

Abstract
The plasminogen activators, urokinase PA (u-PA) and tissue-type PA (t-PA), are believed to play important roles in inflammatory cell infiltration, fibrin deposition, and joint destruction associated with rheumatoid arthritis; however, their precise roles in such processes, particularly u-PA, have yet to be defined. Using gene-deficient mice we examined the relative contribution of the PAs to the chronic systemic collagen-induced arthritis model. Based on clinical and histological assessments, u-PA−/− mice developed significantly milder disease and t-PA−/− mice more severe disease compared with the relevant wild-type mice. Fibrin deposition within joints paralleled disease severity and was particularly pronounced in t-PA−/− mice. Likewise, cytokine levels in the synovium reflected the severity of disease, with interleukin-1β levels in particular being lower in u-PA−/− mice and increased in t-PA−/− mice. The antibody response to type II collagen was normal in both knockouts; however, T cells from u-PA−/− mice had a reduced proliferative response and produced less interferon-γ on antigen stimulation in vitro. These results indicate that the major effect of u-PA in the collagen-induced arthritis model is deleterious, whereas that of t-PA is protective. Our data highlight the complexities of PA function, and suggest that approaches either to target u-PA or to enhance local t-PA activity in joints may be of therapeutic benefit in rheumatoid arthritis.
The plasminogen activators (PAs), namely urokinase (u-PA) and tissue-type PA (t-PA), are serine proteases that cleave the circulating zymogen, plasminogen, to generate the less specific serine protease, plasmin. 1 The PA/plasmin system has been implicated in fibrin removal, as well as in the tissue remodeling and cell migration occurring during both physiological and pathological processes. u-PA binds to a specific cell-surface receptor (u-PAR) 2 and part of its action seems to involve cell-mediated proteolysis of latent matrix-degrading proteases or growth factors. 3,4 A prime function of t-PA is likely to be in fibrin removal (fibrinolysis) because it requires such a surface for optimal activity. 1 This proteolytic system can be harmful or beneficial possibly depending on which component of the extracellular matrix is targeted. 5
Infiltration of inflammatory cells into the synovium and erosion of cartilage and bone are prominent features of rheumatoid arthritis (RA) joints. 6 Enhanced u-PA activity and reduced t-PA activity have been associated with disease severity, 7 and the former has been proposed as possibly being a key component in both the inflammatory and tissue remodeling processes. 8-10 Accumulation of intra-articular fibrin is also a common feature of RA, presumably resulting from an altered balance between coagulation and fibrinolysis; this fibrin accumulation may have adverse effects. 11-13 Several different cell types present in arthritic joints, for example, synoviocytes, chondrocytes, and inflammatory cells, can produce PAs and their inhibitors in vitro, including in response to inflammatory cytokines. 8-10,14-17
The availability of PA-deficient mice 18 makes it possible to begin to delineate the roles of PAs in inflammatory and other disease models. It has been reported recently in the monoarticular antigen-induced arthritis model 19 that u-PA−/− mice had enhanced disease severity that correlated in turn with the extent of fibrin(ogen) deposition in the joint; it was concluded that u-PA had a major role in fibrin removal in this model. Collagen-induced arthritis (CIA) is widely used as a systemic autoimmune model for RA with which it has many immunological and pathological parallels; 20 as an example, in support of its relevance, anti-tumor necrosis factor (TNF)-α therapy is somewhat effective both in this model 21 and in RA. 22 We have previously reported that the CIA model can be easily and conveniently adapted to C57BL/6 mice (H-2b), 23,24 a mouse strain onto which gene-deficient mice are commonly backcrossed. Clinically, histologically, and immunologically, the disease the mice develop resembles that seen in other strains of mice. 24 It has been shown, using DBA/1 mice, that u-PA mRNA in joints increases during CIA development. 25 Because u-PA−/− and t-PA−/− mice, backcrossed onto a C57BL/6 background, are available we were able to test the effect of depletion of the individual PAs in the CIA model because backcrossing to the DBA/1 background is not necessary.
We report here, as opposed to the findings for the monoarticular antigen-induced arthritis model, 19 that u-PA−/− mice develop only mild CIA disease compared with wild-type mice, with little inflammation and joint destruction. In contrast, t-PA−/− mice develop more severe disease than wild-type mice with sustained inflammation and fibrin accumulation within the joints. In general, fibrin levels seemed to correlate with disease severity, as did inflammatory cytokine production. These results suggest that therapies that target u-PA or enhance t-PA activity in joints may be beneficial in the treatment of RA.
Materials and Methods
Mice
u-PA-gene-deficient mice (u-PA−/−), t-PA-gene-deficient mice (t-PA−/−), and control mice (u-PA+/+ and t-PA+/+, respectively), 18 backcrossed for 11 generations onto the C57BL/6 background, were originally provided by Dr. P. Carmeliet (University of Leuven, Leuven, Belgium). C57BL/6 mice (originally obtained from Central Animal Services, Monash University, Clayton, Victoria, Australia) were used as a positive control for the CIA model. Mice, bred in our on-site animal facility, were fed standard rodent chow and water ad libitum and housed in sawdust-lined cages in groups of five. Mice, 8 to 12 weeks of age, were used in all experiments. All experiments were approved by The Royal Melbourne Hospital Research Foundation Animal Ethics Committee.
Collagen-Induced Arthritis
Mice were immunized intradermally in the base of the tail with 100 μg of chick type II collagen (CII) (Sigma, St. Louis, MO) emulsified in an equal volume of complete Freund’s adjuvant (CFA) containing 5 mg/ml of heat-killed Mycobacterium tuberculosis (H37 Ra; Difco, Detroit, MI); this procedure was repeated as a boost 21 days later as previously published. 23,24,26,27 Animals were assessed for redness and swelling of limbs and a clinical score was allocated for each limb using an established scoring system 23,24,26,27 as follows: 0, normal; 1, slight swelling and/or erythema; 2, extensive swelling and/or erythema; 3, severe swelling and/or rigidity. Severity of arthritis is expressed in terms of the mean clinical score (range, 0 to 12 per mouse), number of affected limbs per mouse (range, 0 to 4), and maximum clinical score per limb (range, 0 to 3). The thickness of the hind paws was measured using spring calipers (Mitutoyo, Tokyo, Japan), accurate to 0.01 mm. 23
Histology
At termination, the rear limbs and ankles were removed, fixed, decalcified, and paraffin embedded as previously described. 23,27 Frontal sections (5 μm) were stained with either hematoxylin and eosin to examine joint architecture or with safranin O, fast green, and hematoxylin for proteoglycan loss, and evaluated without knowledge of the experimental groups, using the histological assessment of Joosten and colleagues. 28 Briefly, infiltration of cells, cartilage damage, and bone erosions were all scored separately from 0 (normal) to 3 (severe), and proteoglycan loss (with safranin O stain) from 0 (normal) to 3 (complete loss of staining). These scores were added to give an overall histological score out of 12.
Detection of Fibrin(ogen) by Immunohistochemistry
Fibrin(ogen) deposition was identified in rear limbs as previously described. 19,29 Briefly, paraffin-embedded sections were deparaffinized and incubated with 5% (w/v) bovine serum albumin (BSA, Sigma) and 20% (v/v) normal goat serum for 1 hour. Slides were incubated for 30 minutes with a rabbit anti-mouse fibrinogen serum (a gift from Dr. J. Degen, Division of Development Biology, Children’s Hospital Research Foundation, Cincinnati, OH), diluted 1:1000. After washing, slides were then incubated with a biotinylated goat anti-rabbit IgG (DAKO, Carpinteria, CA), followed by a streptavidin-peroxidase conjugate (DAKO). Endogenous peroxidase activity was blocked with 0.3% (v/v) H2O2 (Sigma) in methanol. Peroxidase activity was demonstrated by incubation with 3,3′-diaminobenzidine/tetrahydrochloride (Sigma)-H2O2 solution. Slides were counterstained with hematoxylin. The specificity of the stain was confirmed using a nonimmune rabbit serum as previously described. 19,29 Joints from hind limbs were scored for fibrin(ogen) staining using a scale of 0 (normal) to 6 (maximum), based on the amount and intensity of staining, as previously described. 29
Anti-CII Enzyme-Linked Immunosorbent Assay (ELISA)
Antibodies to CII were measured in serum by ELISA as previously described. 23,24,27 Horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma) or IgG subclass-specific (IgG1, IgG2a, IgG2b, or IgG3; Southern Biotechnology, Birmingham, AL) detection antibodies were used. A standard curve for anti-CII IgG was constructed from sera of CII-hyperimmunized mice using arbitrary units (U/ml). For each IgG-subclass, the levels of anti-CII antibody were standardized such that u-PA+/+ mice had a mean level of 100 U/ml.
T-Cell Proliferation Assay
Cells from inguinal lymph nodes were isolated at the end of the experiment, and cultured (5 × 10 5 cells/well, 2 to 3 mice/group) for 72 hours, at 37°C (5% CO2), with 0 to 100 μg/ml of denatured CII (boiled for 10 minutes) in RPMI containing 50 μmol/L 2-ME and 5% (v/v) fetal calf serum (200 μl/well). 24,27 Sixteen hours before harvesting cells, 100 μl of supernatant was removed and stored at −20°C for subsequent cytokine detection; 100 μl of media was added to each well and cells were pulsed with 1 μCi of [3H]TdR (Amersham, Buckinghamshire, UK). Cells were harvested using an Inotech cell harvester (Inotech, Lansing, MI) and DNA synthesis measured by [3H]TdR incorporation using a Beckman β-scintillation counter (Beckman Instruments Inc., Irvine, CA). Results are expressed in cpm.
Preparation of Joint Tissue Washouts
After sacrifice, the tendons and synovium from the ankle joints of the hind limbs were dissected free from the surrounding tissue and washed in 200 μl of Dulbecco’s modified Eagle’s medium, supplemented with HEPES (20 mmol/L), l-glutamine (2 mmol/L), and penicillin (50U/ml)/streptomycin (50 μg/ml), and incubated for 1 hour at room temperature to allow the elution of cytokines. 27 Supernatants were then removed and stored at −20°C until assayed.
Cytokine ELISAs
TNF-α and interleukin (IL)-1β levels were measured in ankle joint tissue washouts by ELISA as previously described. 27 IFN-γ levels were measured in the supernatants from in vitro T cell cultures, also by ELISA. The coating and capture antibodies were as follows: TNF-α, anti-ΤNF-α monoclonal antibody (mAb) (G281-2626, Pharmingen), and a biotinylated anti-ΤNF-α mAb (MP6-XT3, Pharmingen); IL-1β, polyclonal anti-IL-1β Ab and a biotinylated anti-IL-1β mAb (Endogen, Woburn, MA); IFN-γ, anti-IFN-γ mAb (R4-6A2, Pharmingen) and a biotinylated anti-IFN-γ mAb (XMG1.2, Pharmingen). To detect the cytokines, a streptavidin-horseradish peroxidase conjugate (Pharmingen), followed by TMB-peroxidase substrate (Kirkegaard and Perry Laboratories, Gaithersburg, MD), was used. A standard curve was constructed using serial dilutions of purified TNF-α, IL-1β, or IFN-γ (Pharmingen) starting at a concentration of 2 ng/ml. Each ELISA was sensitive down to 15 pg/ml.
Statistics
For clinical and histological scores, cytokine levels, anti-CII levels, and T cell proliferation, the Mann-Whitney two-sample rank test was used to determine the level of significance between the means of the knockout and wild-type groups; results are expressed as the mean ± SEM. Correlations between clinical score and histological features were performed using the Spearman correlation coefficient. For other differences, including the number of affected limbs/mouse and the clinical score of individual limbs, the chi-square test was used, with Fisher exact test. P ≤ 0.05 was considered statistically significant.
Results
Reduced Severity of CIA in u-PA-Deficient Mice
To evaluate the requirement for endogenous u-PA for the development of CIA, u-PA−/− mice and control u-PA+/+ mice were immunized with CII in CFA, followed by a boost 21 days later. C57BL/6 mice were also immunized as a positive control for the CIA model. u-PA+/+ mice had a similar day of onset, disease incidence, and severity as C57BL/6 mice (Table 1) ▶ , the strain onto which they had been backcrossed (see Materials and Methods). u-PA−/− mice had significantly milder disease compared with u-PA+/+ mice (P = 0.03) (Table 1 ▶ , Figure 1A ▶ ). In Figure 1B ▶ this reduction is demonstrated in terms of the clinical score of individual limbs. Of the 156 limbs from u-PA−/− mice, only 12% (18 limbs) had a clinical score greater than 1 compared with 31% (38 limbs) of the 124 limbs from u-PA+/+ mice (P = 0.0001, chi-square test) (Figure 1B) ▶ . Of interest, only eight limbs from u-PA−/− mice scored the maximum of 3 and in all cases these limbs were rigid, indicating ankylosis, but were not swollen (see histology below). This appearance was confirmed by measuring the actual paw thickness (Figure 1C) ▶ . Individual limbs from u-PA−/− mice with a clinical score of 3 were significantly less swollen than individual limbs from u-PA+/+ mice with a clinical score of 3 (P = 0.027) (Figure 1C) ▶ . There were no significant differences between u-PA−/− and u-PA+/+ mice with regards to the cumulative incidence of disease or the day of disease onset (Table 1) ▶ .
Table 1.
Comparison of CIA in Wild-Type and Gene-Deficient Mice
| C57BL/6 | u-PA+/+ | u-PA−/− | t-PA+/+ | t-PA−/− | |
|---|---|---|---|---|---|
| Incidence (%)* | 24/33 (73) | 25/31 (81) | 24/39 (62) | 15/22 (68) | 18/19 (95)§ |
| Median (range)† | 3 (1–12) | 3 (1–12) | 2 (1–6) | 3 (1–12) | 9 (1–12)‡ |
| Mean clinical score (day 60) | 2.7± 0.7 | 2.7± 0.7 | 1.0± 0.3¶ | 2.9± 0.8 | 6.3± 1.2‡§ |
| Day of onset | 30.2± 1.6 | 29.0 ± 0.9 | 30.4± 1.2 | 34.3± 2.7 | 30.2± 1.8‡ |
*Cumulative incidence.
†Median (range) of maximum clinical scores obtained for arthritic mice.
‡Data excludes four mice sacrificed at day 21 (see text).
§P < 0.05, t-PA−/− versus t-PA+/+ or C57BL/6.
¶P < 0.05, u-PA−/− versus u-PA+/+ or C57BL/6.
Figure 1.
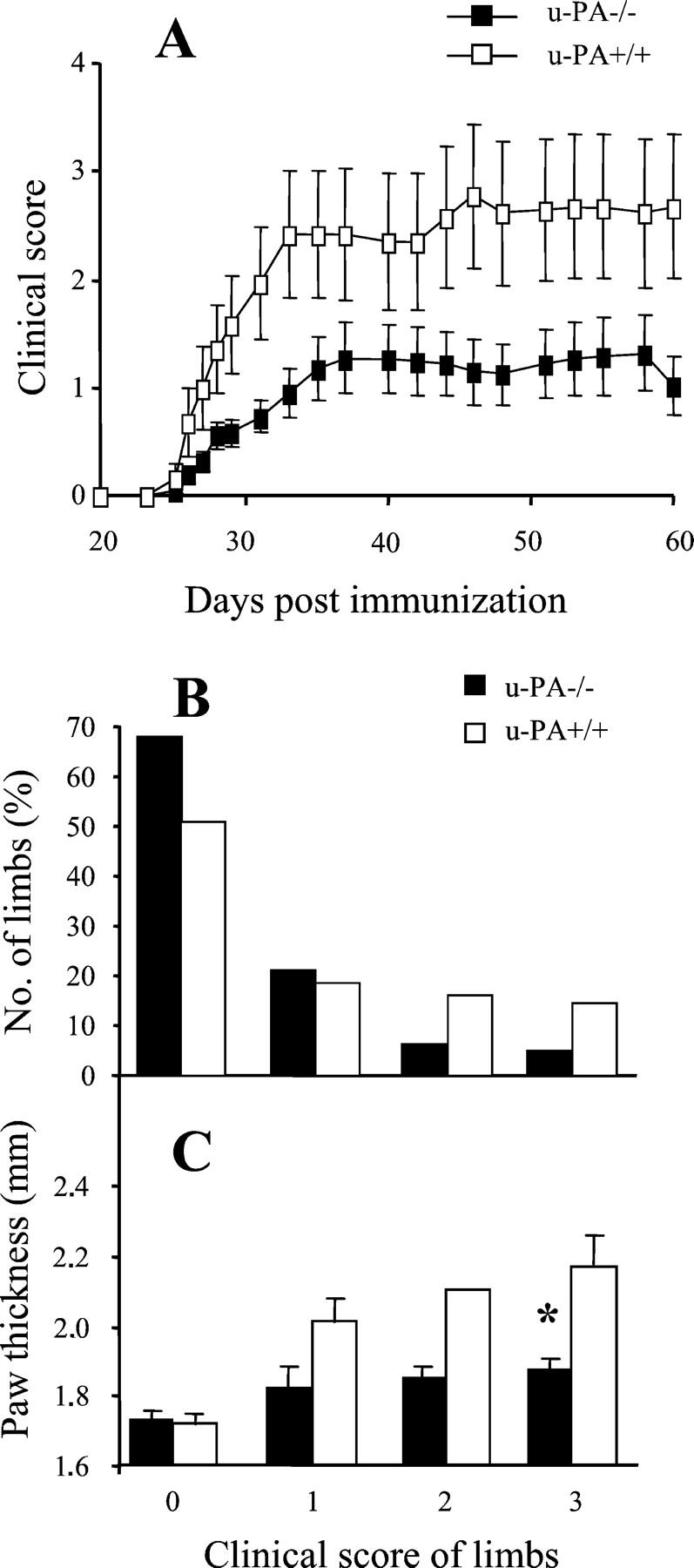
CIA development in u-PA−/− mice and control u-PA+/+ mice. A: Severity. Results are expressed as the mean clinical score ± SEM. There is significantly milder disease in the u-PA−/− mice (n = 39) compared with the u-PA+/+ mice (n = 31) (P = 0.03, Mann-Whitney using the mean clinical score of individual mice averaged throughout days 21 to 60). B: The number (%) of individual limbs with a particular clinical score is presented. For u-PA−/− mice, there are significantly less limbs with clinical scores of 2 or 3 compared with u-PA+/+ mice (P < 0.001, multi-way chi-square). C: Paw thickness for individual limbs with a particular clinical score, expressed as the mean thickness ± SEM. For u-PA+/+ mice, there is a highly significant correlation between the paw thickness and clinical score of individual limbs (r = 0.846, P < 0.001). For u-PA−/− mice, there is a weak, but significant, correlation between the paw thickness and clinical score of individual limbs (r = 0.475, P = 0.002). *, P = 0.027, paws from u-PA−/− mice with a clinical score of 3 are significantly less swollen than paws from u-PA+/+ mice with a clinical score of 3.
Increased Severity of CIA in t-PA-Deficient Mice
To evaluate the requirement for endogenous t-PA for the development of CIA, t-PA−/− mice and control t-PA+/+ mice were immunized as above. t-PA+/+ mice had a similar day of onset, disease incidence, and severity as C57BL/6 mice and u-PA+/+ mice (Table 1) ▶ . Eighteen (95%) of 19 t-PA−/− mice developed arthritis compared with 15 (68%) of 22 t-PA+/+ mice (P = 0.040, Table 1 ▶ ). Four of the t-PA−/− mice had to be sacrificed at day 21, the day of the booster injection, because of severe swelling of all limbs and so have not been included in the subsequent analyses; however, they suggest that a booster injection may not be required for arthritis development in t-PA−/− mice. These results confirmed earlier results using t-PA−/− and t-PA+/+ mice backcrossed to C57BL/6 for only one generation, in which 26 (70%) of 37 t-PA−/− mice developed arthritis compared with 13 (30%) of 44 t-PA+/+ mice (P = 0.0003, data not shown).
t-PA−/− mice had significantly more severe disease compared with t-PA+/+ mice (P = 0.015) (Table 1 ▶ , Figure 2A ▶ ). This increase in severity was seen both as an increase in the number of affected limbs per mouse (Figure 2B) ▶ and higher clinical scores for these affected limbs (Figure 2C) ▶ . For example, 60% (9 of 15) t-PA−/− mice had three or four affected limbs compared with only 18% (4 of 22) t-PA+/+ mice (P = 0.024, chi-square test) (Figure 2B) ▶ . Moreover, 45% (27 limbs) of these individual limbs from t-PA−/− mice reached a maximum score of 3 compared with only 19% (17 limbs) from t PA+/+ mice (P = 0.002, chi-square test) (Figure 2C) ▶ . There were no differences in the degree of swelling, as measured by paw thickness, between limbs from t-PA−/− and t-PA+/+ mice with the same clinical score (data not shown). There was also no difference in the day of disease onset (Table 1) ▶ .
Figure 2.
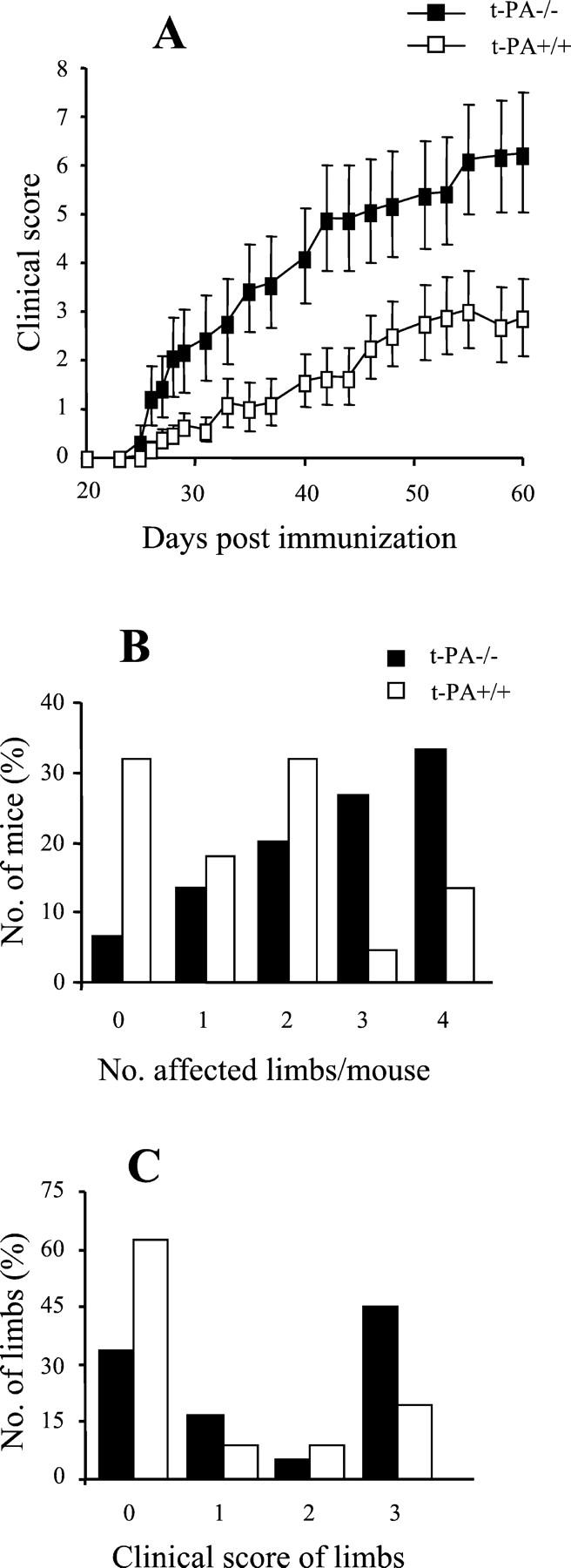
CIA development in t-PA−/− mice and control t-PA+/+ mice. A: Severity. Results are expressed as the mean clinical score ± SEM. There is significantly more severe disease in the t-PA−/− mice (n = 15) compared with the t-PA+/+ mice (n = 22) (P = 0.015, Mann-Whitney using the mean clinical score of individual mice averaged throughout days 21 to 60). B: The number (%) of mice that developed arthritis in a given number of limbs is presented. C: The number (%) of individual limbs with a particular clinical score is shown. For t-PA−/− mice, there are significantly more limbs with a clinical score of 3 compared with t-PA+/+ mice (P < 0.001, multi-way chi-square).
Effect of u-PA and t-PA Deficiency on Joint Pathology in CIA
Both inflammation and joint destruction are aspects of arthritis that are not necessarily linked; 28,30 therefore, it is possible that, whereas some aspects of disease may be affected in u-PA−/− and/or t-PA−/− mice, others may be normal. To investigate the effect of a lack of u-PA and t-PA on joint pathology and to gain possibly more insight into their mode of action, hind limbs were examined by histology for inflammatory cell infiltration, cartilage damage, proteoglycan depletion, and bone erosions. For each strain, representative limbs were taken to assess histology.
u-PA-Deficient Mice
Two hundred seventy-seven joints from 26 hind limbs of u-PA−/− mice and 192 joints from 18 hind limbs of u-PA+/+ mice were examined. There was a significant correlation between the clinical score of each limb and each histological feature for both genotypes (data not shown). By histology, for the eight limbs from u-PA−/− mice given a clinical score of 3, arthritis was primarily confined to the ankle joint, with the majority of joints in the feet being normal.
Figure 3 ▶ shows representative arthritic joints from u-PA−/− (Figure 3, A and C) ▶ and u-PA+/+ (Figure 3, B and D) ▶ mice. The histological features of infiltration, cartilage damage, proteoglycan depletion, and bone erosions were lower in u-PA−/− mice compared with u-PA+/+ mice (Figure 3G) ▶ . When the histological scores for each feature were combined for each strain, the mean total histological score for u-PA−/− mice was found to be significantly lower compared with u-PA+/+ mice (1.6 ± 0.4 versus 3.6 ± 0.8, P < 0.01).
Figure 3.
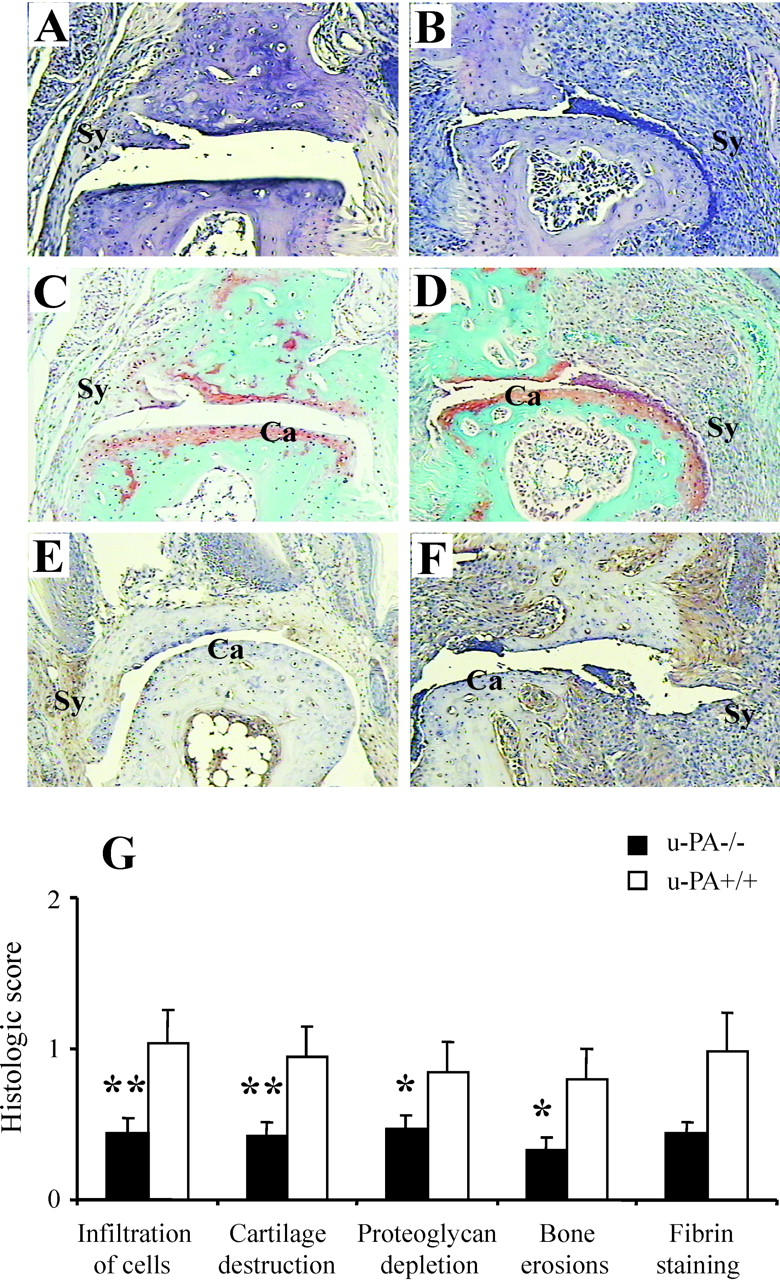
Histological assessment of CIA in u-PA−/− mice. Distal interphalangeal joints from u-PA−/− mice (A, C, and E) and u-PA+/+ mice (B, D, and F). These are representative of average clinical scores in each case. A and B: H&E staining. C and D: Safranin O, fast green staining with a hematoxylin counterstain. E and F: Immunohistochemical detection of fibrin. Note that the arthritis is milder in the joints from the u-PA−/− mice and there is only very weak fibrin staining. Ca, cartilage; Sy, synovium. Original magnifications, ×125. G: Histological scores (mean ± SEM) for each histological feature (n = 26 for u-PA−/− and 18 for u-PA+/+ limbs) and for fibrin staining (n = 10 for u-PA−/− and 10 for u-PA+/+ limbs). Note that scoring for fibrin staining is 0 to 6 and for all other histological features is 0 to 3. *, P < 0.05; **, P < 0.01 u-PA−/− versus u-PA+/+ mice.
t-PA-Deficient Mice
One hundred sixty-six joints from 14 hind limbs of t-PA−/− mice and 202 joints from 15 hind limbs of t-PA+/+ mice were examined. There was a significant correlation between the clinical score of each limb and each histological feature for both genotypes (data not shown). Figure 4 ▶ shows the most severely affected joints from t-PA−/− (Figure 4, A and C) ▶ and t-PA+/+ (Figure 4, B and D) ▶ mice. The histological features of infiltration, cartilage damage, proteoglycan depletion, and bone erosions were higher in t-PA−/− mice compared with t-PA+/+ mice (Figure 4I) ▶ , with some limbs from t-PA−/− mice having massive cell infiltration greater than that seen in any t-PA+/+ mouse. When the histological scores for each feature were combined for each strain, the mean total histological score for t-PA−/− mice was found to be significantly higher compared with t-PA+/+ mice (5.6 ± 0.9 versus 2.9 ± 0.9, P < 0.05).
Figure 4.
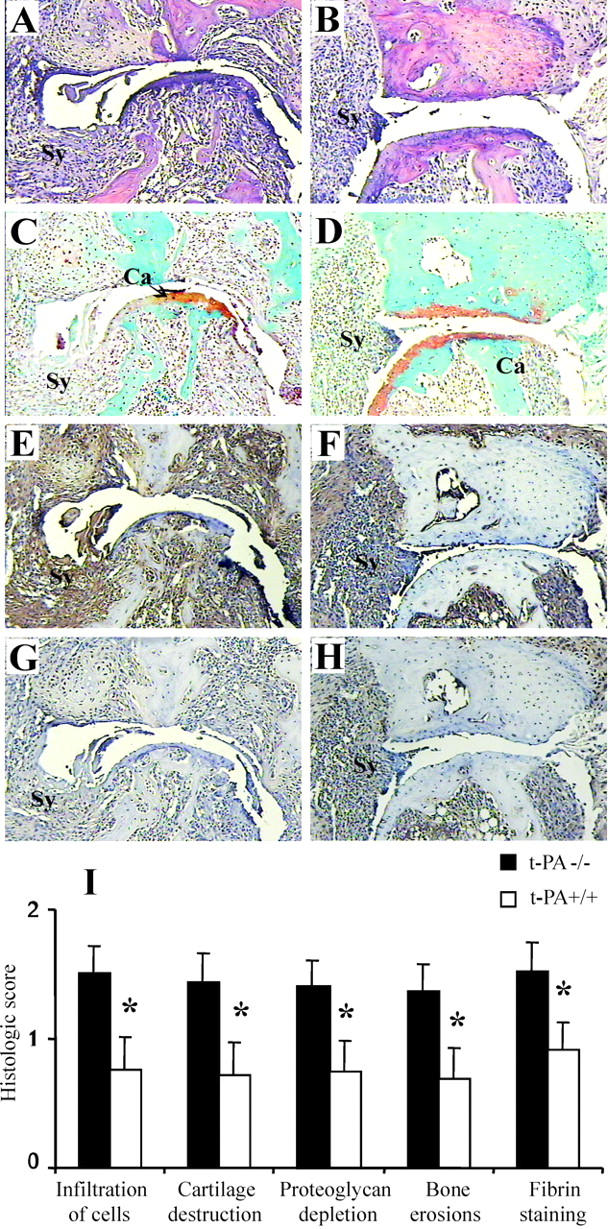
Histological assessment of CIA in t-PA−/− mice. Severely affected metatarsal phalangeal joints from t-PA−/− mice (A, C, E, and G) and t-PA+/+ mice (B, D, F, and H). These represent joints with the most severe arthritis in each case. A and B: H&E staining. C and D: Safranin O, fast green staining with a hematoxylin counterstain. E and F: Immunohistochemical detection of fibrin. G and H: Control staining for fibrin. Note that the arthritis is more severe in the joint from the t-PA−/− mouse and there is more fibrin staining. Ca, cartilage; Sy, synovium. Original magnifications, ×125. I: Histological scores (mean ± SEM) for each histological feature (n = 14 for t-PA−/− and 15 for t-PA+/+ limbs) and for fibrin staining (n = 11 for t-PA−/− and 9 for t-PA+/+ limbs). Note that scoring for fibrin staining is 0 to 6 and for all other histological features is 0 to 3. *, P < 0.05 t-PA−/− versus t-PA+/+ mice.
Fibrin(ogen) Deposition in Arthritic Joints
Recently in both the CIA 29 and the monoarticular antigen-induced arthritis 19 models, the extent of fibrin deposition in the joints correlated with disease severity. In the latter model, based in part on increased fibrin deposition in u-PA−/− mice, it was concluded that u-PA was responsible for fibrin removal. 19 However, t-PA is often primarily responsible for fibrinolysis. 1,5 We therefore monitored fibrin(ogen) accumulation in the various mice under study.
In nonarthritic joints there was minimum fibrin(ogen) staining as previously reported (data not shown). 29 Figure 3 ▶ shows fibrin(ogen) staining in representative arthritic joints from u-PA−/− (Figure 3E) ▶ and u-PA+/+ (Figure 3F) ▶ mice. For u-PA−/− mice and u-PA+/+ mice there was a significant correlation between the amount of fibrin deposition and the histological score of a limb (r = 0.499, P = 0.049 and r = 0.843, P = 0.002, respectively). For u-PA−/− mice there was little fibrin(ogen) staining in the joints, the maximum being a score of 2 of a possible 6 compared with 5 of 6 for u-PA+/+ mice. Thus, there was a trend for more fibrin deposition in the joints of u-PA+/+ mice compared with joints of u-PA−/− mice (Figure 3G) ▶ . This finding of minimal fibrin deposition in the joints of arthritic u-PA−/− mice is contrary to what was found in the monoarticular antigen-induced model, 19 but is in keeping with the correlation with disease severity.
Figure 4 ▶ shows fibrin(ogen) staining in the most severely affected joints from t-PA−/− (Figure 4E) ▶ and t-PA+/+ (Figure 4F) ▶ mice. The fibrin deposition in the joints with severe arthritis was diffuse, covering a large area including the synovium and pannus tissue and even extending into the joint space in some instances (Figure 4E ▶ compared with control staining Figure 4G ▶ ). In all cases, nonspecific background staining was subtracted from fibrin-stained sections. For t-PA−/− mice and t-PA+/+ mice there was a significant correlation between the amount of fibrin deposition and the histological score of a limb (r = 0.848, P = 0.001 and r = 0.842, P = 0.004, respectively). There was significantly more fibrin deposition in t-PA−/− mice compared with t-PA+/+ mice (P = 0.025, Figure 4I ▶ ).
Cellular and Humoral Immune Responses to CII in u-PA−/− and t-PA−/− Mice
CIA development is dependent on both a cellular and humoral immune response to CII, 24,31-33 and alterations in either one may affect arthritis. To investigate this, in vitro T cell responses were examined. T cells from u-PA−/− mice, although still proliferating to CII in a dose-dependent manner, showed significantly reduced responses compared with T cells from u-PA+/+ mice (P = 0.034) (Figure 5A) ▶ . The amount of IFN-γ in CII-specific T cell culture supernatants from u-PA−/− mice was also significantly lower than for u-PA+/+ mice (P = 0.030) (Figure 5B) ▶ . There was no significant difference for the T cells from t-PA−/− and t-PA+/+ mice in the proliferative response to CII (Figure 5C) ▶ or in the amount of IFN-γ produced (Figure 5D) ▶ .
Figure 5.

Effect of u-PA and t-PA deficiency on CII-specific immune responses in CIA. A: Cellular response to CII in u-PA−/− or u-PA+/+ mice. B: IFN-γ levels from the CII-specific T-cell culture supernatants described in A. C: Cellular response to CII in t-PA−/− or t-PA+/+ mice. D: IFN-γ levels from the CII-specific T-cell culture supernatants described in C. Inguinal lymph node cells from pools of three mice immunized for CIA were cultured in the presence of differing concentrations (0 to 100 μg/ml) of denatured CII for 72 hours. Cultures were pulsed with tritiated thymidine ([3H]TdR) for the last 16 hours (see Materials and Methods). Results are expressed as the amount of [3H]TdR incorporation (mean ± SEM) in cpm (A and C). Cells from u-PA−/− mice showed significantly less [3H]TdR incorporation than those of u-PA+/+ mice at all CII concentrations (P = 0.034). IFN-γ levels were measured by ELISA and are expressed as the mean ± SEM (pg/ml) (B and D). Cells from u-PA−/− mice produced significantly less IFN-γ than those from u-PA+/+ mice at all CII concentrations (P = 0.030). E: IgG antibody response to CII measured at the end of each experiment (day 60). Results are expressed as the mean ± SEM. F: IgG1, IgG2b, IgG2c, and IgG3 antibody response to CII in sera from u-PA+/+ and u-PA−/− mice. Results are expressed as the mean ± SEM, where the mean level of antibody for each IgG subclass was standardized such that u-PA+/+ mice had a mean level of 100 U/ml.
Antibodies to CII were measured in sera of mice at the end of each experiment. There was no significant difference in the anti-CII IgG response between u-PA−/− and u-PA+/+ mice, or between t-PA−/− and t-PA+/+ mice (Figure 5E) ▶ . These findings confirmed earlier results using mice backcrossed to C57BL/6 for only one generation (unpublished results). Given that CII is a T-cell-dependent antigen and T cells from u-PA−/− mice had a reduced proliferative response to CII and produced less IFN-γ on antigen stimulation in vitro, levels of circulating anti-CII IgG subclasses were measured in u-PA+/+ and u-PA−/− mice. In DBA/1 mice, antibodies to CII of the IgG2a and IgG2b subclass are particularly linked to the development of arthritis. 34 C57BL/6 mice produce IgG2c and not IgG2a; however, IgG2c is detected by cross-reactivity with the anti-IgG2a reagent used. 24 There were no significant differences in the anti-CII IgG subclass response between u-PA−/− and u-PA+/+ mice (Figure 5F) ▶ .
Proinflammatory Cytokine Levels in Joints of u-PA−/− and t-PA−/− Mice with Arthritis
Given the different degrees of severity of disease of u-PA−/− mice and t-PA−/− mice, which were reflected in the differing levels of inflammation present in the joints, we next determined whether there was a correlation with the levels of the key proinflammatory cytokines, TNF-α and IL-1β.
For u-PA−/− mice, the mean levels of TNF-α (Figure 6A) ▶ and IL-1β (Figure 6B) ▶ detected in ankle joint washouts from arthritic mice were significantly lower than for u-PA+/+ mice (P = 0.030 and P = 0.028, respectively). For t-PA−/− mice, there was a trend for a higher mean level of TNF-α in joint washouts from arthritic mice compared with t-PA+/+ mice (Figure 6A) ▶ , and a significantly higher mean level of IL-1β compared with t-PA+/+ mice (P = 0.001) (Figure 6B) ▶ .
Figure 6.

Effect of u-PA and t-PA deficiency on proinflammatory cytokine levels in joints in CIA. TNF-α (A) and IL-1β (B) levels (ELISA) were measured at sacrifice in washouts from ankle joints of arthritic mice. Results are expressed as the mean level ± SEM for each cytokine (pg/ml). *, P < 0.05, knockout versus wild-type; **, P = 0.001, knockout versus wild-type.
Discussion
Our previous establishment of the CIA model in mice on a C57BL/6 background, which resembles the arthritis seen in DBA/1 mice clinically, histologically, and immunologically, 23,24 enabled us in the present study to conveniently assess the roles of both u-PA and t-PA in this model, using the corresponding gene-deficient mice. The data indicate that overall the lack of expression of either PA has a different outcome with u-PA being deleterious and t-PA protective for the arthritis development in this model, consistent with there being significant differences in key functions for each protease. u-PA+/+ and t-PA+/+ mice immunized for CIA in the same experiment showed no significant differences in their arthritic responses, nor did their response differ from those seen in C57BL/6 mice. For both gene-deficient mice the local levels of fibrin(ogen) and inflammatory cytokines correlated with disease severity. CIA is dependent on the establishment of both cellular and humoral immunity to CII. 24,31-33 Evidence was obtained in u-PA−/− mice for a decrease in the cellular immune response to CII; however, for neither gene-deficient strain was there any evidence for a change in the humoral response to the arthritogenic antigen.
Although u-PA−/− mice developed very mild disease, a small number of limbs were given the maximum clinical score of 3, based on their rigidity. However, these limbs were not swollen, as one would normally expect. It was subsequently confirmed by histology that these mice had arthritis predominately in their ankle joints, making the limb rigid, but had little arthritis in the joints of the foot, accounting for the lack of swelling and inflammation. Therefore, u-PA−/− mice develop even milder disease than is indicated by their overall clinical score. In fact, no joint showed severe disease comparable to that seen in u-PA+/+ mice, contrary to many knockout mouse studies in which a limited number of joints may develop disease just as severe as the appropriate wild-type mouse, even though overall they may have few joints affected. 35
Our results with u-PA−/− mice are in contrast to those reported in the monoarticular antigen-induced arthritis model using mBSA in which it was found that u-PA−/− mice developed more severe disease than control mice; 19 however, we have found similar observations to Busso and colleagues 19 in the acute monoarticular mBSA/IL-1 arthritis model in u-PA−/− mice. 36 These different findings highlight the complex nature of arthritis, the differing roles the same mediator can have, and the need for evaluation of several models of heterogeneous human diseases, such as RA, to delineate the role certain molecules may play clinically.
There could be several reasons for these differing outcomes in u-PA−/− mice. u-PA is implicated in fibrin removal, cell migration, and tissue remodeling. 1 Unlike the two models in which arthritis develops in a single joint, CIA is a chronic systemic model. Therefore, molecules associated with cell trafficking and chemotaxis could be more critical in CIA than in the other monoarticular models that involve direct antigen injection into the joints. There are additional examples of inflammation models in the literature where it has been concluded, based on u-PA−/− mouse data, that u-PA expression in inflammatory cells is important in the migration of cells into the inflamed site. 37 Another possibility is that u-PA expression is more significant for chronic inflammatory reactions than for those of an acute nature. Consistent with this possibility, it has been found that u-PA−/− mice, while mounting a normal early inflammatory response to a pulmonary cryptococcal infection, failed to elicit an effective immune response and clear the organisms after a delayed period of 14 to 21 days, thereby ultimately leading to their death by progressive infection. 38 The reduced IL-1β and TNF-α levels in the joints of u-PA−/− mice could be as a result of fewer cells, for example macrophages, being present there. Endogenously produced u-PA/plasmin has been shown to amplify cytokine levels including those of TNF-α and IL-1β; 3,39,40 therefore it is possible that a lack of u-PA could also directly reduce the levels of these cytokines known to play a role in this model.
It has previously been reported using u-PA−/− mice that u-PA was required for T cell proliferation and activation in vitro, and that its absence led to a reduced Th1-polarized profile of cytokines. 41 Our observations above are consistent with these findings because we found that antigen-challenged T cells from u-PA−/− mice produced less IFN-γ, a Th1-type cytokine, than their wild-type counterparts. CIA has been reported to be a Th1-type disease and suppression of the Th1 response leads to a reduction in disease, similar to what we observed above in the u-PA−/− mice. 42 However, we found no decrease in the levels of IgG2a or IgG2c antibody to CII (Th1) or an increase in the level of IgG1 antibody to CII (Th2) that would suggest this is the case.
It has been shown in DBA/1 mice that u-PA mRNA is increased in arthritic joints and we have confirmed this in the current study using mice on the C57BL/6 background (our unpublished observations). Issues such as its source in the inflamed joints and whether its proteolytic activity is required need to be explored. In support of the possible clinical relevance of our findings above, u-PA activity is increased in RA synovial tissue. 7 In vitro studies have shown that u-PA expression and that of its inhibitors can be modulated in vitro by several relevant human cell types in response to a range of stimuli 8-10,14-17 and several of these sources have been proposed by us in a model depicting a key role for this protease in the pathogenesis of RA. 10
t-PA−/− mice were found to develop more severe CIA than t-PA+/+ mice, with a large accumulation of fibrin(ogen) in the joints. This accumulation suggests that in CIA t-PA, but not u-PA, has an important role in fibrinolysis. In the antigen-induced arthritis model, it was recently concluded that u-PA was the predominant PA in this context; 19 it should be noted that in the CIA model our data indicate that there was minimal accumulation of fibrin(ogen) in the joints of u-PA−/− mice. Accumulation of extravascular fibrin in RA tissues is a common feature of the synovitis, and the amount of fibrin(ogen) accumulation reflects the amount of joint inflammation. 11-13 Decreased t-PA activity has been reported in rheumatoid synovia although the presence of fibrin D-dimers in rheumatoid synovial tissue and fluids is consistent with plasmin activity endeavoring to degrade fibrin clot formation. 7 Continual and unresolved fibrin deposition may provoke permanent chronic joint damage, impede normal synovial nutrition, enhance vascular permeability, result in chemotactic activity of fibrin(ogen) degradation products, promote cellular adherence and migration on the fibrin matrix, and increase fibrin-mediated inflammatory cytokine production by macrophages. 43,44 Consistent with this notion and our data above, it has been shown in the CIA model 29 and the antigen-induced model 19 that intra-articular fibrin(ogen) contributes to the maintenance of synovial inflammation and correlates with disease severity. The enhanced arthritis seen in t-PA−/− mice, both in the CIA model above and in the mBSA/IL-1 monoarticular model, 36 would seem to have some analogy to reports where it was concluded, using a similar approach, that t-PA was the major protective PA for the enhanced renal injury observed in these mice in a crescentic glomerulonephritis model 45 and for the proposed protection by fibrinolysis against axonal degeneration and demyelination after sciatic nerve injury. 5 Thus fibrin(ogen) accumulation correlates widely with the degree of inflammation in all of these various models.
In t-PA−/− mice, the mean level of IL-1β, important in joint destruction and inflammation, 28 was significantly higher than the mean level in t-PA+/+ mice. This increase is in agreement with the associated heightened amount of cartilage damage, bone erosion, and disease severity. It has been shown that fibrin enhances the expression of IL-1β by human peripheral blood mononuclear cells. 44 Furthermore, inhibition of thrombin, which converts fibrinogen into fibrin, resulted in a reduced incidence and severity of CIA, with down modulation of synovial proinflammatory IL-1β and IL-12 p35 cytokine mRNAs. 29
In conclusion, our present results indicate that nonspecific inhibition of PA activity is unlikely to give the best therapeutic outcomes for the treatment of RA. Rather, they suggest that perhaps u-PA should be considered as a therapeutic target in RA; in contrast, approaches to enhance t-PA activity in joints may also be beneficial—in this context, administration of t-PA has been shown to inhibit carrageenan-induced inflammation in rats. 46
Acknowledgments
We thank Jennifer Davis for assistance with the maintenance and care of the mice, P. Carmeliet for the mice, and J. Degen for the anti-mouse fibrinogen serum.
Footnotes
Address reprint requests to Dr. Andrew D. Cook, Arthritis and Inflammation Research Center, University of Melbourne, Department of Medicine, Victoria 3010, Australia. E-mail: adcook@unimelb.edu.au.
Supported by the National Health and Medical Research Council of Australia, including a Senior Principal Research Fellowship (to J. A. H.).
Current address of I. K. C.: Autoimmunity and Transplantation Division, The Walter and Eliza Hall Institute, Parkville, Victoria, Australia.
References
- 1.Irigoyen JP, Munoz-Canoves P, Montero L, Koziczak M, Nagamine Y: The plasminogen activator system: biology and regulation. Cell Mol Life Sci 1999, 56:104-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blasi F, Vassalli JD, Dano K: Urokinase-type plasminogen activator: proenzyme, receptor, and inhibitors. J Cell Biol 1987, 104:801-804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saksela O, Rifkin DB: Cell-associated plasminogen activation: regulation and physiological functions. Annu Rev Cell Biol 1988, 4:93-126 [DOI] [PubMed] [Google Scholar]
- 4.Moscatelli D, Rifkin DB: Membrane and matrix localization of proteinases: a common theme in tumor cell invasion and angiogenesis. Biochim Biophys Acta 1988, 948:67-85 [DOI] [PubMed] [Google Scholar]
- 5.Akassoglou K, Kombrinck KW, Degen JL, Strickland S: Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol 2000, 149:1157-1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris Jr ED: Rheumatoid Arthritis. Philadelphia, W.B. Saunders Company, 1997
- 7.Busso N, Peclat C, So A, Sappino AP: Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints. Ann Rheum Dis 1997, 56:550-557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamilton JA, Stanley ER, Burgess A, Shadduck R: Stimulation of macrophage plasminogen activator by colony stimulating factors. J Cell Physiol 1980, 103:435-445 [DOI] [PubMed] [Google Scholar]
- 9.Hamilton JA, Slywka J: Stimulation of human synovial fibroblast plasminogen activator production by mononuclear cell supernatants. J Immunol 1981, 126:851-855 [PubMed] [Google Scholar]
- 10.Hamilton JA, Campbell IK, Wojta J, Cheung D: Plasminogen activators and their inhibitors in arthritic disease. Ann NY Acad Sci 1992, 667:87-100 [DOI] [PubMed] [Google Scholar]
- 11.Jasani MK: Fibrin: metabolism, immunopathogenesis and significance in rheumatoid arthritis. Immunopathogenesis of Rheumatoid Arthritis. 1978, :p 137 PM Johnson. Surrey, Red Books, Edited by GS Panayi GS [Google Scholar]
- 12.Weinberg JB, Pippen AM, Greenberg CS: Extravascular fibrin formation and dissolution in synovial tissue of patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum 1991, 34:996-1005 [DOI] [PubMed] [Google Scholar]
- 13.Zacharski LR, Brown FE, Memoli VA, Kisiel W, Kudryk BJ, Rousseau SM, Hunt JA, Dunwiddie C, Nutt EM: Pathways of coagulation activation in situ in rheumatoid synovial tissue. Clin Immunol Immunopathol 1992, 63:155-162 [DOI] [PubMed] [Google Scholar]
- 14.Leizer T, Clarris BJ, Ash PE, van Damme J, Saklatvala J, Hamilton JA: Interleukin-1 beta and interleukin-1 alpha stimulate the plasminogen activator activity and prostaglandin E2 levels of human synovial cells. Arthritis Rheum 1987, 30:562-566 [DOI] [PubMed] [Google Scholar]
- 15.Campbell IK, Piccoli DS, Butler DM, Singleton DK, Hamilton JA: Recombinant human interleukin-1 stimulates human articular cartilage to undergo resorption and human chondrocytes to produce both tissue- and urokinase-type plasminogen activator. Biochim Biophys Acta 1988, 967:183-194 [DOI] [PubMed] [Google Scholar]
- 16.Campbell IK, Last K, Novak U, Lund LR, Hamilton JA: Recombinant human interleukin-1 inhibits plasminogen activator inhibitor-1 (PAI-1) production by human articular cartilage and chondrocytes. Biochem Biophys Res Commun 1991, 174:251-257 [DOI] [PubMed] [Google Scholar]
- 17.Hamilton JA, Cheung D, Filonzi EL, Piccoli DS, Wojta J, Gallichio M, McGrath K, Last K: Independent regulation of plasminogen activator inhibitor 2 and plasminogen activator inhibitor 1 in human synovial fibroblasts. Arthritis Rheum 1992, 35:1526-1534 [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC: Physiological consequences of loss of plasminogen activator gene function in mice. Nature 1994, 368:419-424 [DOI] [PubMed] [Google Scholar]
- 19.Busso N, Peclat V, Van Ness K, Kolodziesczyk E, Degen J, Bugge T, So A: Exacerbation of antigen-induced arthritis in urokinase-deficient mice. J Clin Invest 1998, 102:41-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Myers LK, Rosloniec EF, Cremer MA, Kang AH: Collagen-induced arthritis, an animal model of autoimmunity. Life Sci 1997, 61:1861-1878 [DOI] [PubMed] [Google Scholar]
- 21.Williams RO, Feldmann M, Maini RN: Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci USA 1992, 89:9784-9788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feldmann M, Maini RN: Anti-TNFα therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 2001, 19:163-196 [DOI] [PubMed] [Google Scholar]
- 23.Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA: Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol 1998, 161:3639-3644 [PubMed] [Google Scholar]
- 24.Campbell IK, Hamilton JA, Wicks IP: Collagen-induced arthritis in C57BL/6 (H-2b) mice: new insights into an important disease model of rheumatoid arthritis. Eur J Immunol 2000, 30:1568-1575 [DOI] [PubMed] [Google Scholar]
- 25.Salvi R, Peclat V, So A, Busso N: Enhanced expression of genes involved in coagulation and fibrinolysis in murine arthritis. Arthritis Res 2000, 2:504-512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campbell IK, Rich MJ, Bischof RJ, Hamilton JA: The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol 2000, 68:144-150 [PubMed] [Google Scholar]
- 27.Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA: Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res 2001, 3:293-298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joosten LA, Helsen MM, Saxne T, van De Loo FA, Heinegard D, van Den Berg WB: IL-1αβ blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-α blockade only ameliorates joint inflammation. J Immunol 1999, 163:5049-5055 [PubMed] [Google Scholar]
- 29.Marty I, Peclat V, Kirdaite G, Salvi R, So A, Busso N: Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest 2001, 107:631-640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulherin D, Fitzgerald O, Bresnihan B: Clinical improvement and radiological deterioration in rheumatoid arthritis: evidence that the pathogenesis of synovial inflammation and articular erosions may differ. Br J Rheumatol 1996, 35:1263-1268 [DOI] [PubMed] [Google Scholar]
- 31.Seki N, Sudo Y, Yoshioka T, Sugihara S, Fujitsu T, Sakuma S, Ogawa T, Hamaoka T, Senoh H, Fujiwara H: Type II collagen-induced murine arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. J Immunol 1988, 140:1477-1484 [PubMed] [Google Scholar]
- 32.Svensson L, Jirholt J, Holmdahl R, Jansson L: B cell-deficient mice do not develop type II collagen-induced arthritis (CIA). Clin Exp Immunol 1998, 111:521-526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corthay A, Johansson A, Vestberg M, Holmdahl R: Collagen-induced arthritis development requires alpha beta T cells but not gamma delta T cells: studies with T cell-deficient (TCR mutant) mice. Int Immunol 1999, 11:1065-1073 [DOI] [PubMed] [Google Scholar]
- 34.Watson WC, Townes AS: Genetic susceptibility to murine collagen II autoimmune arthritis. Proposed relationship to the IgG2 autoantibody subclass response, complement C5, major histocompatibility complex (MHC) and non-MHC loci. J Exp Med 1985, 162:1878-1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mori L, Iselin S, De Libero G, Lesslauer W: Attenuation of collagen-induced arthritis in 55-kDa TNF receptor type 1 (TNFR1)-IgG-treated and TNFR1-deficient mice. J Immunol 1996, 157:3178-3182 [PubMed] [Google Scholar]
- 36.Yang YH, Carmeliet P, Hamilton JA: Tissue-type plasminogen activator deficiency exacerbates arthritis. J Immunol 2001, 167:1047-1052 [DOI] [PubMed] [Google Scholar]
- 37.Lluis F, Roma J, Suelves M, Parra M, Aniorte G, Gallardo E, Illa I, Rodriguez L, Hughes SM, Carmeliet P, Roig M, Munoz-Canoves P: Urokinase-dependent plasminogen activation is required for efficient skeletal muscle regeneration in vivo. Blood 2001, 97:1703-1711 [DOI] [PubMed] [Google Scholar]
- 38.Gyetko MR, Chen GH, McDonald RA, Goodman R, Huffnagle GB, Wilkinson CC, Fuller JA, Toews GB: Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans. A murine transgenic model. J Clin Invest 1996, 97:1818-1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsushima K, Taguchi M, Kovacs EJ, Young HA, Oppenheim JJ: Intracellular localization of human monocyte associated interleukin 1 (IL 1) activity and release of biologically active IL 1 from monocytes by trypsin and plasmin. J Immunol 1986, 136:2883-2891 [PubMed] [Google Scholar]
- 40.Sitrin RG, Shollenberger SB, Strieter RM, Gyetko MR: Endogenously produced urokinase amplifies tumor necrosis factor-alpha secretion by THP-1 mononuclear phagocytes. J Leukoc Biol 1996, 59:302-311 [DOI] [PubMed] [Google Scholar]
- 41.Gyetko MR, Libre EA, Fuller JA, Chen GH, Toews G: Urokinase is required for T lymphocyte proliferation and activation in vitro. J Lab Clin Med 1999, 133:274-288 [DOI] [PubMed] [Google Scholar]
- 42.Mauri C, Williams RO, Walmsley M, Feldmann M: Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur J Immunol 1996, 26:1511-1518 [DOI] [PubMed] [Google Scholar]
- 43.Barnhart MI, Riddle JM, Bluhm GB: Immunocytology in arthritic joints. Ann Rheum Dis 1967, 26:281-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perez RL, Roman J: Fibrin enhances the expression of IL-1 beta by human peripheral blood mononuclear cells. Implications in pulmonary inflammation. J Immunol 1995, 154:1879-1887 [PubMed] [Google Scholar]
- 45.Kitching AR, Holdsworth SR, Ploplis VA, Plow EF, Collen D, Carmeliet P, Tipping PG: Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med 1997, 185:963-968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stringer KA, Bose SK, McCord JM: Antiinflammatory activity of tissue plasminogen activator in the carrageenan rat footpad model. Free Radic Biol Med 1997, 22:985-988 [DOI] [PubMed] [Google Scholar]