins, including cdc6, Cdt1, and MCMs. Cellular geminin, which is expressed during S phase, associates with Cdt1 and thus prevents relicensing of the TR origins to prevent overreplication, a mechanism similar to the control of replication of host genomic DNA.
Our study, to determine whether the low-affinity LANA binding site (LBS2) is important for replication, suggests that a single LANA binding site (LBS1) along with the RE is sufficient for replication. Deletion of LBS2 from MRE reduced replication ability by ∼40%, which was comparable to the reduction (a 49% reduction) in replication efficiency obtained in the context of the entire TR unit (24). The increased replication of a plasmid containing both the LANA binding sites, LBS1/2, may be due to the fact that the binding of LANA bends the DNA, which may facilitate the binding of cellular replication complexes at the DNA (61). In terms of the replication element, even though the RE is critical for the replication of plasmids and also showed some binding affinity to ORC2, it was unable to support replication independently of the LBS. This suggests that not only the RE but also the surrounding sequences are required for replication function.
Since the RE and the MRE-containing plasmids showed some binding with ORCs even in the absence of LANA in our chromatin immunoprecipitation assay, it is possible that the ORCs may have some affinity to the RE region and that LANA helps in stabilizing the association of replication proteins. Further work to understand the recruitment and the dynamics of cellular replication protein at the TR is ongoing in our laboratory.
Recent studies have suggested that LANA is bound to the chromatin throughout all the phases of the cell cycle, and we have previously shown that LANA binds to ORCs (6, 57). This suggests that ORCs are bound to the chromatin of the KSHV episome at the TR throughout the cell cycle (56, 57). The licensing of the replication origins of the TR is determined similarly to that of the cellular DNA during the S phase of the cell cycle.
Acknowledgments
This work was supported by Public Health Service grants from NCI (CA091792) and NIDCR (DE014136 and DE017338) (to E.S.R.). S.C.V. is supported by the Lady Tata Memorial Trust. E.S.R. is a scholar of the Leukemia and Lymphoma Society of America.
We thank Carlo Franco Balane-Bolivar for technical help. We sincerely thank Bruce Stillman (Cold Spring Harbor Laboratories, NY) for providing us with the cDNA of human geminin and Hideko Nishitani (Kyushu University, Fukuoka, Japan) for the pCDNAhCdt1.
Footnotes
Published ahead of print on 6 December 2006.
REFERENCES
- 1.Adams, A. 1987. Replication of latent Epstein-Barr virus genomes in Raji cells. J. Virol. 61:1743-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aiyar, A., C. Tyree, and B. Sugden. 1998. The plasmid replicon of EBV consists of multiple cis-acting elements that facilitate DNA synthesis by the cell and a viral maintenance element. EMBO J. 17:6394-6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bajaj, B. G., S. C. Verma, K. Lan, M. A. Cotter, Z. L. Woodman, and E. S. Robertson. 2006. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 351:18-28. [DOI] [PubMed] [Google Scholar]
- 4.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 5.Ballestas, M. E., and K. M. Kaye. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250-3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbera, A. J., J. V. Chodaparambil, B. Kelley-Clarke, V. Joukov, J. C. Walter, K. Luger, and K. M. Kaye. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856-861. [DOI] [PubMed] [Google Scholar]
- 7.Bell, S. P., and A. Dutta. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71:333-374. [DOI] [PubMed] [Google Scholar]
- 8.Blow, J. J., and B. Hodgson. 2002. Replication licensing—defining the proliferative state? Trends Cell Biol. 12:72-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 10.Chang, Y. 1997. Kaposi's sarcoma and Kaposi's sarcoma associated herpesvirus (human herpesvirus 8): where are we now? J. Natl. Cancer Inst. 89:1829-1831. [DOI] [PubMed] [Google Scholar]
- 11.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 12.Chaudhuri, B., H. Xu, I. Todorov, A. Dutta, and J. L. Yates. 2001. Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 98:10085-10089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 14.Cotter, M. A., II, C. Subramanian, and E. S. Robertson. 2001. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 291:241-259. [DOI] [PubMed] [Google Scholar]
- 15.Dhar, S. K., K. Yoshida, Y. Machida, P. Khaira, B. Chaudhuri, J. A. Wohlschlegel, M. Leffak, J. Yates, and A. Dutta. 2001. Replication from oriP of Epstein-Barr virus requires human ORC and is inhibited by geminin. Cell 106:287-296. [DOI] [PubMed] [Google Scholar]
- 16.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dupin, N., T. L. Diss, P. Kellam, M. Tulliez, M. Q. Du, D. Sicard, R. A. Weiss, P. G. Isaacson, and C. Boshoff. 2000. HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406-1412. [PubMed] [Google Scholar]
- 18.Dupin, N., C. Fisher, P. Kellam, S. Ariad, M. Tulliez, N. Franck, E. van Marck, D. Salmon, I. Gorin, J. P. Escande, R. A. Weiss, K. Alitalo, and C. Boshoff. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 96:4546-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fejer, G., M. M. Medveczky, E. Horvath, B. Lane, Y. Chang, and P. G. Medveczky. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J. Gen. Virol. 84:1451-1462. [DOI] [PubMed] [Google Scholar]
- 20.Friborg, J., Jr., W. Kong, M. O. Hottiger, and G. J. Nabel. 1999. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 402:889-894. [DOI] [PubMed] [Google Scholar]
- 21.Fujimuro, M., F. Y. Wu, C. ApRhys, H. Kajumbula, D. B. Young, G. S. Hayward, and S. D. Hayward. 2003. A novel viral mechanism for dysregulation of beta-catenin in Kaposi's sarcoma-associated herpesvirus latency. Nat. Med. 9:300-306. [DOI] [PubMed] [Google Scholar]
- 22.Gao, S. J., M. Alsina, J. H. Deng, C. R. Harrison, E. A. Montalvo, C. T. Leach, G. D. Roodman, and H. B. Jenson. 1998. Antibodies to Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in patients with multiple myeloma. J. Infect. Dis. 178:846-849. [DOI] [PubMed] [Google Scholar]
- 23.Gao, S. J., Y. J. Zhang, J. H. Deng, C. S. Rabkin, O. Flore, and H. B. Jenson. 1999. Molecular polymorphism of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) latent nuclear antigen: evidence for a large repertoire of viral genotypes and dual infection with different viral genotypes. J. Infect. Dis. 180:1466-1476. [DOI] [PubMed] [Google Scholar]
- 24.Garber, A. C., J. Hu, and R. Renne. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401-27411. [DOI] [PubMed] [Google Scholar]
- 25.Godfrey, A., J. Anderson, A. Papanastasiou, Y. Takeuchi, and C. Boshoff. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510-2518. [DOI] [PubMed] [Google Scholar]
- 26.Grundhoff, A., and D. Ganem. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 28.Hsieh, D. J., S. M. Camiolo, and J. L. Yates. 1993. Constitutive binding of EBNA1 protein to the Epstein-Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J. 12:4933-4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu, J., A. C. Garber, and R. Renne. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 76:11677-11687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu, J., and R. Renne. 2005. Characterization of the minimal replicator of Kaposi's sarcoma-associated herpesvirus latent origin. J. Virol. 79:2637-2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hung, S. C., M. S. Kang, and E. Kieff. 2001. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proc. Natl. Acad. Sci. USA 98:1865-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kellam, P., C. Boshoff, D. Whitby, S. Matthews, R. A. Weiss, and S. J. Talbot. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J. Hum. Virol. 1:19-29. [PubMed] [Google Scholar]
- 33.Komatsu, T., M. E. Ballestas, A. J. Barbera, B. Kelley-Clarke, and K. M. Kaye. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225-236. [DOI] [PubMed] [Google Scholar]
- 34.Krishnan, H. H., P. P. Naranatt, M. S. Smith, L. Zeng, C. Bloomer, and B. Chandran. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krithivas, A., M. Fujimuro, M. Weidner, D. B. Young, and S. D. Hayward. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J. Virol. 76:11596-11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lagunoff, M., and D. Ganem. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147-154. [DOI] [PubMed] [Google Scholar]
- 37.Lennette, E. T., D. J. Blackbourn, and J. A. Levy. 1996. Antibodies to human herpesvirus type 8 in the general population and in Kaposi's sarcoma patients. Lancet 348:858-861. [DOI] [PubMed] [Google Scholar]
- 38.Lim, C., H. Sohn, D. Lee, Y. Gwack, and J. Choe. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J. Virol. 76:10320-10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lygerou, Z., and P. Nurse. 2000. Cell cycle. License withheld—geminin blocks DNA replication. Science 290:2271-2273. [DOI] [PubMed] [Google Scholar]
- 40.McGarry, T. J., and M. W. Kirschner. 1998. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell 93:1043-1053. [DOI] [PubMed] [Google Scholar]
- 41.Mendez, J., X. H. Zou-Yang, S. Y. Kim, M. Hidaka, W. P. Tansey, and B. Stillman. 2002. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell 9:481-491. [DOI] [PubMed] [Google Scholar]
- 42.Meselson, M., and F. W. Stahl. 1958. The replication of DNA in Escherichia coli. Proc. Natl. Acad. Sci. USA 44:671-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore, P. S., and Y. Chang. 2003. Kaposi's sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu. Rev. Microbiol. 57:609-639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishitani, H., and Z. Lygerou. 2002. Control of DNA replication licensing in a cell cycle. Genes Cells 7:523-534. [DOI] [PubMed] [Google Scholar]
- 45.Nishitani, H., S. Taraviras, Z. Lygerou, and T. Nishimoto. 2001. The human licensing factor for DNA replication Cdt1 accumulates in G1 and is destabilized after initiation of S-phase. J. Biol. Chem. 276:44905-44911. [DOI] [PubMed] [Google Scholar]
- 46.Norio, P., C. L. Schildkraut, and J. L. Yates. 2000. Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein-Barr virus chromosome after infection of established cell lines. J. Virol. 74:8563-8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radkov, S. A., P. Kellam, and C. Boshoff. 2000. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 6:1121-1127. [DOI] [PubMed] [Google Scholar]
- 48.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S. J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reisman, D., J. Yates, and B. Sugden. 1985. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol. 5:1822-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ritzi, M., K. Tillack, J. Gerhardt, E. Ott, S. Humme, E. Kremmer, W. Hammerschmidt, and A. Schepers. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell Sci. 116:3971-3984. [DOI] [PubMed] [Google Scholar]
- 51.Schaarschmidt, D., J. Baltin, I. M. Stehle, H. J. Lipps, and R. Knippers. 2004. An episomal mammalian replicon: sequence-independent binding of the origin recognition complex. EMBO J. 23:191-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schepers, A., M. Ritzi, K. Bousset, E. Kremmer, J. L. Yates, J. Harwood, J. F. Diffley, and W. Hammerschmidt. 2001. Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein-Barr virus. EMBO J. 20:4588-4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shinohara, H., M. Fukushi, M. Higuchi, M. Oie, O. Hoshi, T. Ushiki, J. Hayashi, and M. Fujii. 2002. Chromosome binding site of latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus is essential for persistent episome maintenance and is functionally replaced by histone H1. J. Virol. 76:12917-12924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos, et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 55.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stedman, W., Z. Deng, F. Lu, and P. M. Lieberman. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566-12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verma, S. C., T. Choudhuri, R. Kaul, and E. S. Robertson. 2006. Latency-associated nuclear antigen (LANA) of Kaposi's sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 80:2243-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verma, S. C., K. Lan, T. Choudhuri, and E. S. Robertson. 2006. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through its cis-acting elements within the terminal repeats. J. Virol. 80:3445-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verma, S. C., and E. S. Robertson. 2003. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 222:155-163. [DOI] [PubMed] [Google Scholar]
- 60.Wohlschlegel, J. A., B. T. Dwyer, S. K. Dhar, C. Cvetic, J. C. Walter, and A. Dutta. 2000. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290:2309-2312. [DOI] [PubMed] [Google Scholar]
- 61.Wong, L. Y., and A. C. Wilson. 2005. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 79:13829-13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wysokenski, D. A., and J. L. Yates. 1989. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol. 63:2657-2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yates, J. L., and N. Guan. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65:483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye, F. C., F. C. Zhou, S. M. Yoo, J. P. Xie, P. J. Browning, and S. J. Gao. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121-11129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]