

Abstract
IκBα is the major regulator of transcription factor NF-κB function. The ankyrin repeat region of IκBα mediates specific interactions with NF-κB dimers, but ankyrin repeats 1, 5 and 6 display a highly dynamic character when not in complex with NF-κB. Using chemical denaturation, we show here that IκBα displays two folding transitions: one, a non-cooperative conversion under weak perturbation, and another major cooperative folding phase upon stronger insult. Taking advantage of a native Trp in ankyrin repeat (AR) 6 and engineered Trps in AR2, AR4 and AR5 we identified that the cooperative transition involves AR2 and AR3 while the non-cooperative transition involves AR5 and AR6. The major structural transition can be affected by single amino acid substitutions converging to the ‘consensus’ ankyrin repeat sequence, significantly increasing the native state stability. We further characterized the structural and dynamical properties of the native state ensemble of IκBα and the stabilized mutants by H/2H exchange mass spectrometry and NMR. The solution experiments were complemented with molecular dynamics simulations to elucidate the microscopic origins of the stabilizing effect of the ‘consensus’ substitutions, which can be traced to the fast conformational dynamics of the folded ensemble.
The NF-κB/IκB system is a core element of transcriptional regulation in all eukaryotic cells playing roles in development, cell growth and apoptosis 1. This signaling system is misregulated in diseases such as cancer, arthritis, asthma, diabetes, AIDS and viral infections 2. NF-κB is an inducible transcription factor whose subcellular localization and transcriptional activity are regulated by a family of inhibitor of kappa-B (IκB) proteins 1. Despite the extensive sequence similarity between the IκB family members, each individual protein has different NF-κB inhibition efficiencies, has a different degradation rate, and responds differently to NF-κB inducing signals 3–5.
IκBα, the major inhibitor of NF-κB function, is a single polypeptide whose sequence consists of two distinct regions: an N-terminal ~60 amino acids termed the ‘signal response region’, and a C-terminal ankyrin repeat (AR) region that encompasses ~220 amino acids. This latter AR region mediates the specific interaction with NF-κB dimers, as shown in the co-crystal structure of IκBα in complex with the p50/p65 heterodimer (Figure 1a)6,7. The NF-κB/IκBα interface involves contacts with several subdomains of NF-κB, mediated by the different ankyrin repeats and a PEST sequence at the C-terminus of IκBα. The surface area of the interaction is extensive, burying more than 4000 Å2, and all six ankyrin repeats are involved in the formation of a noncontiguous contact surface (Figure 1b) 6,7.
Figure 1.
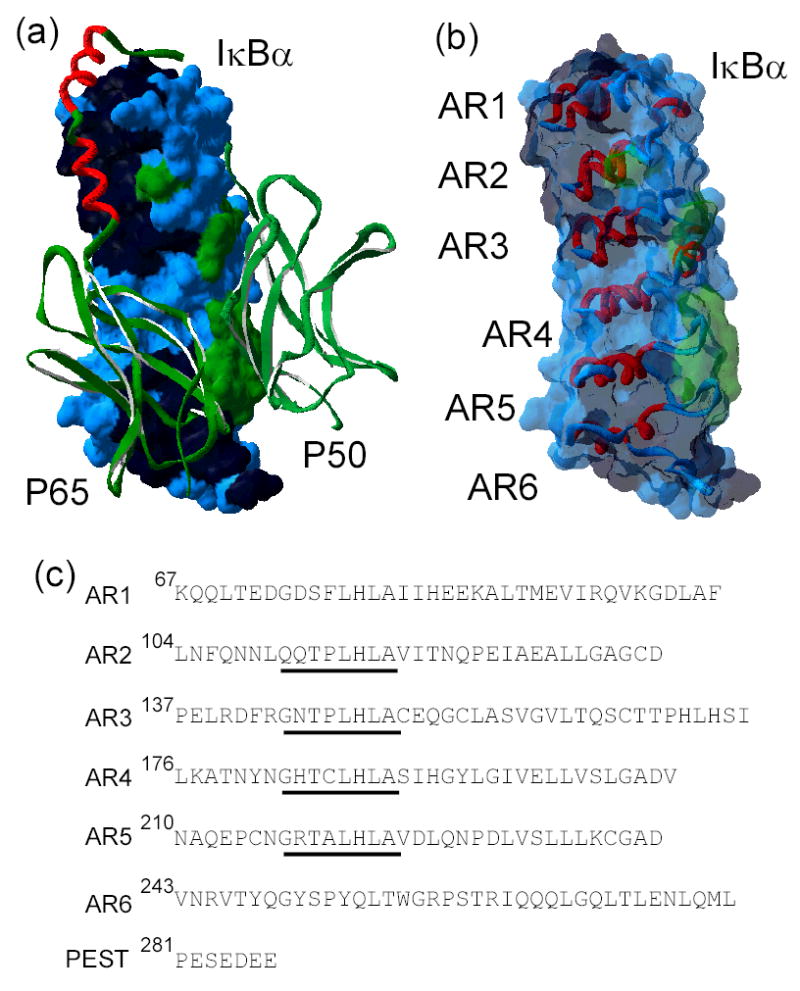
a) The x-ray crystal structure of IκBα (surface representation) in complex with NF-κB (ribbon representation) (pdb accession number 1NFI) 7. The dimerization domains of NF-κB p50 and p65 are shown in green and the NF-κB p65-NLS polypeptide in red. The surface of IκBα is colored according to the NF-κB contact regions: p65 contacting residues in black, p50 contacting residues in green, non-contacting residues in blue. b) The high resolution structure of only IκBα from the structure of the complex in the same orientation as in a), with a ribbon representation of the ankyrin repeats (AR), and the same coloring scheme as in a) for the translucent surface. c) Amino acid sequence of the ankyrin repeat domain of IκBα(67 to 287), aligned according to each ankyrin repeat. The ‘consensus’ GxTPLHLA motif is underlined.
Attempts to crystallize IκBα were unsuccessful in the absence of the NF-κB binding partner (G. Ghosh personal communication), and the AR domain has a strong tendency to aggregate when isolated at physiological temperature 8. The first, fifth, and sixth ARs of IκBα display a highly dynamic character when not complexed with NF-κB, as evidenced by the extent of amide H/2H exchange 8. Thus, it has been suggested that in the ‘free state’ of IκBα, parts of the molecule do not form a compact fold, but rather resemble a ‘molten globule’ 8. Since this is not a characteristic of all ankyrin repeat proteins, it is unclear what characteristics of its sequence determine the folding and stability of IκBα, and how these modulate NF-κB binding and, ultimately, signaling.
Ankyrin repeat (AR) proteins owe their name to the cytoskeletal protein Ankyrin, which contains 24 tandem copies of similar repetitions of ~33 amino acids. Since first being discovered, over 6000 non-redundant AR proteins have been identified. Some of these contain as few as four repeats, or as many as 29 9. Family members act as signaling proteins, cytoskeletal constituents or adaptor proteins and may be localized in the nucleus, cytoplasm, or may be membrane-bound or secreted 10. The structures of 20 naturally occurring AR proteins and of five designed AR proteins have been solved 11. In all cases, the AR domains adopt a highly similar fold: the repeats stack against each other in a linear fashion by folding into two antiparallel α-helices connected by a short loop, followed by a β-hairpin that protrudes away from the helical stack. This non-globular fold is stabilized by both intra and inter-repeat interactions. Interhelical interactions both within and between repeats are predominantly hydrophobic, while H-bonding interactions occur in the β-hairpin loop region of one repeat with the adjacent ones. This architecture results in the formation of a right-handed solenoid with a large solvent accessible surface area 12.
Natural AR proteins are composed of degenerate repeating sequences, where no position remains strictly invariant. However, the AR fold can be specified by the probability of amino acid occurrence at each position of the known protein sequences 13,14. Using this approach, consensus designed AR proteins have been successfully synthesized. These consensus proteins display a compact AR fold and have a high thermodynamic stability 15,16,14,17. Kohl et al. have noted that the variation of residues outside of the consensus positions also influence stability 16.
Despite the apparently modular architecture of AR domains, the equilibrium folding mechanisms of most AR models can usually be described by a two-state folding transition, which assumes that only the denatured and fully folded species are significantly populated (Main et al., 2003 and 2005 and references therein 18,19). Both experiments and simulations suggest that the two-state character of the transition can be understood if the AR domains fold up by a mechanism that is reminiscent of a nucleation-propagation growth 20,21. According to this model, once initial nucleation takes place, the different structural modules fold up over it in a highly cooperative fashion. Subtle variations in the interactions between modules, however, may result in decoupling of the individual elements, giving rise to more complicated folding scenarios where partially folded intermediates can be detected 22,23,20,24. Still, our understanding of the relationship between repeat number, domain stability, inter-module coupling and protein function is incomplete.
In the case of IκBα, native topology-based models using the structure of IκBα taken from the structure in complex with NF-κB, predict that two separate folding events are necessary to attain complete folding, each encompassing the folding of roughly three consecutive ARs 20. The folding appears to nucleate at AR2-AR3 and propagates outward to include AR1 and AR4. Folding of AR5 and AR6 occurs in the second folding transition. In this work, we present experimental evidence that IκBα displays two structural transitions: one, a non-cooperative transition under weak perturbation, and another major cooperative unfolding phase upon stronger insult. Furthermore, we show that the major structural conversion can be significantly affected by single amino acid substitutions converging to the consensus AR sequence and that they increase the native state stability. Finally, we complement the solution experiments with molecular dynamics simulations to elucidate the microscopic origins of the stabilizing effect, which can be traced to the fast conformational dynamics of the folded ensemble.
Results
Equilibrium folding behavior of wild type Iκ Bα
The ankyrin repeat (AR) domain of IκBα (amino acids 67 to 287) was expressed in soluble form and purified as previously described 8. The purified protein displayed high α-helical secondary structure content, as indicated by the far-UV circular dichroism (CD) spectrum (Figure 2a). The absolute value of the CD signal was in the range expected based on the co-crystal structure of IκBα and NF-κB 6,7. Moreover, it has been shown that this ‘free’ state has as much helical signal as the NF-κB bound form 8. The free IκBα displayed a single sharp transition upon thermal denaturation, with a midpoint of 45.3 °C (inset to Figure 2a, Table 1). During thermal denaturation, a significant CD change took place before the major transition occurred, accounting for about one quarter of the total ellipticity difference. Unfortunately, the thermal transition was irreversible, owing to the formation of a higher-order oligomer 8, precluding a deeper thermodynamic analysis of the data. Therefore, we report only the apparent Tm for the major phase when comparing mutant forms of IκBα.
Figure 2.
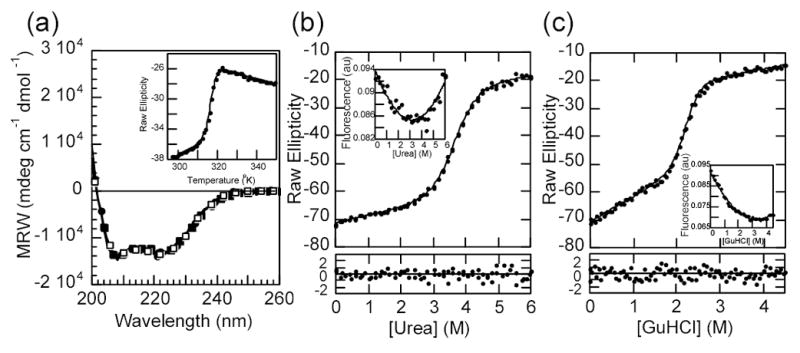
a) The far UV circular dichroism spectra of IκBα and selected mutants: wild type, continuous line; Q111G, open circles; C186P·A220P, closed circles, Q111G·C186P·A220P, closed squares. Inset: thermal unfolding of IκBα wild type followed by the CD signal at 225 nm. b) Urea denaturation curve of IκBα wild type at 3 μM total protein concentration, (conditions described in Materials and Methods) followed by the CD signal at 225nm. The line represents the best fit of the data to a two-state folding model with a linear drift in the native ensemble baseline. The residuals of the fit are shown in the bottom panel. Inset: intrinsic fluorescence of IκBα collected during the same experiment. The line is drawn through the points merely to guide the eye. c) GuHCl denaturation curve of IκBα wild type in the same conditions as in a). Insert: intrinsic fluorescence of IκBα collected during the same experiment.
Table 1.
Equilibrium Folding Parameters of IκBα
| denaturant | Temp | urea | GuHCl | ||||
|---|---|---|---|---|---|---|---|
| Protein | Tm (ºC) | ΔGH2O (kcal mol−1) | m kcal mol−1 M−1) | baseline (deg M−1) | ΔGH2O (kcal mol−1) | m kcal mol−1 M−1) | baseline (deg M−1) |
| Wild type (67-287) | 45.3 ± 0.5 | 6.8 ± 0.5 | 1.9 ± 0.2 | 3.1 ± 0.2 | 8.1 ± 0.5 | 3.7 ± 0.3 | 6.0 ± 0.3 |
| Q111G | 50.1 ± 0.5 | 9.1 ± 0.8 | 2.0 ± 0.2 | 2.5 ± 0.2 | 12.2 ± 0.9 | 4.6 ± 0.3 | 4.2 ± 0.2 |
| A133W | 36.9 ± 0.5 | 3.7 ± 0.4 | 1.3 ± 0.1 | 3.2 ± 0.8 | ND | ND | ND |
| A133W1 | ND | 3.5 ± 0.2 | 1.2 ± 0.07 | NA | ND | ND | ND |
| C186P | 49.8 ± 0.5 | 8.9 ± 1.1 | 2.4 ± 0.3 | 2.8 ± 0.3 | 9.7 ± 0.8 | 4.2 ± 0.3 | 5.4 ± 0.4 |
| L205W | 38.8 ± 0.5 | 3.1 ± 1.0 | 1.5 ± 0.6 | 2.9 ± 1.6 | ND | ND | ND |
| A220P | 46.8 ± 0.5 | 7.0 ± 0.6 | 2.0 ± 0.2 | 3.0 ± 0.3 | 8.6 ± 0.5 | 4.0 ± 0.2 | 6.2 ± 0.3 |
| C239W | 42.8 ± 0.5 | 5.6 ± 0.8 | 1.8 ± 0.3 | 2.1 ± 0.5 | ND | ND | ND |
| C186P·A220P | 51.9 ± 0.5 | 8.3 ± 0.8 | 2.2 ± 0.2 | 2.4 ± 0.2 | 10.9 ± 0.7 | 4.8 ± 0.3 | 4.2 ± 0.3 |
| C186P·A220P·A133W | 43.2 ± 0.5 | 5.7 ± 0.7 | 1.7 ± 0.2 | 2.6 ± 0.5 | 7.4 ± 0.7 | 3.6 ± 0.3 | 7.7 ± 0.4 |
| C186P·A220P·A133W1 | ND | 5.7 ± 0.1 | 1.71 ± 0.03 | NA | 7.1 ± 0.5 | 3.4 ± 0.2 | NA |
| C186P·A220P·Q111G2 | 56.8 ± 0.5 | 11.3 ± 1.4 | 2.3 ± 0.3 | 1.9 ± 0.2 | 11.1 ± 0.4 | 4.2 ± 0.2 | 1.0 ± 0.2 |
| Wild type (67-206) | 45.1 ± 0.5 | 6.7 ± 0.6 | 2.1 ± 0.2 | 0.8 ± 0.3 | 7.7 ± 0.3 | 4.0 ± 0.1 | 1.9 ± 0.2 |
determined by Trp fluorescence signal.
data below 1M GuHCl was excluded from the fit of the data.
To gain insight into the equilibrium folding mechanism of IκBα, we sought conditions where the folding transitions were fully reversible. At pH 7.5, in 50 mM NaCl, 1mM DTT, 25 mM Tris-HCl buffer, the refolding upon chemical denaturation was >95% reversible, as determined by CD and size exclusion chromatography. Denaturation curves were obtained using either urea or guanidinium hydrochloride (GuHCl) as denaturants (Figure 2 b, c). The CD signal changed upon addition of denaturant and displayed a sharp transition, typical of a cooperatively folded unit. Again, as in the thermal denaturation, a significant part of the ellipticity change took place before the major transition occurred. This linear change accounted for 20% of the total amplitude when urea was used as a denaturant, and about 30% if the denaturant was GuHCl.
Despite the repeating protein architecture and the apparent complications of the folding transitions, the CD change could be fitted to a simple two-state folding model, for which a linear change of the folding free energy with denaturant concentration may be assumed (Figure 2 b, c) 25. Similar behavior has been observed for other repeat proteins (Main et al., 2003 and references therein 18), which would suggest that only the unfolded and the fully folded ensembles are significantly populated at any given point. The ellipticity changes taking place at low denaturant concentrations were included as a linear effect on the pre-transition baseline in the global fit (see Materials and Methods). From this fit, it was possible to extract the folding free energy in water (ΔGH2O), the cooperativity parameter (m-value) and the pre-transition slope, which are reported in Table 1. The values for all of these parameters are in line with those reported for other natural AR proteins of similar sizes 18.
In parallel with the sloping pre-transition, there was a small but measurable change in the intrinsic fluorescence of IκBα which is due to the single tryptophan in AR6 (inserts to Figures 2 b, c). About 90% of the amplitude of the fluorescence change occurred below 1 M denaturant, clearly below the concentration at which the major CD change occurred. Attempts to eliminate the pre-transition with the use of additives (salt, glycerol, TMAO), or by lowering the temperature, that often stabilize native structures or increase the cooperativity of transitions, had little or no affect on the fluorescence change, which remained non-cooperative (data not shown). The majority of the fluorescence signal was not reporting on the major cooperative folding transition, but instead was reporting local changes in AR6, the location of the Trp residue.
In summary, unlike other reported ankyrin repeat proteins, the ‘free state’ of IκBα displays two structural transitions: one, a non-cooperative transition, and another major cooperative unfolding phase. We sought, therefore, to probe how each of these folding transitions is affected by mutational perturbation.
Design and folding properties of consensus mutants
Unlike globular proteins, repeat proteins consist of several tandemly arranged structural units that adopt a similar fold. As a result, the local environments around the individual positions are expected to be structurally similar along the different repeats if the same amino acid sequence is strictly repeated 14. However, IκBα, as any other natural AR protein, consists of repeats with different sequences (Figure 1c) and as such the resulting folding mechanism may depend on subtle differences of the amino acid sequence and/or the protein microenvironment 20,24. In the case of repeat proteins, it has been shown that synthetic proteins that are coded by the conserved ‘consensus’ sequence are highly stable 15,13,17. Given the marginal thermodynamic stability of IκBα, mutations to the consensus residues were introduced at those sites that deviate from the strongest signatures 18. The most prevalent signature in the ankyrin consensus is the GXTPLHLA motif at the beginning of each repeat (see Figure 1c). When folded, this motif forms the framework of the β-turn protruding from the α-helical stack. We engineered single amino acid substitutions in AR2, AR4 and AR5 to conform to the consensus signature and then analyzed the folding stablity and dynamics of the resulting mutant IκBα proteins.
The mutant proteins were expressed and purified in the same way as the wild type, and eluted as single peaks in size exclusion chromatography corresponding to monomers. The far UV CD spectra of the proteins were superimposable with those of the wild type protein, indicating that the substitutions did not perturb the overall secondary structure content (Figure 2a). The single site substitutions to the GXTPLHLA consensus were all slightly stabilizing, leading to increases in apparent Tm of up to 5 °C (Table 1). Similar to wild-type IκBα, thermal denaturation of all the mutant IκBα proteins was irreversible and led to formation of higher-order oligomers suggesting a similar irreversible thermal denaturation mechanism.
In addition to thermal denaturation, chemical denaturation experiments were performed for each protein with either urea or GuHCl. The curves were fitted to the described model, and the extracted parameters are presented in Table 1. Although the thermal transition is irreversible, the apparent Tm correlates with the extracted free energy of unfolding extrapolated from the chemical denaturation experiments. This suggests that the formation of higher-order oligomers is somehow related to the overall stability of the domain. Similarly, the unfolding free energies calculated for either chemical denaturant were related, even though the absolute values differed (Table 1). The stronger denaturant effect of GuHCl as compared to urea has often been attributed to the marked ionic strength change upon GuHCl perturbation that may well affect the folding transitions 25. As for the wild type, all the mutants showed a significant slope in the pre-transition baseline. The absolute value of the pre-transistion baseline is slightly affected by the amino acid substitutions (Table 1), but we found no significant correlation between the slope of the baseline and the unfolding free energy.
Wild type IκBα contains 10 prolines, three of which lie in the conserved GXTPLHLA positions of AR2, AR3 and AR6 (Figure 1c). Together, the mutations that converged to the consensus positions in AR4 and AR5 caused a stabilization of about ~2.7 kcal/mol (C186P·A220P; Table 1). Analysis of the single site substitutions shows that the majority of the stabilizing effect is brought about by the substitution in AR4 (C186P), since this single mutant retains most of the energy gained by the double substitution (Table 1). Interestingly, the most stabilizing substitution, Q111G, is located in AR2 (Table 1). This is the only AR of the wild type IκBα that does not have a Gly in that relative position of the consensus sequence.
Complete replacement converging to the consensus signature in AR2 through AR5 is established by combining the three described substitutions, Q111G·C186P·A220P. This mutant showed a remarkable increase in stability when compared to the wild type protein (Table 1). The apparent Tm was more than 10 °C higher than that observed for the wild type protein, and chemical denaturation indicated that it gained 4.5 kcal/mol in the free energy of unfolding. While most of the contributions to this change could be attributed to the Q111G mutation, we note that the free energies of unfolding of the individual substitutions were additive within error for the urea denaturations but not for the GuHCl denaturations. Remarkably, there was a qualitative change in the folded ensemble baseline in the GuHCl denaturation curve (Figure 3a). In this case, two separable components of the pre-transition were observed. A transition between 0 and 1M GuHCl was now separated by a flat region that accompanied the major cooperative unfolding transition that had shifted to significantly higher GuHCl concentration. Fitting these data to a three-state model gave the same ΔGH2O and m value for the major transition as were obtained from fitting to a two-state model using only the data for the major transition (see Table 1).
Figure 3.
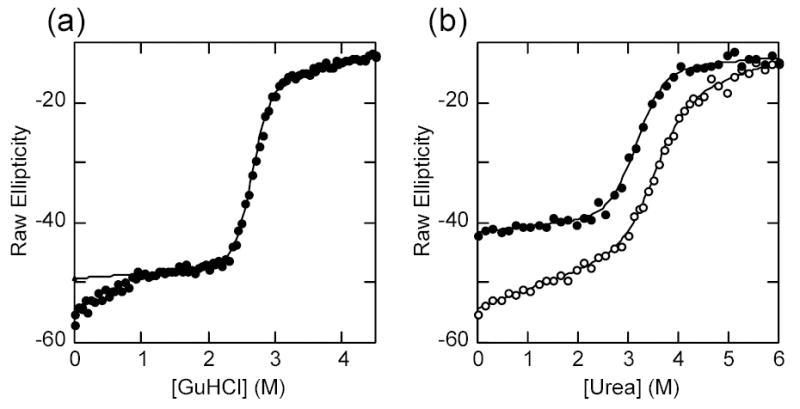
a) GuHCl denaturation curve of IκBα Q111G·C186P·A220P at 2 μM under standard conditions. The line represents the best fit to a two-state folding model. The data points below 1M were left out of the fit. b) Urea denaturation curve of IκBα(67-206) (closed circles) with the urea denaturation curve for the wild type protein shown for comparison (open circles). The line represents the best fit to a two-state folding model.
1-anilinonaphtalene-8-sulfonate (ANS) is an extrinsic probe that is used for characterizing conformational states of proteins. It is generally thought to monitor conformational changes and the presence of ‘partially folded’ states because of its ability to bind to relatively hydrophobic pockets of proteins, but not to the native or denatured states 26. Protein species capable of binding ANS are usually indicative of a ‘molten-globular’ state, which are thought to preserve a protein core and native-like secondary structure, but lack the tight packing of side chains, and are more flexible than the native state 27,28. Under native conditions, IκBα binds ANS with a stoichiometry of about five ANS molecules per protein 8. In contrast, the stabilized mutants clearly bound less ANS, corresponding to about one less ANS binding site (data not shown). Thus, it is reasonable to assume that the stabilizing effect is related to the native state ensemble properties.
Folding properties of a deletion mutant
Unlike globular domains, repeat-based architecture suggests that large sequence deletions might be done without severely disturbing the overall fold. Terminal deletions of this kind have been assayed in some ankyrin repeat proteins, and the resulting fragments shown to retain a cooperatively folded structure 21,29. In order to dissect how the C-terminal repeats of IκBα contribute to the pre-transition baselines of the folding curves, we engineered C-terminal deletion mutants. Most of the deletions resulted in insoluble protein expression (not shown), but a fragment encompassing only the first four ankyrin repeats of IκBα (amino acids 67-206) was soluble and could be purified to homogeneity 30. The thermal denaturation profile of IκBα67-206 showed a cooperative, irreversible, unfolding transition with a midpoint of 45 °C comparable to that of wild-type protein except that the pre-transition baseline was flat. Reversible chemical denaturations with either urea or GuHCl gave the same ΔGH2O and m values as the wild type protein except again the pre-transition baseline was flat (Table 1, Figure 3b). When the chemical denaturation was done under the same conditions, the absolute value of the CD signal lost in the pre-transition baseline for the wild type protein could be readily ascribed to the deleted terminal ankyrin repeats (Figure 3b).
Spectroscopic probes
In order to develop a local probe for the folding of the N-terminal ankyrin repeats, we introduced a Trp residue in the second helix of AR2. We chose to replace Ala 133 because it is located at a non-conserved position of the ankyrin fold and is expected to be protected from solvent and hence display a fluorescence change upon unfolding. The resulting protein (A133W) was expressed and purified, and the fluorescence emission spectra of both proteins are shown in Figure 4a. The intrinsic fluorescence signal of the wild type IκBα emanates mainly from a single Trp residue, present in the AR6 at position 258. Its emission spectrum showed a maxima around 348 nm, indicating that the electronic environment of this Trp residue is not strongly apolar 31. Its emission does, however, shift to higher wavelength upon denaturation, consistent with further exposure to a polar environment. On the other hand, the signal from the A133W mutant, which contains the additional Trp in AR2, displayed a high intensity that was quenched upon unfolding (Figure 4a). The emission maximum shifts to a similar extent in both proteins, indicating that the local environment of W133 is as polar as the one for W258.
Figure 4. Probing the local folding of AR2-AR3.
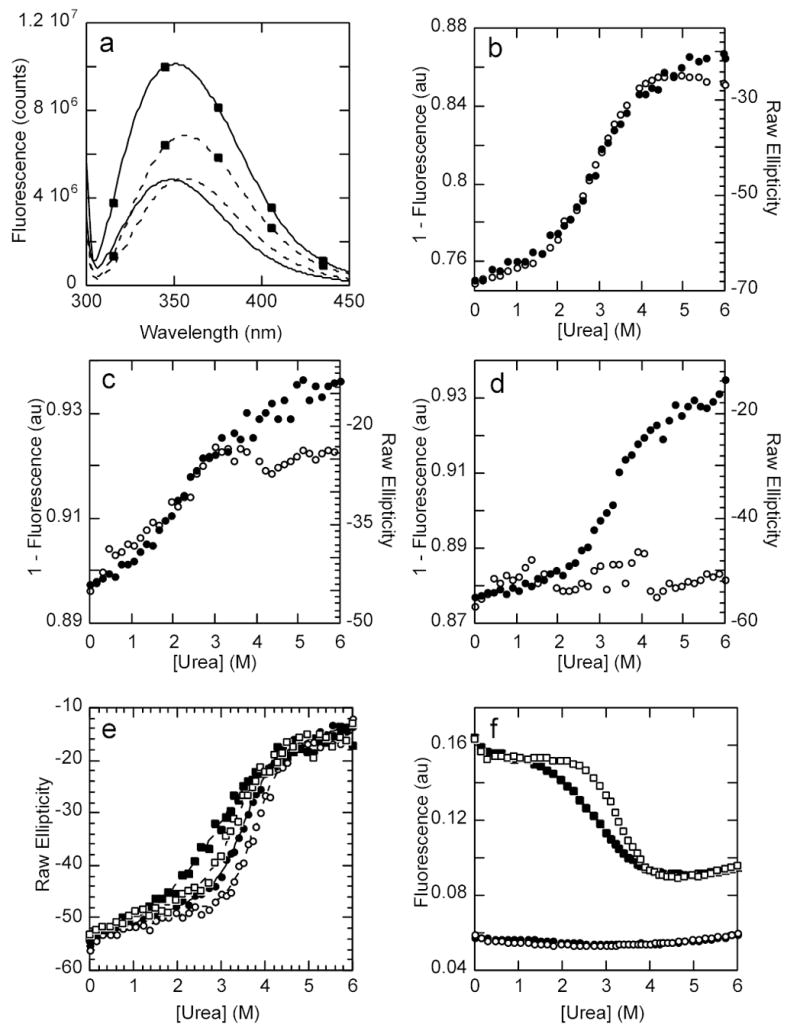
a) Fluorescence emission spectra of wild type IκBα at 1 μM in standard conditions and the A133W mutant (marked by closed squares) under the same conditions. The continuous line shows the emission spectra under native conditions and the dashed line shows the emission spectra of the same proteins in 6M urea. The excitation wavelength was set to 295 nm. b) Urea denaturation curve of IκBα A133W showing the CD signal (closed circles) and the fluorescent signal (open circles). c) Urea denaturation curve of IκBα L205W showing the CD signal (closed circles) and the fluorescent signal (open circles). d) Urea denaturation curve of IκBα C239W showing the CD signal (closed circles) and the fluorescent signal (open circles). e) Urea denaturation curves followed by the CD signal at 225nm of IκBα wild type (closed circles), A133W (closed squares), C186P·A220P (open circles) and A133W·C186P·A220P (open squares). f) Urea denaturation curves followed by the fluorescent signal of the same proteins, with the same symbols as in e).
The urea induced unfolding of A133W monitored by CD qualitatively resembled that of the wild type protein: a major cooperative transition with a significant pre-transition slope (Figure 4b). In contrast to what was observed for the wild type protein where the Trp is located only in AR6, the fluorescence change of the A133W mutant unequivocally paralleled the CD change (Figure 4b). The folding parameters extracted upon fitting either set of data to the same folding model were identical (Table 1). The urea induced denaturations indicated that the mutation caused a destabilization of about ~3 kcal/mol (Table 1). GuHCl denaturation was also performed, but the destabilization caused by the mutation precluded confident fitting of the data due to the lack of a proper baseline (data not shown). Taken together, these data strongly suggest that the major cooperative folding transition involves AR2, which can be perturbed by a local mutation and monitored by an independent probe, namely, the fluorescent change of the incorporated Trp.
To further point to the contributions of the C-terminal ankyrin repeats, Trp mutations were independently introduced in AR4 and AR5, in the analogous positions to A133W. Both proteins were soluble and could be purified, and thermal and chemical denaturation experiments were performed (Table 1, Figure 4c–d). Similar to what was observed for A133W, the mutation L205W causes a general destabilization of the fold, and its fluorescent signal change parallels the CD change (Figure 4c). In contrast, the introduction of a Trp in AR5 (C239W) does not strongly affect the folding profile and the change in fluorescence signal with denaturant did not follow the CD change (Figure 4d). Together with the deletion mutant described in the previous section, the results obtained for these mutants suggest that AR5 and AR6 do not strongly participate in the major cooperative folding phase observed for the wild type protein.
To quantify the effects of distal mutations, we modified IκBα with both C186P·A220P in the A133W background. This triple mutant had an apparent Tm that was similar to the wild type protein presumably because the destabilizing effect of the A133W is now compensated by the stabilizing C186P·A220P substitutions (Table 1). The urea denaturation profiles of this and the related single and double mutants are shown in Figure 4. In all cases, the CD signal displayed a single cooperative transition with a significant pre-transition baseline (Figure 4e). The curves all fit well to a two-state model accounting for the pre-transition baseline (Table 1). The fluorescence change was mainly associated with W133, since it overwhelmed the signal of W258 when the experiment was done under the same conditions (Figure 4f). Using the fluorescence of W133, we observed a stabilization of the pre-transition slope caused by the proline substitutions in AR4 and AR5. For the cooperative transition, the parameters derived from the fitting of the fluorescence data closely matched the values derived from the CD signal, indicating that both probes were likely reporting the same folding event (Table 1). In this case, the free energy change of the triple mutant could be approximated as the additive calculation of the separated contributions within experimental error. In summary, the effect caused by mutation to the GXTPLHLA consensus motif in AR4 and AR5 had distinctive effects on AR2 that were detected by fluorescence changes in the Trp introduced in AR2 as well as by the major cooperative transition detected by CD.
Conformational dynamics probed by H/2H exchange
Amide H/2H exchange experiments followed by mass spectrometry were used to probe the solvent accessibility of different regions of wild type IκBα and selected mutants. Similar to our previous results for IκBα(67-317) 8, digestion of the IκBα(67-287) proteins with pepsin resulted in 19 peptides that covered 74% of the sequence. It is interesting to note that since IκBα is comprised of repeated structural elements, pepsin cleavage results in peptides spanning similar secondary structural elements for each AR, allowing direct comparison between them 8. Wild type IκBα and the C186P·A220P, A133W·C186P·A220P and Q111G·C186P·A220P mutants were incubated for various deuteration times, and kinetic plots of the increase in deuteration over time were generated. Figure 5 compares the amide H/2H exchange results for selected regions of the various mutants. Considering that ankyrin repeat proteins form elongated non-globular structures, the exchange rate of IκBα is notably asymmetric 8. After 2 min of exchange, the amides of AR2 and AR3 incorporate less than one fifth of the possible deuterons, while the equivalent regions of AR1 and AR5 incorporate four fifths. All mutants showed the same general exchange pattern as the wild type however quantifiable data on the β-hairpin of AR1 could not be obtained due to a new peptide from the C186P·A220P mutation that overlapped (Figure 5). There was a small but measurable decrease in the exchange of overlapping peptides corresponding to the β-hairpin of AR5 in the C186P·A220P mutant but no difference was observed in AR4 (Figure 5d vs. 5c). After accounting for the fact that mutation to proline removes one possible deuteration site, the difference corresponded to 2 amides that were no longer exchanging. The Q111G mutation in AR2 showed a small decrease in amide H/2H exchange in the β-hairpin of AR2 (Figure 5e). The A133W mutation, also in AR2, showed small increases in exchange in the β-hairpin of AR2 as well as in the variable loop of AR1 (Figure 5e and 5f). Although the deuteration of AR6 was not quantifiable in the triple mutants, the peptide mass envelopes showed no differences in deuteration (data not shown). Thus, the mutations caused small local effects on the backbone amide exchange, but did not cause any long-range effects.
Figure 5.
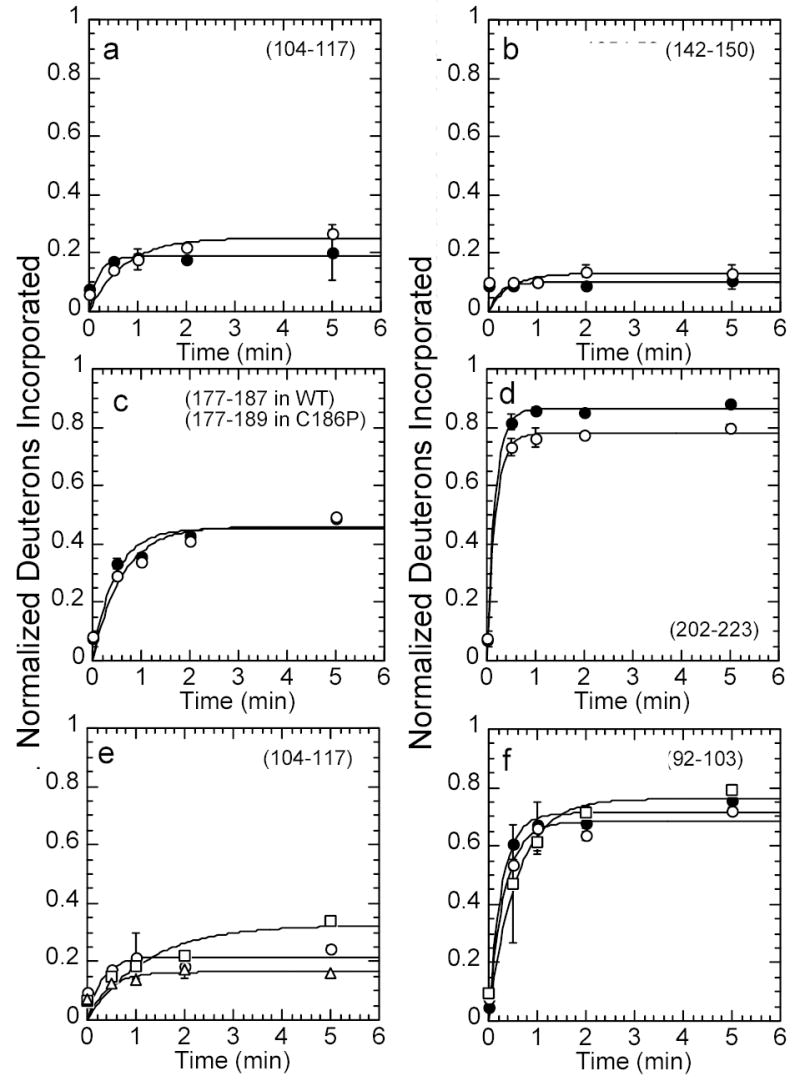
Kinetic plots of amide H/2H exchange (fit to a single exponential) in wild type IκBα, IκBα C186P·A220P, IκBα A133W·C186P·A220P, and IκBα Q111G·C186P·A220P. Deuterium incorporation was compared in wild type IκBα (filled circles) and IκBα C186P·A220P (open circles) in (a) the β-hairpin loop in AR2 (residues 104-117, 12 amides), (b) the β-hairpin loop in AR3 (residues 142-150, 7 amides), (c) the β-hairpin loop in AR4 (residues 177-187, 10 amides in wild type IκBα, residues 177-189, 11 amides in C186P), (d) the β-hairpin loop in AR5 and the end of the variable loop in AR4 (residues 202-223, 19 amides in wild type IκBα, 18 amides in A220P). Deuterium incorporation was also compared in IκBα C186P·A220P (open circles), IκBα A133W·C186P·A220P (open squares), and IκBα Q111G·C186P·A220P (open triangles) in (e) the β-hairpin loop in AR2 (residues 104-117, 12 amides), and (f) the variable loop in AR1 (residues 92-103, 11 amides). The amide H/2H exchange in Q111G·C186P·A220P was the same as that in C186P·A220P, within error, so it was omitted for clarity in panel (f).
Conformational dynamics probed by NMR
The 1H, 15N heteronuclear single quantum coherence (HSQC) spectrum of the wild type IκBα showed only 146 out of the possible 208 cross-peaks and widely varying line-widths and intensities indicative of conformational exchange (Figure 6, red). The large number of resonances observed at random coil chemical shifts was unusual when compared to results from other AR proteins 32–34. The observed spectrum was consistent with substantial conformational heterogeneity, reflecting motions on the chemical shift timescale 35,36. The 1H, 15N HSQC spectrum of the C186P·A220P mutant (Figure 6a, red) was collected under the same conditions and compared to the spectrum of the wild type protein (Figure 6, black). The proline substitutions dramatically improved the quality of the NMR spectrum. In this case, 198 of the 206 expected cross peaks were observed. Most of the cross peaks had narrower line-widths compared to the wild type protein with larger decreases seen in AR4 (Table 2). The 1H, 15N HSQC spectrum of the Q111G·C186P·A220P mutant showed 202 of the 206 expected cross peaks, similar to the C186P·A220P mutant (data not shown). These observations strongly suggest that the stabilized mutants of IκBα adopt a somewhat more compactly folded structure, having reduced conformational fluctuations.
Figure 6.
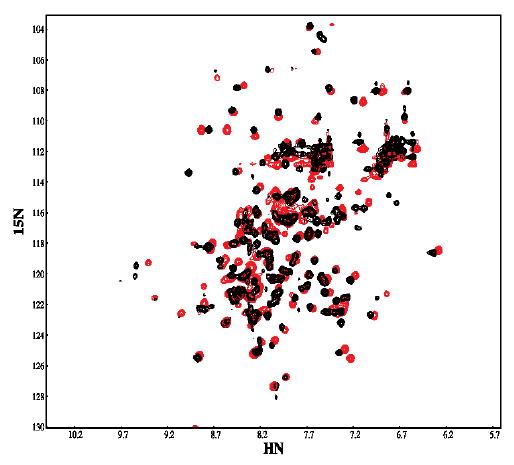
The 1H 15N HSQC NMR spectrum of the wild type protein (black) overlaid with the spectrum of the C186P·A220P mutant (red). Spectra were recorded at 800 MHz in buffer containing 25 mM Tris , 50 mM NaCl, 50 mM arginine, 50 mM glutamic acid, 1 mM DTT, 5mM CHAPS and 0.5 mM EDTA in 90% H2O 10% D2O at pH 7.5 and 15 °C.
Table 2.
Width at half-height for assigned residues in the 1H.15N HSQC spectrum of wild type vs. C186P+A220P IκBα
| Ankyrin Repeat | Secondary structure | residue | peak width wild type (Hz) | peak width C186P·A220P (Hz) | difference |
|---|---|---|---|---|---|
| 1 | β-hairpin | G74 | 36.1 | 23.0 | 13.1 |
| 1 | helix | A81 | 27.2 | 23.4 | 3.8 |
| 1 | loop | G99 | 31.7 | 20.9 | 10.8 |
| 2 | helix | A118 | 29.2 | 26.4 | 2.8 |
| 2 | loop | G134 | 35.2 | 22.5 | 12.7 |
| 3 | β-hairpin | G144 | 37.6 | 22.8 | 14.8 |
| 4 | β-hairpin | G185 | 45.5 | 32.3 | 13.2 |
Molecular Dynamics simulation of Iκ Bα conformational dynamics
Detailed atomistic computer simulations can help investigate the complex interactions that underlie protein dynamics 37,38. Aiming at a molecular description of the effects caused by making the consensus substitutions in IκBα, we undertook the simulation of the fast conformational dynamics of these proteins using an empirical force field 39. The analysis focused on a comparison of the wild type and the C186P·A220P mutant.
The initial coordinates of IκBα were taken from the high resolution structure of the co-crystal of IκBα in complex with NF-κB 7. The molecular dynamics were performed by simulating the 213 amino acid protein in explicit solvent at a constant temperature of 300 K. The same protocol was followed for simulating the dynamics of IκBα C186P·A220P, for which we introduced the mutations in the designed positions. After initial equilibration, three independent trajectories were started for each protein and followed for 5 nsec. The root mean square deviation (RMSD) of the backbone atoms for the last two nsec of the trajectories showed significant deviations from the crystal structure (Figure 7a). In all cases, the structures relaxed to an RMSD between 2 and 4 Å, and fluctuated around that position during the last nsecs of the trajectories (Figure 7a). To identify if particular regions of IκBα displayed different magnitudes of fluctuation, the RMSD of each residue was calculated (Figure 7b). In general, the larger fluctuations were found in residues 70-80 (AR1) and residues 250-287 (AR6), with AR6 showing the largest displacements. This can also be seen in Figure 8 which depicts snapshots taken from the last nsec of each trajectory. Although the helical content is not strongly affected, the 3D structure of the protein diffuses away from its initial configuration and samples a wider conformational space. This is most prevalent at the terminal repeats, with AR6 displaying the largest fluctuations (Figure 7b). In the case of the wild type protein, three additional areas fluctuated more than 2 Å from the initial structure. These regions correspond to the loop between AR1 and AR2 (residues 95 - 105), and the β-hairpin regions of AR5 and AR6 (residues 210-220 and residues 245-255, respectively). These later regions showed somewhat reduced deviations when the dynamics of the C186P·A220P mutant were simulated (Figure 7b, Figure 8), and can be considered to be a local effect of the substitutions.
Figure 7.
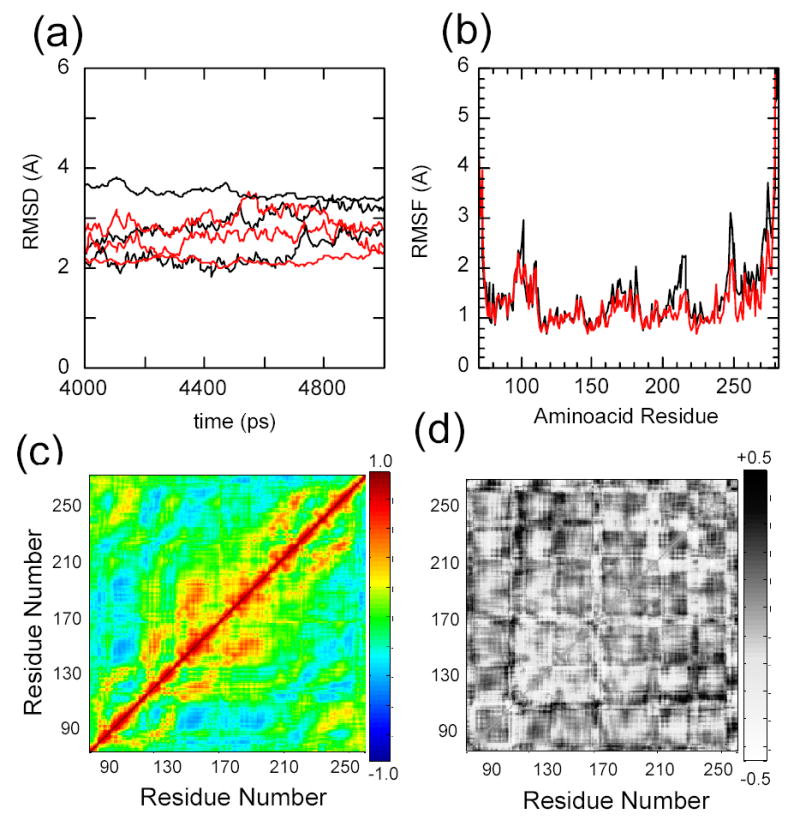
Molecular dynamics simulations of IκBα with the initial coordinates taken from the co-crystal structure of IκBα in complex with NF-κB (pdb accession 1NFI, chain E), in explicit solvent, relaxed for 5 nsec (black). The C186P·A220P mutations were computationally introduced prior to the relaxation (red). Three independent trajectories were run for each protein a) Root mean square deviation of the proteins relative to the co-crystal form. b) Average root mean square fluctuations of the individual amino acids. c) Average Cα covariance matrix of wild type IκBα. d) Cα covariance difference between wild type IκBα and the C186P·A220P mutant.
Figure 8.
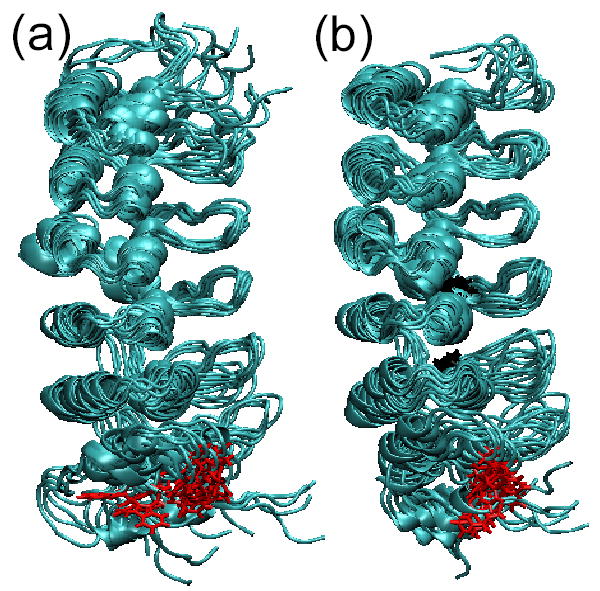
Three snapshots of the last nsec of each of the trajectories of the all-atom MD simulations superimposed on residues 112-198. a) Results for the wild type IκBα ensemble, b) Results for the IκBα C186P·A220P mutant ensemble. The Trp 258 is shown in red and Pro 186 and Pro 220 are shown in black.
Correlated motions between residues that are not close in 3D space were identified by computing the covariation in residue motion of each Cα with every other Cα. After removing the contributions of the rotational and translational degrees of freedom, the covariance calculations were performed for the ensemble of structures over the last nsec of the trajectories (Figure 7c, d). For the wild type protein, the correlations reveal a central cluster of positively covarying residues (120 to 210), corresponding to AR2, AR3, AR4 (Figure 7c). The covariance matrix of the mutant displays small changes overall. The largest differences are noticeable as an increase in the long range correlation between residues in AR2 and AR5-AR6 (110-140, 220-270) (Figure 7d)
Discussion
IκBα is one of the major regulators of NF-κB function. Our goal in this work was to probe the structural and dynamical properties of the native state ensemble of IκBα. Aiming at a molecular interpretation of IκBα function, we characterized the folding of the IκBα ankyrin repeat region and attempted to relate it to the biological properties of its native state ensemble.
Folding to the native state ensemble of Iκ Bα
It has been previously suggested that, unlike other ankyrin repeat proteins, only some of the ankyrin repeats of IκBα form a compactly folded unit when not in complex with NF-κB 8,40. Native topology-based landscape models based on the co-crystal structure of the IκBα/NF-κB complex predicted an initial folding transition which involved only the first three and a half ankyrin repeats 20. A second folding transition involved the rest of the fourth, the fifth and the sixth repeats, and we speculated that this folding transition would only occur upon binding to NF-κB. We report here that the experimental folding landscape of IκBα involves two transitions: a minor non-cooperative conversion upon subtle perturbation, and a major cooperative folding event (Figure 2).
Using stabilizing mutations and tryptophan substitutions, we were able to identify the major cooperative transition as involving the second and third ankrin repeat as predicted by the computational modeling 20. Indeed, the major cooperative folding event of IκBα can be related to the formation of a stable "core" which corresponds to the compact folding of the most stable repeating elements. Others have shown that individual repeats may differ in stability, and that after formation of the "core", less stable repeats may be added via a nucleation-propagation mechanism 22,20,21. For IκBα, this corresponds to a region that encompasses at least AR2, AR3 and AR4, since a fluorescent probe located at the AR2-AR3 interface or at the AR3-AR4 interface parallels the major CD folding transition (Figure 4). These substitutions also cause a significant decrease in the overall stability of the domain (Table 1). This region also corresponds to the repeats that show the least extent of H/2H exchange in the native state (Figure 5, and 8). In contrast, an analogous Trp substitution at the interface AR5-AR6 reports only a weak non-cooperative transition and does not have much affect on the protein stability.
Support for the contention that only the first four ARs contribute to the cooperative transition was obtained from studies of a protein in which all of AR5 and AR6 were deleted (IκBα(67-206)). This truncated protein displays a single cooperative folding transition, with parameters comparable to that of the wild-type protein, strongly suggesting that most of the stability of the native protein can be ascribed to the first four ankyrin repeats.
Stabilization of the Iκ Bα fold by convergence to the GXTPLHLA consensus
Compact, crystallizable and highly stable AR proteins have been synthesized by evaluating the probability of amino acid occurrence at each position of the known AR protein sequences 13,41,42,16,18,14. This ‘consensus’ approach identified the motifs that specify the ankyrin fold, where the most prevalent signature is the GXTPLHLA motif 18. Suggestion that this motif was important for stability came from Barrick and coworkers, where mutation of the consensus proline in each repeat of the Notch ankyrin domain resulted in a similar decreasing stability of ~1 kcal/mol 23. This motif is only strictly present in AR3 of IκBα, and differs by a single amino acid in AR2, AR4 and AR5. Introduction of the consensus motif in any of the repeats stabilized the overall fold, with the mutation to Gly in AR2 contributing most strongly (Table 1). Changes in stability upon substitution to the consensus in AR4 and AR5 yielded similar results when they were probed globally by CD or locally with a fluorescent signal in AR2 (Figure 4, Table 1).
The high conservation of the GXTPLHLA motif in natural AR proteins hints at the functional importance of this motif in AR evolution. High resolution structures have shown that this motif forms the basis of an interrepeat H-bond network that connects one ankyrin repeat to the next 16. Here, we have shown that mutations in IκBα that converge to the GXTPLHLA consensus sequence have several effects. H/2H exchange, NMR data and MD simulations all show that when the entire consensus is present, the amplitude of the dynamic fluctuations is lower than in native IκBα (Figures 6 – 8). Recalling that the GXTPLHLA sequence forms an interrepeat H-bond network, mutations that strengthen this network are likely to both increase the overall stability of the protein and decrease dynamic fluctuations. These results strongly support the previous suggestion that this motif performs a key architectural role in coupling the interaction energies between different folding elements 16.
Both experiments and simulations show that repeat proteins are likely to fold via a nucleation-propagation mechanism 20,43,21 and according to these models, the inter-element coupling is one of the most crucial parameters that describes the stability and cooperative behavior of the overall system 44. In an elegant series of experiments, Barrick and colleagues have shown that both the stability and cooperativity of the Notch AR domain are strongly influenced by the number of repeats and the coupling between them 22,21,45. As the number of repeats increases, both the cooperativity of the folding transition and the native state stability increases. We observed that introduction of the Pro in AR4 had a stronger effect than in AR5 (Table 1). This observation suggests that the consensus signature in AR4 consecutively increases the stability of the domain by contributing additional interactions between AR4 and AR2-AR3. Similarly, the C186P·A220P mutant could have additional interactions between AR5 and the rest of the repeats in the "core". Furthermore, the quality of the 1H, 15N HSQC spectrum was dramatically improved for the stabilized C186P·A220P protein, showing nearly all of the expected cross peaks (Figure 6).
Results from folding studies on the stabilized mutants, the Trp mutants, and IκBα(67-206) together strongly support the conclusion that the observed non-cooperative transition that occurred at low denaturant concentrations is due to AR5 and AR6 whereas the cooperative folding unit in IκBα corresponds to AR1-AR4. Linear non-cooperative transitions have also been observed in other proteins and in some cases these indicate downhill folding events 46. Introduction of all three stabilizing mutations in AR2–5, caused the cooperative unfolding of the now strongly stabilized main "core" to be separable from the non-cooperative transition (Figure 3a). Indeed, the sequence of AR6 is the most divergent from the consensus with only the G and P retained in the GXTPLHLA signature. This AR was not even identified as an ankyrin repeat by sequence comparisons, but was first identified when the structures of IκBα in complex with NF-κB were obtained 6,7. The MD simulations also show that AR6 displays large fluctuations even in the stabilized C186P·A220P mutant.
Functional implications of the folding landscape of Iκ Bα
NF-κB contacts IκBα in three discontinuous patches (Figure 1a). The p65-NLS polypeptide contacts parts of AR1, AR2 and AR3, and the p50 and p65 dimerization domains contact AR4, 5 and 6 (Figure 1b). However, the high resolution structures do not adequately explain the binding specificity of IκBα towards NF-κB dimers, or why mutations of interface residues have little affect on the binding affinity 40. In addition, IκBα has similar affinity towards both hetero and homodimeric forms of NF-κB, which may be related to the folding of the partners upon binding 47. When not in complex with IκB, the p65-NLS does not have a defined electron density 48,49. Here, we identified AR2-AR4 as the folded "core" of IκBα and this is precisely where the NLS contacts IκBα and folds into a helical structure as seen in the crystal structure of the complex (Figure 1). On the other hand, AR5 and AR6, which we identified here as the ‘molten’ regions of IκBα contact the dimerization domain of NF-κB, which is a compact immunoglobulin-like fold. Indeed, regions of both NF-κB and IκBα appear to fold upon binding 8,40. This mutually coupled folding / binding scenario opens the possibility of a multi-state binding reaction, where the folding depends on the intermolecular interactions 50. Since the folding of IκBα is exquisitely dependent on the coupling of distal regions, the different IκB isoforms may present large differences in binding, despite having similar contacting residues. For example, IκBβ has an insertion of about 30 amino acids between AR3 and AR4, that might alter how the ARs are coupled and therefore significantly affect the NF-κB binding energetics 51,52.
In a biological framework, the regulation of the folding events (by covalent modifications and/or binding to other molecules) could correspond to regulatory checkpoints of the signaling system. The low thermodynamic stability of the folded state may be related to the extremely short in vivo half life of IκBα 5, which is a crucial parameter of the overall NF-κB response 53. We showed here that the folding landscape of IκBα can be modified by point mutations, and it will be interesting to analyze the effects these have on binding activity, degradation rates, and the subsequent signaling response.
Materials and Methods
Reagents and chemicals
All the reagents used were purchased from Sigma (Sigma Aldrich, St Louis, MO, USA) and were the maximum purity available. DNA oligonucleotides were purchased from IDT (Integrated DNA Technologies, Coralville, IA, USA). Restriction enzymes, DNA ligase and DNA polymerase was purchased from New England Biolabs (Beverly, MA, USA).
Protein expression and purification
IκBα(67-287) was recombinantly expressed in E. coli and purified as described 8. Mutation of the IκBα gene was done by the inverse PCR method 54, and the entire protein coding sequence was checked by DNA sequencing. All the mutant proteins were expressed and purified in the same manner as the wild type protein, yielding between 5 and 20 mg per liter of bacterial culture. Protein concentration was determined by spectrophotometry using an extinction coefficient of 12950 M−1cm−1 for the proteins with a wild type background, and 18450 M−1cm−1 for the proteins in A133W background 55 and 2980 M−1cm−1 for IκBα(67-206).
Circular dichroism
Circular dichroism measurements were performed with an Aviv 202 spectropolarimeter (Aviv Biomedical, Lakewood, NJ, USA). Mean residual ellipticity was calculated as: MRW = deg / (10 x L x M x (#bonds))
where deg is the measured ellipticity, L is the pathlength in centimeters, M is the protein concentration in Molar, and #bonds is the number of peptide bonds. All the spectra were collected at a constant temperature of 25°C, unless otherwise stated.
Fluorescence
The fluorescence emission spectra were acquired in an Fluoromax-2 fluorimeter (Jobin Yvon, Edison, NJ, USA), using a 1cm pathlength quartz cuvette, at a constant temperature of 25°C.
Equilibrium folding experiments
Folding curves were performed in an Aviv202 spectropolarimeter equipped with a Hamilton Microlab 500 titrator (Hamilton, Reno, NV, USA). A 1cm fluorescence quartz cuvette containing 3 ml of 1–3 μM of the native protein in 25 mM Tris-HCl, 50 mM NaCl, 1 mM DTT, 0.5 mM EDTA, pH 7.5 and was titrated with denatured protein (7.3 – 7.8 M urea or 5.3 – 5.8 M GuHCl in 25 mM Tris-HCl, 50 mM NaCl, 1 mM DTT, 0.5 mM EDTA, pH 7.5), in 30 to 40 injection steps. Samples were equilibrated with constant stirring at 80 rpm for 180 to 300 seconds at each point prior to data collection. The CD signal was collected at 225 nm, averaged over 10 seconds, and the fluorescence signal was collected through a 320 cut-off filter with an excitation wavelength of 280nm, averaged over 5 seconds.
Folding curves were fit to a two state folding model, assuming a linear dependence of the folding free energy on denaturant concentration 56. The pre (native) and post (unfolded) transition baselines were treated as linearly dependent on denaturant concentration. The data was globally fit to:
where Sobs is the observed signal, p1 and p2 are the pre and post transition baselines, a1 and a2 their corresponding y-intercepts. ΔG is the folding free energy in water and m is the cooperativity parameter (m-value). Occasionally, the data were also fit to a three-state model as described by Hecky and Muller, 2005 57. The data were fit using a non-linear least square fitting algorithm with the Kaleidagraph program (Synergy Software, Reading, PA, USA).
Amide H/2H exchange
The exchange reaction for the free IκBα protein was initiated by diluting 130 μM wild type IκBα, IκBα C186P·A220P, IκBα Q111G·A220P·C186P, or IκBα A133W·A220P·C186P, in 50 mM Tris, 150 mM NaCl, 1 mM DTT (pH 7.5) 10-fold into D2O. The reaction proceeded for 0, 0.5, 1, 2, or 5 minutes at ambient temperature, and then the reaction was quenched by ten-fold dilution with 0.1% TFA at 0 °C (sample pH = 2.2). A 150 μL aliquot of the exchange reaction was immediately transferred to 25 μL of immobilized pepsin (Pierce Biotechnology), where digestion proceeded for 5 min, and 10 μL aliquots of each digestion were immediately frozen in liquid N2, and stored at −80 °C until analysis. Exchange reactions were performed in triplicate. Peptic peptides were sequenced using matrix-assisted laser desorption ionization tandem mass spectrometry (4800 MALDI Tof-Tof (Applied Biosystems) or a Q-STAR XL hybrid quadrupole time-of-flight mass spectrometer with an orthogonal MALDI source (Applied Biosystems).
Samples were analyzed by MALDI mass spectrometry using a Voyager DE-STR mass spectrometer (Applied Biosystems) as described previously58, except the matrix concentration was 4.5 mg/ml and the matrix was adjusted to pH 2.2. To minimize back exchange, each sample was analyzed individually. IκBα spectra were analyzed, as described previously58, to determine the average number of deuterons incorporated into each peptic peptide. Side-chain contributions due to residual deuterium (4.5 %) were subtracted from the total number of deuterons incorporated, and only the backbone deuteration of each peptide is reported. Data were corrected for back-exchange loss of deuterons during analysis, as described previously59,58, using the peptide of m/z 1374.77 from wild type IκBα after exchange for >24 hours as a reference. Back exchange was 40%.
Nineteen peptides that cover 74% of the sequence were analyzed for the wild type IκBα protein. For the C186P·A220P mutant, 13 peptides were analyzed yielding 66% coverage. For the A133W·C186P·A220P and Q111G·C186P·A220P mutants, 10 peptides yielded 56% coverage.
Nuclear Magnetic Resonance
NMR experiments were performed on a Bruker AMX-800 spectrometer. Uniformly 15N-labeled protein samples were used at concentrations of 0.2 mM. The samples contained 25 mM Tris , 50 mM NaCl, 50 mM arginine, 50 mM glutamic acid, 1 mM DTT, 5mM CHAPS and 0.5 mM EDTA in 90% H2O 10% D2O at pH 7.5 and 15°C.
Molecular Dynamics
To explore the dynamics of IκBα at a microscopic level, we performed explicit solvent molecular dynamics (MD) simulations of the wild type and a modeled C186P·A220P mutant. The starting coordinates of IκBα were obtained from the co-crystal structure of NF-κB bound to IκBα 7. The C186P·A220P mutant was generated using the MMTSB Tool Set 60 by substituting the wild type residues with prolines at positions 186 and 220. We solvated the wild type and mutant structures of IκBα with TIP3P water molecules in a box of dimensions 10 Å greater than the protein itself. The resulting dimensions of the cells were 95 Å x 54 Å x 57 Å for the wild type and 91 Å x 57 Å x 54 Å for the mutant. The final solvated protein systems contained 3,264 protein atoms and 26,622 water molecules for the wild type and 3,271 protein atoms and 26,628 water molecules for the mutant.
The all-atom CHARMM force field 61 was used for all MD simulations of wild type IκBα and the C186P·A220P mutant using a standard protocol under periodic boundary conditions. The SHAKE algorithm fixed all bonds involving hydrogens, allowing the step size of 2 fs during the course of the MD simulations. The nonbonded interactions were truncated at 12 Å using the force shift and switch methods to smooth the electrostatic and Lennard-Jones interactions, respectively, with the Lennard-Jones switching function turned on at 10 Å. The nonbonded atom list was maintained to an interatomic distance of 14 Å and it was updated heuristically. To equilibrate the water molecules around the protein, we began by harmonically constraining the protein with a force constant of 50 kcal/mol/Å2 and minimizing the system for 20 steps using steepest descent (SD) algorithm, then relaxing the force constant to 20 kcal/mol/Å2 and minimizing the system for 50 steps using the Adopted Basis Newton Raphson (ABNR) algorithm. Then, we applied 50 ps of initial MD starting at 48K and ending at 298K. For the full production run, we released the harmonic constraints on the protein and performed 5 ns of MD simulations at 300K. Three independent simulations were performed for each protein.
Acknowledgments
We thank Charles Brooks III and Ilya Khavrutskii for helpful discussions. DUF is a Jane Coffin Childs Fellow, SMET is a Cancer Research Foundation Fellow. This work was supported by NIH grant GM071862.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Ann Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Ann Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 6.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the IkappaBalpha/NF-kappaB complex reveals mechanisms of NF-kappaB inactivation. Cell. 1998;95:759–70. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 7.Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–58. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- 8.Croy CH, Bergqvist S, Huxford T, Ghosh G, Komives EA. Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 2004;13:1767–77. doi: 10.1110/ps.04731004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Letunic I, Copley RR, Schmidt S, Ciccarelli FD, Doerks T, Schultz J, Ponting CP, Bork P. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 2004;32(Database issue):D142–4. doi: 10.1093/nar/gkh088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 1999;24:311–6. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- 11.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mosavi LK, Cammett TJ, Desrosiers DC, Peng ZY. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004;13:1435–48. doi: 10.1110/ps.03554604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 14.Mosavi LK, Minor DL, Jr, Peng ZY. Consensus-derived structural determinants of the ankyrin repeat motif. Proc Nat Acad Sci U S A. 2002;99:16029–34. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P, Grutter MG, Pluckthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat Biotechnol. 2004;22:575–82. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 16.Kohl A, Binz HK, Forrer P, Stumpp MT, Pluckthun A, Grutter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc Nat Acad Sci U S A. 2003;100:1700–5. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosavi LK, Peng ZY. Structure-based substitutions for increased solubility of a designed protein. Protein Eng. 2003;16:739–45. doi: 10.1093/protein/gzg098. [DOI] [PubMed] [Google Scholar]
- 18.Main ER, Jackson SE, Regan L. The folding and design of repeat proteins: reaching a consensus. Curr Opin Struct Biol. 2003;13:482–9. doi: 10.1016/s0959-440x(03)00105-2. [DOI] [PubMed] [Google Scholar]
- 19.Main ER, Lowe AR, Mochrie SG, Jackson SE, Regan L. A recurring theme in protein engineering: the design, stability and folding of repeat proteins. Curr Opin Struct Biol. 2005;15:464–71. doi: 10.1016/j.sbi.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Ferreiro DU, Cho SS, Komives EA, Wolynes PG. The energy landscape of modular repeat proteins: topology determines folding mechanism in the ankyrin family. J Mol Biol. 2005;354:679–92. doi: 10.1016/j.jmb.2005.09.078. [DOI] [PubMed] [Google Scholar]
- 21.Mello CC, Barrick D. An experimentally determined protein folding energy landscape. Proc Nat Acad Sci U S A. 2004;101:14102–7. doi: 10.1073/pnas.0403386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bradley CM, Barrick D. Limits of cooperativity in a structurally modular protein: response of the Notch ankyrin domain to analogous alanine substitutions in each repeat. J Mol Biol. 2002;324:373–86. doi: 10.1016/s0022-2836(02)00945-2. [DOI] [PubMed] [Google Scholar]
- 23.Bradley CM, Barrick D. Effect of multiple prolyl isomerization reactions on the stability and folding kinetics of the notch ankyrin domain: experiment and theory. J Mol Biol. 2005;352:253–65. doi: 10.1016/j.jmb.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 24.Tang KS, Guralnick BJ, Wang WK, Fersht AR, Itzhaki LS. Stability and folding of the tumour suppressor protein p16. J Mol Biol. 1999;285:1869–86. doi: 10.1006/jmbi.1998.2420. [DOI] [PubMed] [Google Scholar]
- 25.Fersht AR. Structure and Mechanism in Protein Science. Freeman; New York: 1999. [Google Scholar]
- 26.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the "molten globule" intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–28. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 27.Brooks CL, 3rd, Gruebele M, Onuchic JN, Wolynes PG. Chemical physics of protein folding. Proc Nat Acad Sci U S A. 1998;95:11037–8. doi: 10.1073/pnas.95.19.11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 1995;21:167–95. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B, Peng Z. A minimum folding unit in the ankyrin repeat protein p16(INK4) J Mol Biol. 2000;299:1121–32. doi: 10.1006/jmbi.2000.3803. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Joo SH, Tsai MD. An NF-kappaB-specific inhibitor, IkappaBalpha, binds to and inhibits cyclin-dependent kinase 4. Biochem. 2003;42:13476–83. doi: 10.1021/bi035390r. [DOI] [PubMed] [Google Scholar]
- 31.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum; New York: 1999. [Google Scholar]
- 32.Yang Y, Nanduri S, Sen S, Qin J. The structural basis of ankyrin-like repeat function as revealed by the solution structure of myotrophin. Structure. 1998;6:619–26. doi: 10.1016/s0969-2126(98)00063-x. [DOI] [PubMed] [Google Scholar]
- 33.Yang Y, Rao NS, Walker E, Sen S, Qin J. Nuclear magnetic resonance assignment and secondary structure of an ankyrin-like repeat-bearing protein: myotrophin. Protein Sci. 1997;6:1347–51. doi: 10.1002/pro.5560060625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan C, Li J, Mahajan A, Poi MJ, Byeon IJ, Tsai MD. Solution structure of the human oncogenic protein gankyrin containing seven ankyrin repeats and analysis of its structure--function relationship. Biochem. 2004;43:12152–61. doi: 10.1021/bi049116o. [DOI] [PubMed] [Google Scholar]
- 35.Cliff MJ, Williams MA, Brooke-Smith J, Barford D, Ladbury JE. Molecular recognition via coupled folding and binding in a TPR domain. J Mol Biol. 2005;346:717–32. doi: 10.1016/j.jmb.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 36.Dyson HJ, Wright PE. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv Protein Chem. 2002;62:311–40. doi: 10.1016/s0065-3233(02)62012-1. [DOI] [PubMed] [Google Scholar]
- 37.Brooks CL, 3rd, Onuchic JN, Wales DJ. Statistical thermodynamics. Taking a walk on a landscape. Science. 2001;293:612–3. doi: 10.1126/science.1062559. [DOI] [PubMed] [Google Scholar]
- 38.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 39.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comp Chem. 1983;4:187–217. [Google Scholar]
- 40.Huxford T, Mishler D, Phelps CB, Huang DB, Sengchanthalangsy LL, Reeves R, Hughes CA, Komives EA, Ghosh G. Solvent exposed non-contacting amino acids play a critical role in NF-kappaB/IkappaBalpha complex formation. J Mol Biol. 2002;324:587–97. doi: 10.1016/s0022-2836(02)01149-x. [DOI] [PubMed] [Google Scholar]
- 41.Devi VS, Binz HK, Stumpp MT, Pluckthun A, Bosshard HR, Jelesarov I. Folding of a designed simple ankyrin repeat protein. Protein Sci. 2004;13:2864–70. doi: 10.1110/ps.04935704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrer P, Binz HK, Stumpp MT, Pluckthun A. Consensus design of repeat proteins. Chembiochem. 2004;5:183–9. doi: 10.1002/cbic.200300762. [DOI] [PubMed] [Google Scholar]
- 43.Kajander T, Cortajarena AL, Main ER, Mochrie SG, Regan L. A new folding paradigm for repeat proteins. J Am Chem Soc. 2005;127:10188–90. doi: 10.1021/ja0524494. [DOI] [PubMed] [Google Scholar]
- 44.Zimm BH, Bragg JK. Theory of the phase transition between helix and random coil in polypeptide chains. J Chem Phys. 1959;31:526–35. [Google Scholar]
- 45.Zweifel ME, Barrick D. Studies of the ankyrin repeats of the Drosophila melanogaster Notch receptor. 2 Solution stability and cooperativity of unfolding. Biochem. 2001;40:14357–67. doi: 10.1021/bi011436+. [DOI] [PubMed] [Google Scholar]
- 46.Oliva FY, Munoz V. A simple thermodynamic test to discriminate between two-state and downhill folding. J Am Chem Soc. 2004;126:8596–7. doi: 10.1021/ja048309w. [DOI] [PubMed] [Google Scholar]
- 47.Bergqvist S, Croy CH, Kjaergaard M, Huxford T, Ghosh G, Komives EA. Thermodynamics reveal that helix 4 in the NLS of NF-kB anchors IkBa forming a very stable complex. J Mol Biol. 2006;360:421–34. doi: 10.1016/j.jmb.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang DB, Huxford T, Chen YQ, Ghosh G. The role of DNA in the mechanism of NFkappaB dimer formation: crystal structures of the dimerization domains of the p50 and p65 subunits. Structure. 1997;5:1427–36. doi: 10.1016/s0969-2126(97)00293-1. [DOI] [PubMed] [Google Scholar]
- 49.Huxford T, Malek S, Ghosh G. Preparation and crystallization of dynamic NF-kappa B.Ikappa B complexes. J Biol Chem. 2000;275:32800–6. doi: 10.1074/jbc.M006037200. [DOI] [PubMed] [Google Scholar]
- 50.Shoemaker BA, Portman JJ, Wolynes PG. Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc Nat Acad Sci U S A. 2000;97:8868–73. doi: 10.1073/pnas.160259697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malek S, Chen Y, Huxford T, Ghosh G. IkappaBbeta, but not IkappaBalpha, functions as a classical cytoplasmic inhibitor of NF-kappaB dimers by masking both NF-kappaB nuclear localization sequences in resting cells. J Biol Chem. 2001;276:45225–35. doi: 10.1074/jbc.M105865200. [DOI] [PubMed] [Google Scholar]
- 52.Malek S, Huang DB, Huxford T, Ghosh S, Ghosh G. X-ray crystal structure of an IkappaBbeta x NF-kappaB p65 homodimer complex. J Biol Chem. 2003;278:23094–100. doi: 10.1074/jbc.M301022200. [DOI] [PubMed] [Google Scholar]
- 53.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:829–839. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clackson T, Detlef G, Jones P. PCR, a Practical Approach. In: McPherson M, Quirke P, Taylor G, editors. PCR, a Practical Approach. IRL Press; Oxford: 1991. p. 202. [Google Scholar]
- 55.Pace CN, Vajdos F, Lanette F, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–23. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–80. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 57.Hecky J, Muller KM. Structural perturbation and compensation by directed evolution at physiological temperature leads to thermal stabilization of b-lactamase. Biochem. 2005;44:12640–12654. doi: 10.1021/bi0501885. [DOI] [PubMed] [Google Scholar]
- 58.Mandell JG, Falick AM, Komives EA. Measurement of amide hydrogen exchange by MALDI-TOF mass spectrometry. Anal Chem. 1998;70:3987–3995. doi: 10.1021/ac980553g. [DOI] [PubMed] [Google Scholar]
- 59.Hughes CA, Mandell JG, Anand GS, Stock AM, Komives EA. Phosphorylation causes subtle changes in solvent accessibility at the interdomain interface of methylesterase CheB. J Mol Biol. 2001;307:967–76. doi: 10.1006/jmbi.2001.4523. [DOI] [PubMed] [Google Scholar]
- 60.Feig M, Karanicolas J, Brooks CL., 3rd MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model. 2004;22:377–95. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 61.MacKerell JAD, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher IWE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics Studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]