

Abstract
Our previous article reported that retroviral transduction of human type I consensus interferon-coding sequence into two human melanoma cells increased their susceptibility to cisplatin-induced apoptosis. Importantly, primary melanoma cells were significantly more sensitive to cisplatin-induced apoptosis with respect to metastatic melanoma cells. The aim of this study was to elucidate the subcellular mechanisms involved in this interferon-induced apoptotic proneness. Our results indicate that 1) cisplatin-induced apoptosis can be referred to as the type II apoptosis, ie, to the mitochondrially driven cascade; 2) treatment of interferon-producing melanoma cells with other type II apoptotic stimuli, such as radiation or staurosporine, also resulted in massive apoptosis, whereas type I stimuli, ie, anti-Fas, were ineffective; 3) interferon sensitization involved the caspase cascade in primary melanoma cells and the alternative pathway represented by cathepsin-mediated apoptosis in metastatic melanoma cells; 4) interferon production sensitizes cells to apoptosis by inducing, as the earliest event, mitochondrial membrane hyperpolarization. These results suggest that constitutive production of type I interferon by melanoma cells can act as an intracellular booster capable of increasing cell proneness to apoptosis by specifically modifying mitochondrial homeostasis and independently from the apoptotic cascade involved.
The balance between cell proliferation and cell death by apoptosis is a key factor in the maintenance of tissue homeostasis. Hence, for many years the neoplastic transformation has been associated with the disturbance of the mechanisms underlying this balance. In recent years cell death by apoptosis has thus gathered the particular attention of physicians specifically devoted to the understanding of the pro-apoptotic activities of anti-tumor drugs and their relative mechanisms of action. 1 As a general rule, whereas cells that die from damage typically swell and burst (necrosis), apoptosis is a rigorously controlled and highly ordered process characterized by specific morphological and functional changes. 2 Executors of apoptosis are a family of cysteinyl aspartate-specific proteases, ie, caspases, whose overexpression is sufficient to cause apoptosis. 3,4 By contrast, examples of caspase-independent commitment to cell death can also be found in several models of apoptosis, including glucocorticoid-induced cell death of thymocytes, growth factor withdrawal-induced apoptosis of lymphoid cells or Bax-mediated cell death. 5,6 However, these complex cascades of events seemed to depend on, or to be associated with, the type of trigger that activates the process. 7 Two pathways, called type I and type II, which are referred to different initiation patterns, have been proposed. 8 The activation of caspase-8 or -9 has, respectively, been suggested as main primus movens of the death process in the two pathways. Importantly however, only type II apoptotic machinery seems to involve mitochondrial-associated caspase cascade in the early phases. 8 It has in fact emerged that mitochondrial function may play a pivotal role in determining cellular commitment to apoptosis. 6,9,10 Thus, during apoptotic signaling, mitochondrial changes result in enhanced production of reactive oxygen species (ROS) and collapse of the mitochondrial membrane potential, 11 which has been thought to represent a point of no return in committing the cell to apoptosis. In turn, mitochondrial integrity has been proposed to be regulated by pro- and anti-apoptotic members of the Bcl-2 family. 12,13 These molecules act as regulators of cell survival or apoptosis and seem to target a number of aspects of mitochondrial function, including mitochondrial permeability and ROS homeostasis. 14 This leads to a specific cascade characterized by the release of factors, eg, cytochrome c, that promote activation of effector caspases, eg, caspase-9, that, together with other components (eg, downstream caspases, apaf-1, caspase-independent endonuclease, and so forth) or with the intervention of cathepsin protease activity, induce apoptosis. 15-17
As mentioned above, regarding drug-mediated apoptosis, one of the major interests in the field of cancer research is represented by the cell susceptibility to drugs of widespread use in clinical practice. Among these, the drug cisplatin (cisPt) gained the attention of physicians in view of its powerful activity against the growth of a plethora of different neoplastic tissues. 18 In recent years cytokine gene transfer into tumor cells has received particular attention as a potentially useful approach for cancer therapy. 19 Studies by our group, as well as by other groups, have for instance shown that type I interferon (IFN) gene transfer into different tumor cell lines resulted in 1) a marked loss of tumorigenicity, 20 2) a down-regulation of oncogene expression and induction of tumor suppressor genes contributing to the anti-proliferative activity, 21-24 and 3) an increase in MHC class I expression that can enhance immune recognition. 20,25,26 Moreover, it has been reported that IFN-α gene transfer sensitizes human tumor cells to apoptosis induced by cytotoxic agents, and that a better therapeutic effect could be achieved when the treatment of tumor-bearing mice with IFN-expressing cells was combined with chemotherapy. 20 We have recently shown that the retroviral transduction of human consensus IFN (CIFN) coding sequence into two human melanoma cell lines resulted in a strong augmentation of cisplatin-induced apoptosis, associated with an IFN-dependent increase in p53 expression. 22 Similar effects, although less marked, were also observed after cultivation of parental melanoma cells in the presence of exogenous IFN. Cisplatin administration to nude mice bearing IFN-producing tumors resulted in complete tumor regression, whereas only partial tumor inhibition was observed after cisplatin treatment of mice with control tumors. 22 In the present work we partially address the mechanisms underlying the IFN-induced modulation of melanoma cell susceptibility to cisPt, pointing to the mitochondrial activity and related redox homeostasis as “supervisors” of cell susceptibility to apoptotic triggering.
Materials and Methods
Cell Lines
The HLA-A2 1B6 and 8863 melanoma cell lines were obtained from the tissues of two patients as previously reported. 22 The 1B6 clone was isolated from the M10538 cell line, established from a primary skin melanoma lesion, 27 whereas the 8863 cell line was derived from a metastatic melanoma. 28 The HLA haplotype of the two patients was previously identified. 27,28 Both cell lines were cultivated in RPMI-1640 supplemented with 50 μg/ml penicillin, 50 μg/ml streptomycin (BioWhittaker, Verviers, Belgium), and 10% fetal calf serum (Sebam, Berlin, Germany).
Recombinant Retrovirus Production and Infection of Melanoma Cells
The LXSN retroviral vector 29 containing the neomycin resistance gene under the control of the SV40 promoter, was obtained from AD Miller (Fred Hutchinson Cancer Research Center, Seattle, WA). The LCIFNSN retroviral vector was constructed by cloning of the EcoRI-BalHI fragment of plasmid pIFNSS 30 into the LXSN vector. The pIFNSS construct, kindly provided by Amgen (Thousand Oaks, CA), contains a sequence encoding a signal peptide, corresponding to that most commonly found in human IFN subtypes, followed by the sequence encoding CIFN, a synthetic IFN whose amino acid residues are common to naturally occurring human IFN subtypes (pIFNSS). 30 Both LXSN and LCIFNSN recombinant retroviruses were obtained following standard trans-infection procedures in GP+E86 31 and Gp+envAm12 32,33 packaging cells as previously reported. 22 LXSN and LCIFNSN retroviruses were used for infection of 1B6 and 8863 melanoma cells, as previously described. 22 After selection into G418-containing medium, control transduced cells (LXSN) or cells producing ∼1000 IU/ml IFN (LCIFNSN) were isolated from both 1B6 and 8863 cell lines. 22
IFN Titration
IFN was titrated on HeLa cells as described elsewhere. 22 IFN titers are expressed as IU. Human recombinant CIFN had a specific activity of 1 × 10 9 U mg/ml of protein and was kindly provided by Amgen.
Treatments
1B6 and 8863 parental melanoma cells, cultivated in the absence or in the presence of 1000 IU/ml of CIFN, or transduced (LXSN and LCIFNSN) melanoma cells were grown at 5 × 10 4 cells/ml density. After 48 hours of incubation at 37°C in a 5% CO2 atmosphere cells were treated as follows: 1) exposed for 1 hour to 50 μmol/L of cisPt, washed, and cultivated in fresh medium for up to 24 hours (early events studies) or 48 hours (late events studies); 2) exposed to 10 μmol/L of staurosporine (STS, Sigma Chemical Co., St. Louis, MO) for 6 hours; 3) irradiated with 660 J/m 2 UVB, 34 washed, and cultivated for up to 24 hours; 4) incubated with 500 ng/ml of anti-Fas antibodies (clone CH11; Upstate Biotechnology, Lake Placid, NY) for 48 hours. Moreover, dimethyl sulfoxide (STS vehicle, Sigma Chemical Co.) or mouse IgM (control for Fas-triggering) were also considered in our experiments. To characterize the apoptotic cascade we used specific caspase (Bouty, Milan, Italy) or cathepsin (ICN Biochemical Inc., Milan, Italy) inhibitors. Four hours before treatment with the apoptotic stimuli, we added 100 μmol/L of DEVD-CHO (cell permeant caspase-3 inhibitor), LEHD-CHO (cell permeant caspase-9 inhibitor), ZVAD-CHO (cell permeant pan-caspase inhibitor), or 20 IU of cystatin C (cathepsin inhibitor) directly to the culture medium. Cells treated with DEVD-CHO, LEHD-CHO, ZVAD-CHO, or cystatin C alone for the same time were considered as control. To investigate the specific involvement of mitochondria in apoptotic phenomenon, cells were pretreated for 1 hour with 10 μmol/L of cyclosporin A (CyA, an inhibitor of mitochondrial pore transition; Sigma Chemical Co.) and than exposed to different apoptotic stimuli. Results obtained by CyA were confirmed by using bongkrekic acid (BA; a generous gift of Prof. A. Toniniello, University of Padova, Padova, Italy) as previously reported. 35 Like CyA, BA is able to modulate mitochondrial pore transition. Cells treated with CyA or BA alone were considered as controls in this series of experiments. In consideration of the similar behavior exerted by BA toward cisPt-induced apoptosis, only the results obtained by CyA are shown. At the end of treatment 1B6 and 8863 cell lines were analyzed for apoptosis quantification, mitochondrial transmembrane potential evaluation, and caspase activity as specified.
Evaluation of Apoptosis
Quantitative evaluation of apoptosis was performed by using the following flow and static cytometry methods: 1) TdT incorporation of labeled nucleotides into DNA strand breaks [terminal dUTP nick-end labeling (TUNEL)-fluorescein isothiocyanate (FITC); Boehringer Mannheim, Milan, Italy]. Cells fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 15 minutes were washed and then permeabilized with 70% ice-cold ethanol for 5 minutes at 4°C. After washing the cells were incubated with TUNEL reaction mixture according to the manufacturer’s instructions. 2) Double staining was done by using the Annexin V-FITC apoptosis detection kit (Eppendorf s.r.l., Milan, Italy). By using this technique, cells that have lost membrane integrity (therefore considered as necrotic cells) will show nuclear red staining with propidium iodide (40 μg/ml) so that they will be easily distinguishable from the living cells. 3) Staining with chromatin dye Hoechst (Molecular Probes, Eugene, OR) was as previously described. 36
Mitochondrial Membrane Potential (ΔΨ) and Mitochondrial Mass
The ΔΨ of control and treated cells was studied by using the probe JC-1. According to this method, cells were stained with 10 μmol/L of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine iodide (JC-1, Molecular Probes). JC-1 is a metachromatic probe able to enter selectively into the mitochondria. It exists in a monomeric form (in the green channel, FL1) but, depending on the membrane potential, JC-1 can form J-aggregates that are associated with a large shift in the emission range (in the orange channel, FL2). 37 JC-1 is both qualitative (considering shift from green to orange) and quantitative (considering the pure fluorescence intensity). Results obtained by JC-1 were confirmed by using 3,33-dihexyloxacarbocyanine iodide probe (DiOC6, Molecular Probes) as previously reported. 38 As a methodological control, cells were also treated with increasing concentration (from 0.1 to 10 μg/ml) of the K+ ionophore valinomycin (Sigma Chemical Co.) that dissipates mitochondrial membrane potential but not pH gradient. According to literature data 37 we observed a dose-dependent decrease in FL2 signals after valinomycin incubation (data not shown). For analysis of mitochondrial mass, cells were incubated at 37°C for 30 minutes with 5 μmol/L of nonylacridine orange (Molecular Probes). After washing, samples were immediately analyzed by flow cytometry. To verify cell viability, parallel tubes were incubated with propidium iodide (40 μg/ml for 15 minutes at 37°C) before analyses.
Caspase Enzyme Assay
Activity of caspase-3, -8, and -9 was measured by using a colorimetric protease assay kit (Chemicon International, Inc., Temecula, CA). Protein (50 to 200 μg) by cytosolic extract was incubated with 200 μmol/L of the DEVD-p-NA (for caspase-3), LEHD-p-NA (for caspase-9), or IETD-p-NA (for caspase-8). The assay is based on spectrophotometric detection of the chromophore p-nitroanilide (p-NA) after cleavage from the labeled substrates. The p-NA light emission can be quantified using a microtiter plate reader at 405 nm. Comparison of the absorbance of p-NA from apoptotic samples with an uninduced control allows determination of the fold increase in caspase activity. 39
Immunofluorescence
Static Cytometry
Control and treated melanoma cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 minutes at room temperature. After washing in the same buffer, cells were permeabilized with 0.5% Triton X-100 (Sigma Chemical Co.) in PBS for 5 minutes. For localization of cytochrome c and apaf-1, samples were incubated at 37°C for 1 hour with monoclonal antibody to cytochrome c (Pharmingen, San Diego, CA) or to apaf-1 (Santa Cruz Biotechnology, Santa Cruz, CA). Cells were then incubated with anti-mouse IgG FITC-conjugate (Sigma Chemical Co.). After washing, samples were mounted with glycerol-PBS (2:1) and observed by intensified video microscopy with a Nikon Microphot fluorescence microscope, as described elsewhere. 40 Images were captured by a color-chilled 3CCD camera (Hamamatsu, Japan). Normalization and background subtraction were performed for each image. Figures were obtained by Optilab (Graftek, France) software for image analysis. For quantitative analyses of cytochrome c intracellular localization and distribution of at least 50 microscopic fields, for a total amount of 500 cells for each sample, were counted in quadruplicate at high magnification (×500) by a fluorescence microscope.
Flow Cytometry
For evaluation of intracellular antigens, control and cisPt-treated melanoma cells (1B6 and 8863) were pelleted and fixed in 70% ice-cold methanol. After washing with cold PBS, samples were stained with monoclonal antibodies to cytochrome c (Pharmingen), Hsp70 (Transduction Laboratories, Lexington, KY), apaf-1 (Santa Cruz Biotechnology), and rabbit polyclonal antibody to cathepsin B (Calbiochem). Negative controls were incubated with mouse IgG1 or total rabbit serum. After 1 hour at 4°C, samples and isotypic controls were incubated for 30 minutes at 37°C with FITC-labeled anti-mouse or FITC-labeled anti-rabbit (Sigma Chemical Co.) antibodies. After washing, cells were analyzed on a flow cytometer. The median values of the cytofluorimetric histograms obtained are reported in the present work. To verify the expression of CD95 on the cell surface of 1B6 and 8863 cells, a monoclonal antibody to CD95 conjugated to R-phycoerythrin (Becton Dickinson, Mountain View, CA) was used. No significant variation in the expression of this receptor (P > 0.05) was found among 1B6 and 8863 parental melanoma cells, cultivated in the absence or in the presence of 1000 IU/ml of CIFN or in transduced melanoma cells (LXSN and LCIFNSN).
Measurements of ROS
Control and treated cells (5 × 105) were harvested and incubated in 495 μl of Hanks’ balanced salt solution (pH 7.4) with 5 μl of hydroetidine (Molecular Probes) or dihydrorhodamine 123 (DHR 123, Molecular Probes) in polypropylene test tubes for 15 minutes at 37°C. The final concentration of hydroetidine and DHR 123 were 1 μmol/L and 10 μmol/L, respectively. Hydroetidine is a chemically reduced ethidium derivative. It is a nonfluorescent membrane-permeable dye that can be oxidized directly to the red fluorescent ethidium bromide by O2− generated inside the cells after different treatments. DHR 123 is a dye freely diffusing into cells and oxidized primarily by H2O2 in a myeloperoxidase-dependent reaction to green fluorescence. 41 As DHR 123 is accumulated by mitochondria, it is possible to detect ROS production at mitochondrial levels. 42
Western Blotting Analysis
Whole-cell extracts were prepared by suspending the cell pellets in lysis buffer (10 6 cells/50 μl) containing 50 mmol/L Tris-HCl, pH 7.5, 2 mmol/L ethylenediaminetetra-acetic acid, 100 mmol/L NaCl, 1 mmol/L Na3Vo4, 1% Nonidet P-40, 10 μg/ml leupeptin, 5 μg/ml aprotin, and 10 μg/ml phenylmethyl sulfonyl fluoride. After incubation at 4°C for 15 minutes, the protein content of the supernatant was determined by Bio-Rad protein assay (Bio-Rad Laboratories, Munich, Germany). For each sample, 25 μg of protein extract were solubilized by boiling in sodium dodecyl sulfate sample buffer with reducing agents and applied to 8% or 10% sodium dodecyl sulfate-polyacrylamide gels, together with the prestained molecular weight markers (Amersham, Arlington Heights, IL). The proteins were then electrophoretically transferred to polyvinylidene difluoride membrane filters (Bio-Rad, Hercules, CA). Blotted membranes were blocked with 5% dried milk in Tris-buffered saline for 2 hours at room temperature and incubated for 1 hour in the same blocking solution containing specific antibodies to: 1) apaf-1 (mouse monoclonal anti-human apaf-1 antibody, Santa Cruz Biotechnology); 2) caspase-3 (mouse monoclonal anti-human antibody, Transduction Laboratories); 3) cathepsin B (rabbit polyclonal anti-human cathepsin B antibody; Calbiochem-Novabiochem Co, Darmstadt, Germany). As a control, the membranes were incubated with specific antibodies to β-tubulin (monoclonal anti-human, clone TUB 2.1; Amersham). Band detection was performed by using the enhanced chemiluminescence system (Amersham).
Data Analysis and Statistics
Data Analysis
Regarding flow cytometry studies, all of the samples were analyzed with a FACScan flow cytometer (Becton Dickinson) equipped with a 488 argon laser. At least 20,000 events have been acquired. Data were recorded and statistically analyzed by a Macintosh computer using CellQuest Software. All data reported in this study are the mean of at least four separate experiments ± SD. Calculation of fluorescence (expressed as median values) was performed after conversion of logarithmically amplified signals into values on a linear scale.
Statistics
Statistical analysis of apoptosis data were performed by using Student’s t-test. Statistical significance of flow cytometry studies was calculated by using the parametric Kolmogorov-Smirnov test. Concerning correlation tests, analysis of variance and regression analyses were performed by using the Statview software program for Macintosh. As a general rule, only P values <0.01 were considered as significant.
Results
Role of Mitochondria in the Enhanced Susceptibility to cisPt-Induced Apoptosis
One of the main markers of apoptosis-associated modification is represented by changes occurring in mitochondria, ie, of the mitochondrial membrane potential (ΔΨ). 6,42 Therefore, specific flow-cytometry analyses were performed 48 hours after cisPt administration to measure ΔΨ by using the JC-1 probe by flow cytometry technique. The results obtained are reported in Figure 1, A ▶ (1B6 cells) and B (8863 cells), where both the percentage of cells undergoing apoptosis and the percentage of cells with depolarized mitochondria are shown. We found a dramatic decrease in ΔΨ in a significant percentage of CIFN-producing cells (LCIFNSN, both in 1B6 and 8863 cell lines) after cisPt treatment; and that this decrease in mitochondrial membrane potential was significantly lower (P < 0.01) in parental and LXSN control cells after the same treatment (Figure 1, A and B) ▶ . In addition, a significantly (P < 0.01) lower percentage of cells with depolarized mitochondria was detected in CIFN-treated cells (data not shown) with respect to CIFN-producing (LCIFNSN) cells. Although this phenomenon was present in both cell lines (8863 and 1B6), our results clearly show that 1B6 primary melanoma cells (Figure 1A) ▶ were significantly (P < 0.01) more sensitive to cisPt-induced apoptosis with respect to the 8863 metastatic melanoma cell line (Figure 1B) ▶ . Furthermore, we found a statistically significant correlation between the apoptotic events (Figure 1, A and B ▶ , solid columns) and the percentages of cells displaying the mitochondrial membrane depolarization phenomenon (Figure 1, A and B ▶ , open columns). This positive correlation was detected for all 1B6 and 8863 cell lines considered: parental, LXSN (control-transfected cells), and CIFN-producing (LCIFNSN) cells after cisPt treatment for 48 hours. In particular, regression analysis indicated a highly significant correlation between the two distributions (8863 cells: r = 0.978, R 2 = 0.956, P < 0.0001; 1B6 cells: r = 0.984, R 2 = 0.969, P < 0.0001).
Figure 1.

Quantitative and qualitative analysis of mitochondrial membrane potential during cisPt-induced apoptosis. A and B: Cytofluorimetric quantitative evaluation of 1B6 (A) and 8863 (B) apoptotic cells as assessed by TUNEL reaction (hatched columns) and cells with depolarized mitochondria (open columns) as assessed by using JC-1 probe. C: Qualitative and quantitative cytofluorimetric analyses of mitochondrial membrane potential in both LCIFNSN (CIFN-producing) 1B6 (left) and 8863 (right) cells before and after 24 hours of cisPt treatment. Boxed areas in C indicate the percentage of cells that express high red fluorescence (corresponding to J-aggregates that typically increase when mitochondrial membrane becomes more polarized). A representative experiment of four is reported.
To search for events upstream of mitochondrial depolarization, we analyzed ΔΨ changes at earlier stages of apoptosis (Figure 1C) ▶ , namely at 24 hours after addition of cisPt (ie, 24 hours before the apoptosis evaluation shown in Figure 1, A and B ▶ ). At this time, no decrease in ΔΨ was found and the percentage of cells with depolarized mitochondria was negligible (<10%). Strikingly, unlike other cell lines, a marked increase of mitochondrial transmembrane potential, ie, a hyperpolarization of the inner mitochondrial membrane, was detected in CIFN-producing cells. In fact, both 1B6 and 8863 CIFN-producing (LCIFNSN) cell lines, showed, after 24 hours of exposure to cisPt, an increase of fluorescence emission in FL2 channel (corresponding to J-aggregates that typically increase when mitochondrial membrane becomes more polarized). A specific quantitative analysis indicated that only a small percentage (30.1% and 27.2% in 1B6 and 8863 cells, respectively) of untreated LCIFNSN cells was detectable in the boxed area (high red fluorescence) of the plotted graph (Figure 1C ▶ , first row). These values were similar to those found in all other cell lines (parental, exogenous CIFN-treated, LXSN, not shown). By contrast, at 24 hours after cisPt addition, this percentage significantly (P < 0.01) increased up to 78.3% and 61.0% in CIFN-producing 1B6 cells and 8863 cells, respectively. Considering that the mitochondria hyperpolarization phenomenon was described as an event occurring during the very early apoptotic phases, 43,44 we hypothesized a relationship between the increased apoptosis in CIFN-producing cells (LCIFNSN) at 48 hours after cisPt addition and the hyperpolarization state of their mitochondrial membrane observed at earlier times (24 hours) of cisPt treatment. In fact, statistical analysis indicated a positive correlation between the two events (IFN-producing 1B6 cells: r = 0.960, P < 0.001; IFN-producing 8863 cells: r = 0.928, P < 0.001).
The megapore formation is a very important event for those stimuli inducing the mitochondrial apoptotic pathway. 45,46 To verify the importance of this mitochondrial event in cisPt-induced apoptosis, we used CyA, an agent capable of inhibiting or reversing pore opening, consequently preventing mitochondrial membrane permeability transition. 47 Our results, shown in Figure 2 ▶ , clearly indicated that the addition of CyA significantly inhibited both cisPt-induced apoptosis (Figure 2, A and B) ▶ and mitochondria depolarization (Figure 2, C and D) ▶ in parental and control 1B6 and 8863 cell lines (P < 0.01) as well as in CIFN-treated and CIFN-producing counterparts (P < 0.0001). Importantly, when cisPt treatment was performed in the presence of CyA, the CIFN-induced enhancement of the percentage of apoptotic cells and cells with depolarized mitochondria was completely abrogated in CIFN-treated 1B6 and 8863 cells as well as in CIFN-producing 1B6 cells, and was substantially inhibited in CIFN-producing 8863 cells. Similar results were obtained by using BA as an alternative inhibitor of mitochondrial pore opening (data not shown). The observation that in the CIFN-producing 8863 cell line treated with cisPt in the presence of CyA or BA, the percentage of both apoptotic cells and cells with depolarized mitochondria remained significantly (P < 0.01) higher than that detected, after similar treatments, in parental, LXSN, and exogenous CIFN-treated cells, suggested that mechanisms other than mitochondrial pore opening were involved in CIFN-induced potentiation of cisPt effects (see below).
Figure 2.

Quantitative evaluation of apoptosis and ΔΨ in the presence of mitochondrial membrane permeability transition inhibitor (CyA). A and B: CyA significantly (P < 0.01) protects both 1B6 (A) and 8863 (B) cells from cisPt-induced apoptosis. Treatment with CyA alone did not induce any appreciable cytotoxic effect in either cell line. C and D: Percentages of 1B6 (C) and 8863 (D) cells with depolarized mitochondria were significantly lower in cisPt-treated cells in the presence of CyA with respect to control cells without CyA (P < 0.01).
Analysis of Apoptosis Cascade
We first considered upstream caspases, ie, those caspases that are involved in the initiation phase of apoptosis: caspase-8 (mainly involved in receptor-mediated apoptosis) and caspase-9 (mainly involved in mitochondria-mediated apoptosis). 8 Results reported in Figure 3A ▶ clearly show that in 1B6 cells 24 hours of cisPt treatment induced the activation of caspase-9 but not of caspase-8. This activation was significantly (P < 0.01) higher in LCIFNSN cells than in transfected control cells (LXSN). Notably, in the 8863 cell line neither caspase-9 nor caspase-8 was active at this time (24 hours of cisPt exposure, Figure 3A ▶ ). Importantly, activation of these two caspases in 8863 cells was undetectable also prolonging cisPt treatment up to 96 hours (data not shown).
Figure 3.
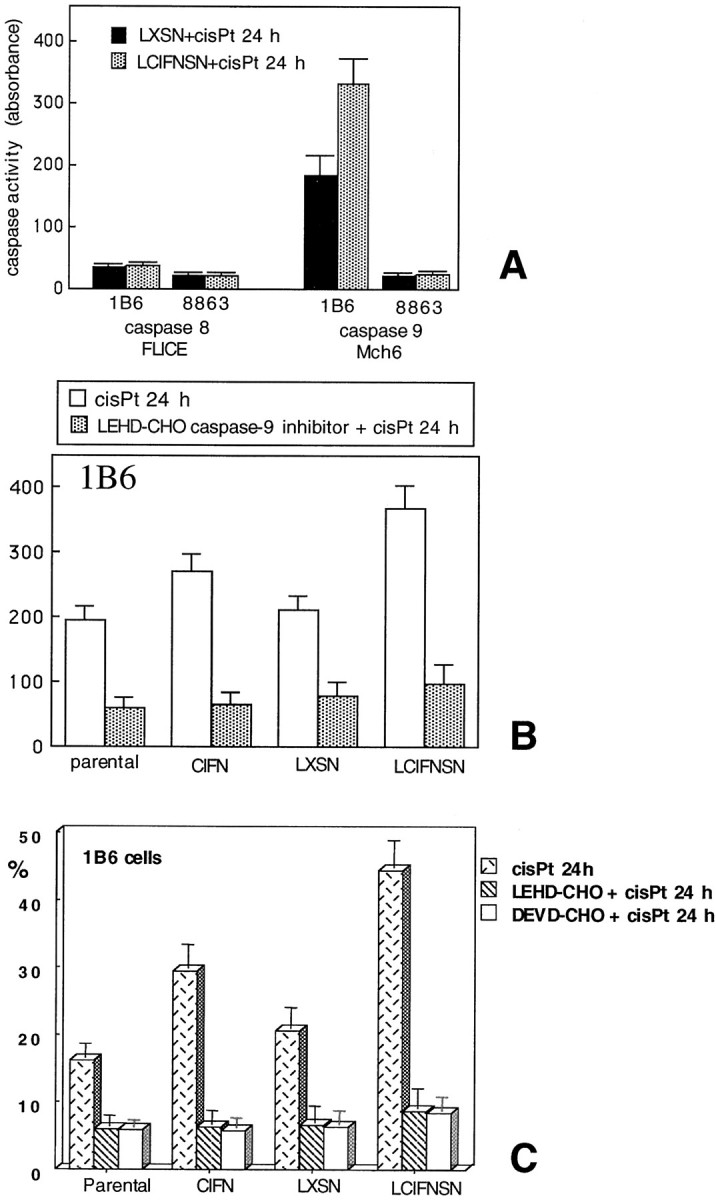
Analysis of apoptotic cascade. A: Activity of caspase-8 and caspase-9 in 1B6 and 8863 IFN-producing (LCIFNSN) and control-transduced (LXSN) cells at 24 hours after cisPt administration as assessed by colorimetric assay. B: Caspase-9 activity in 1B6 cell lines [parental, treated or not with exogenous CIFN, control-transduced (LXSN), and IFN-producing (LCIFNSN)] at 24 hours after cisPt addition, with or without LEHD-CHO caspase-9 inhibitor. C: Percentages of apoptotic cells obtained by annexin V/propidium iodide double staining in the presence or absence of LEHD-CHO and DEVD-CHO, caspase-9 and caspase- inhibitors, respectively.
To better evaluate the involvement of caspase-9 in cisPt-induced apoptosis in 1B6 cells, we used a specific cell permeant inhibitor of this caspase (LEHD-CHO). As shown in Figure 3 ▶ B, 24 hours of cisPt treatment induced the activation of caspase-9 in all 1B6 cell lines considered (Figure 3 ▶ B, open columns). However, according to apoptosis data (reported in Figure 3 ▶ C), this activity was significantly higher (P < 0.01) in LCFINSN cells with respect to other cell lines. Also in parental cells treated with exogenous CIFN caspase-9 activity was significantly (P < 0.01) increased with respect to control cells (parental and LXSN cells). However, the cisPt-induced sensitization to apoptosis [in terms of both percentage of apoptosis (Figure 3C ▶ , dotted columns) and caspase-9 activity (Figure 3B) ▶ ] was significantly (P < 0.01) less pronounced in parental cells cultivated in the presence of exogenous CIFN with respect to CIFN-producing cells (LCIFNSN). Figure 3, B and C ▶ , clearly shows that LEHD-CHO (caspase-9 inhibitor) was able to significantly prevent both caspase-9 activation (Figure 3B) ▶ and apoptosis (Figure 3C ▶ , hatched columns). Notably, in the presence of LEHD-CHO no significant difference (P > 0.05) was revealed, in terms of apoptosis (Figure 3C) ▶ and caspase-9 activity (Figure 3B) ▶ , between parental, parental cultivated in the presence of exogenous CIFN, LXSN, and LCIFNSN 1B6 cell lines. These results clearly indicate that caspase-9 plays a key role in mediating cisPt-induced apoptosis in 1B6 cells. We obtained similar results in 1B6 cells by using DEVD-CHO, a specific inhibitor of caspase-3, a caspase acting downstream to caspase-9 (Figure 3C ▶ , open columns). We thus decided to analyze the activity of caspase-3 in both 1B6 (Figure 4 ▶ ; A to C) and 8863 (Figure 4D) ▶ cell lines. Our analyses revealed a high activity of this caspase in 1B6 cells. In particular, 24 hours after cisPt administration, a powerful caspase-3 activity was already detected in LCIFNSN cells (Figure 4A) ▶ . By contrast, in parental cells, a caspase-3 activity was reached only later (48 hours after cisPt administration, Figure 4A ▶ ). Western blot analysis of 1B6 cells confirmed these results. In fact, as shown in Figure 4C ▶ , the appearance of the mature form of caspase-3 was detectable 48 hours after cisPt administration (represented by the band at 20 kd of molecular weight as a consequence of its activation). By contrast, as revealed by colorimetric assay (not shown) and by Western blot analysis (shown in Figure 4D ▶ ), in 8863 cells caspase-3 (as well as caspase-9) was not active at all times of cisPt treatment considered (up to 96 hours, not shown).Taken together these results clearly indicated that: 1) after cisPt administration, a higher caspase-9 activity was found in CIFN-producing 1B6 cells with respect to their control counterpart (LXSN cells) and 2) that caspase-9 acts in 1B6 cell line as an initiator-caspase. By contrast, 3) in 8863 cell line apoptosis, caspase-8, -9, and -3 seem to be not involved. Accordingly, pan-caspase inhibitor ZVAD-CHO was ineffective in these cells.
Figure 4.

Analysis of apoptotic cascade. A: Caspase-3 activity in 1B6 parental and IFN-producing (LCIFNSN) cells at 24 and 48 hours after cisPt administration obtained by colorimetric assay. B: Caspase-3 activity in 1B6 parental cells, parental cells treated with exogenous CIFN, control-transduced (LXSN), and IFN-producing (LCIFNSN) cells at 24 hours after cisPt addition in the presence or absence of DEVD-CHO caspase-3 inhibitor. C: Western blot analysis of caspase-3 in 1B6 cell lines. Note cleaved caspase-3 in cisPt-treated cells. D: Western blot analysis of caspase-3 in 8863 cell lines shows that caspase-3 was inactive in these cells independently from cisPt treatment.
As caspase-9 is the critical apical caspase activated in cells once cytochrome c is released from the mitochondria, 48 we analyzed cytochrome co-localization in 1B6 cells before and after cisPt addition by intensified video microscopy (Figure 5A) ▶ . In all 1B6 cisPt-untreated cells (parental, control LXSN, CIFN-treated, and LCIFNSN) cytochrome c appeared distributed throughout the cell cytoplasm as a dot-spot (Figure 5A ▶ , left; LCIFNSN-untreated cells are shown). An early event induced by cisPt administration (24 hours) was represented by a complete redistribution of cytochrome c. In particular, a perinuclear localization of this molecule was observed in 1B6 LCIFNSN cells (Figure 5A ▶ , right, arrows). A quantitative analysis of this phenomenon, performed by counting at least 500 cells as specified in Materials and Methods, revealed that this cytochrome c redistribution occurred in 8.3 ± 6.5% of 1B6 LCIFNSN cells, while at the same exposure time (24 hours) only 4.3 ± 2% of control parental cells and 2.6 ± 3.1% of LXSN cells (control-transfected cells) underwent cytochrome c rearrangement. Notably, a significantly (P < 0.01) lower percentage of CIFN-treated cells showing cytochrome c rearrangement was found (22.3 ± 3.2%) with respect to CIFN-producing cells.
Figure 5.
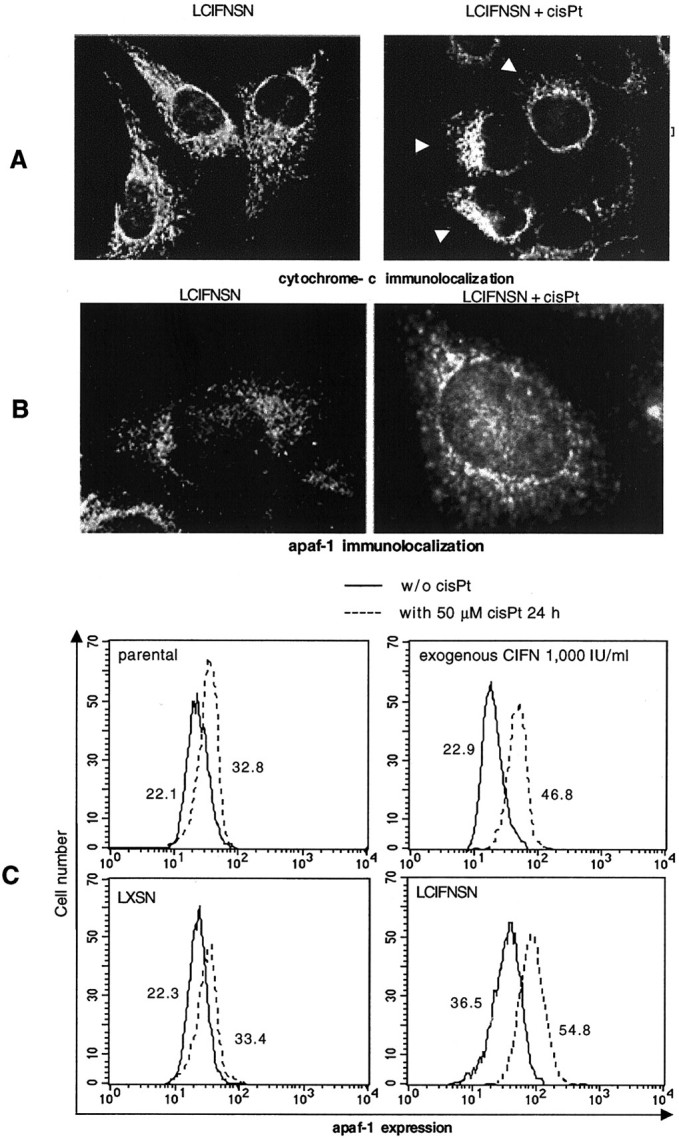
Analytical cytology analyses. A: Cytochrome c intracellular localization in 1B6 CIFN-producing cells (LCIFNSN) before (left) and after (right) 24 hours of cisPt treatment. Original magnification, ×1600. B: Intracellular localization of apaf-1 in 1B6 IFN-producing cells (LCIFNSN) before (left) and after (right) 24 hours cisPt treatment. Original magnification, ×2500. C: Quantitative analysis performed by flow cytometry of apaf-1 in 1B6 cells in the absence (solid line) and after (dotted line) 24 hours of cisPt treatment. Numbers represent the median values of fluorescence histogram. A representative experiment of four is shown.
Once released from mitochondria, cytochrome c interacts with apaf-1 to constitute apoptosome structure 48,49 and to activate pro-caspase-9. As for cytochrome c, intensified video microscopy analyses were thus conducted (Figure 5B) ▶ . Apaf-1 underwent an early appreciable rearrangement, after 24 hours of cisPt exposure, in the LCIFNSN cells only. In particular, it appeared scattered throughout the cell cytoplasm (Figure 5B ▶ , left) before cisPt treatment, whereas it appeared to be localized in the perinuclear region 24 hours after cisPt treatment (Figure 5B ▶ , right). In addition, a significant (P < 0.01) quantitative increase in apaf-1 expression was found in 1B6 melanoma cells treated for 24 hours with cisPt with respect to untreated counterparts (Figure 5C) ▶ . Although significant in all cisPt-treated 1B6 cell lines (as compared to the respective untreated controls), the increase in apaf-1 was higher in CIFN-treated parental cells and in CIFN-producing cells (LCIFNSN) with respect to parental and control LXSN cells, as clearly shown by the median values of cytofluorimetric histograms. More importantly, CIFN-producing 1B6 cells also displayed significantly (P < 0.01) higher basal values of apaf-1 expression (with respect to the other 1B6 cell lines considered) independently from cisPt administration.
Regarding the other cell model system considered in the present work, ie, the 8863 cell line, to analyze the CIFN-mediated sensitization to apoptosis a different approach was followed. In fact, caspases seemed not to be involved in cisPt-induced apoptosis in these cells (see above). Hence, we decided to evaluate the alternative pathway represented by the cathepsin cascade. 50 We thus analyzed the expression and the activation state of cathepsin B by both flow cytometry and Western blot in 8863 cells. Our analyses revealed a low basal expression of this enzyme in all 8863 cell lines considered (Figure 6A ▶ , solid lines). Nevertheless, a significantly (P < 0.01) higher expression of intracellular cathepsin B was observed both in CIFN-treated cells and, more markedly, in CIFN-producing cells with respect to parental and LXSN cells (Figure 6A ▶ , solid lines). Importantly, at 24 hours after cisPt addition, a further, significant increase (P < 0.01) of cathepsin B expression was observed in cells treated with exogenous CIFN as well as in CIFN-producing cells (LCIFNSN) (Figure 6A ▶ , dotted lines). Moreover, according to the results obtained on apoptotic susceptibility (see values in Figure 1B ▶ ), this cisPt-induced increase was more evident in LCIFNSN (CIFN-producing) 8863 cells in which the median value of the cytofluorimetric histogram were more than tripled with respect to LXSN control cells (Δ = +70.3%). In light of these results parallel control experiments were also conducted by using 1B6 cells. As clearly shown by the median values reported in the flow cytometry histograms in Figure 6B ▶ , higher basal expression levels of cathepsin B (analyzed by flow cytometry and Western blot) were found in all 1B6 cell lines with respect to 8863 cell counterparts. However, no difference in the basal expression of cathepsin B was observed between different 1B6 melanoma cell lines considered in this work (parental, LXSN, CIFN, LCIFNSN; Figure 6B ▶ , solid lines). However, importantly, no increase was registered after cisPt addition (Figure 6B ▶ , dotted lines).
Figure 6.
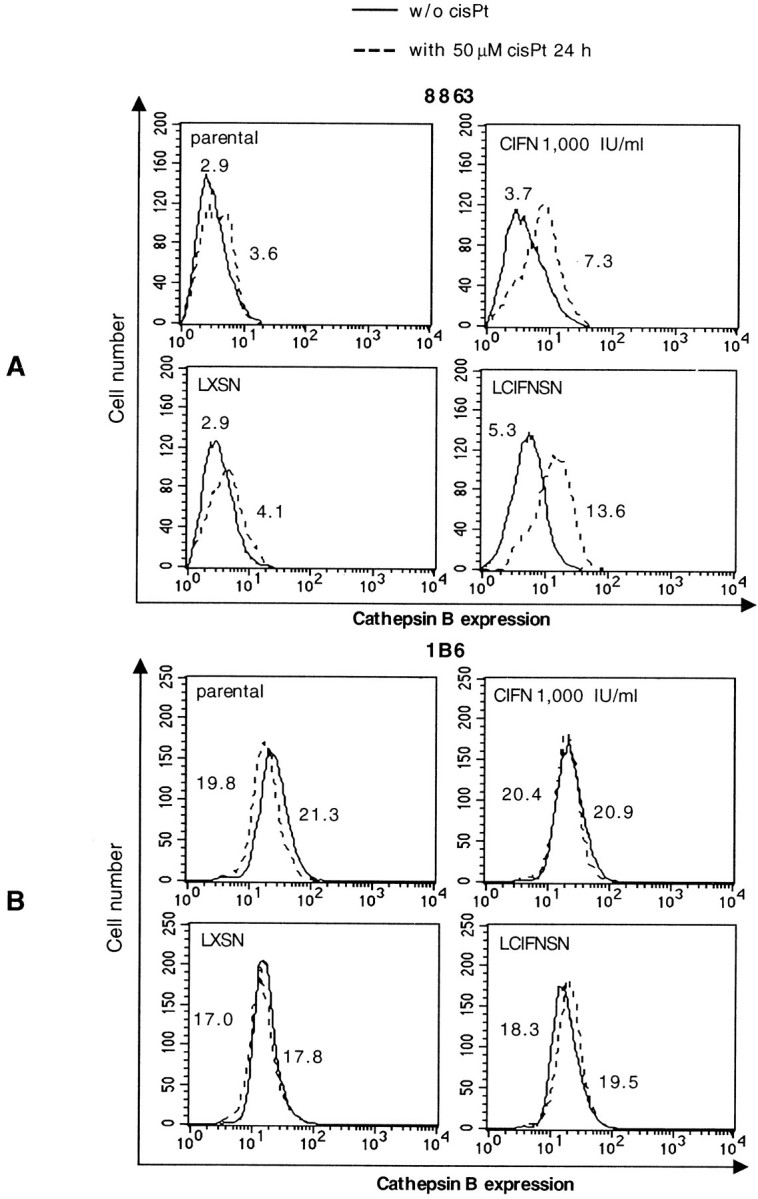
Cathepsin B expression analyses. Cathepsin B expression, as measured by cytofluorimetric analysis in 8863 (A) and 1B6 (B) cell lines in the presence (dotted line) or absence (solid line) of cisPt. Numbers represent the median values of fluorescence histograms. One experiment representative of four is shown.
To verify the involvement of cathepsin B in cisPt-induced apoptosis we also performed specific experiments by using cathepsin B inhibitor cystatin C in both 8863 (Figure 7A) ▶ and 1B6 (Figure 7B) ▶ cell lines. We found that pretreatment with cystatin C was able to significantly (P < 0.01) prevent apoptosis in all 8863 cell lines considered (parental, CIFN-treated, LXSN, and CIFN-producing cells). In fact, in these cells, a significant inhibition of apoptosis was observed at both 24 and 48 hours after cisPt administration (Figure 7A) ▶ . Moreover, a significant inhibition of apoptosis was also found in 1B6 cells (parental, CIFN-treated, LXSN, and LCIFNSN; ∼30%, P < 0.01) at 24 hours after cisPt treatment (Figure 7B) ▶ . Interestingly, the protection from apoptosis exerted by cystatin C in 1B6 cells was lost at 48 hours after exposure to cisPt. These results seem to suggest that cathepsin B, although of relevance, is not primarily involved in cisPt-induced apoptosis in 1B6 cell lines, which preferentially use the caspase-dependent cell death pathway. By contrast, in 8863 cells cathepsin B seems to be of fundamental importance for the death program triggered by cisPt as well as in the CIFN-induced sensitization to cisPt.
Figure 7.
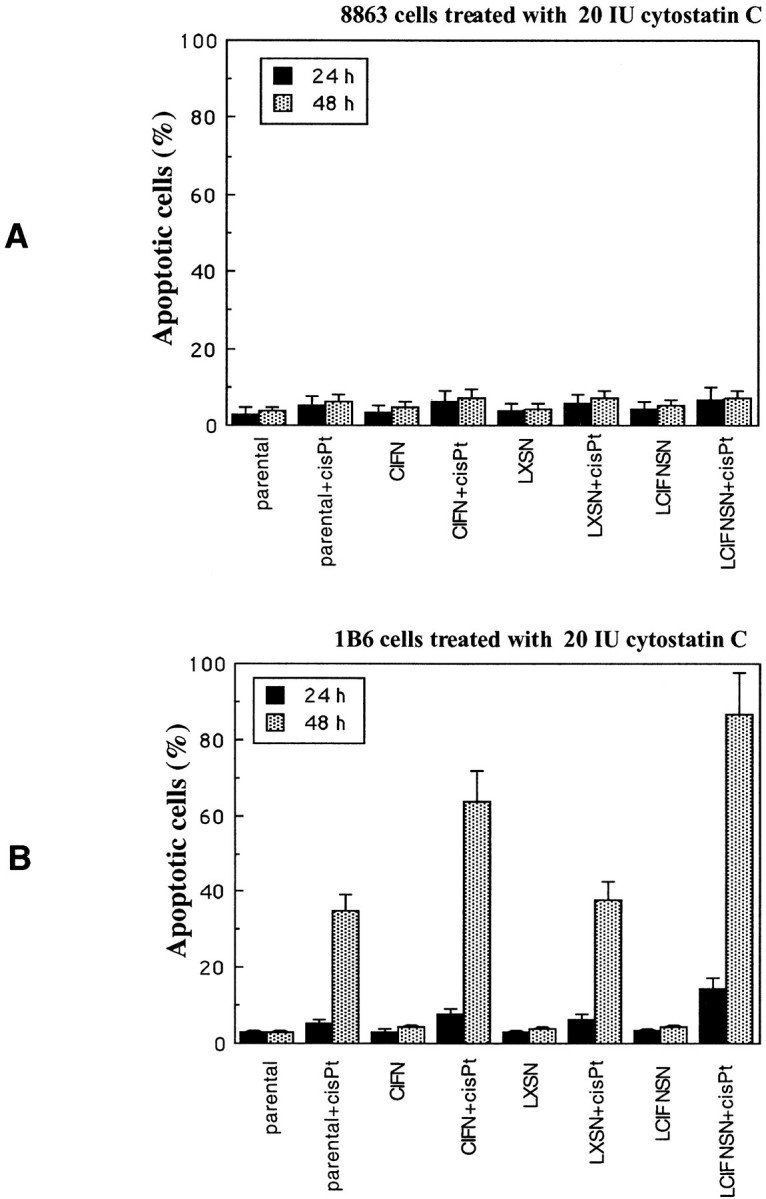
Cathepsin B and apoptosis. Percentage of apoptotic cells at 24 hours (double-staining annexin V/propidium iodide) and at 48 hours (by TUNEL reaction) after cisPt treatment in both 8863 (A) and 1B6 (B) cell lines pretreated with cystatin C as specific inhibitor of cathepsin B. Cystatin C exerted only a partial protection in 1B6 cells after 24 hours of cisPt treatment. By contrast, a significant protective effect was detected in 8863 cells at both 24 and 48 hours after cisPt treatment.
Redox Balance Analysis
Previous studies demonstrated that cell commitment to death is associated with ROS production. 42,51 Thus, in consideration of the well-known relationships between ROS production and mitochondrial function, 43 we also evaluated cell homeostasis in terms of redox balance and ROS production.
Mitochondrial Mass
Because a different number of mitochondria can influence both ROS production and mitochondrial membrane potential measurements, we compared the mitochondrial mass of different 8863 and 1B6 cell lines considered here. To this purpose, the probe nonylacridine orange that preferentially recognizes the mitochondrial structural phospholipid cardiolipin was used. Table 1 ▶ , A and B, clearly shows that there were no significant differences in mitochondrial mass among the different 8863 and 1B6 cell lines independently of cisPt treatment.
Table 1.
Quantification, by Flow Cytometry, of Some Parameters Related to the Redox State of the Cells: 1) Mitochondrial Mass (by Using NAO); 2) H2O2 Production (by Using DHR123); 3) O2 Production (by Using DHE); 4) Hsp70 Expression (by Using Specific mAb); and 5) Ubiquitin Expression (by Using Specific mAb) Both in 8863 (A) and 1B6 (B) Cell Lines
| Parental | CIFN | LXSN | LCIFNSN | |||||
|---|---|---|---|---|---|---|---|---|
| w/o cisPt | with cisPt | w/o cisPt | with cisPt | w/o cisPt | with cisPt | w/o cisPt | with cisPt | |
| A. 8863 | ||||||||
| NAO | 102 ± 13 | 105 ± 11 | 99 ± 9 | 101 ± 7 | 104 ± 9 | 98 ± 7 | 100 ± 6 | 103 ± 8 |
| H2O2 | 89 ± 8 | 94 ± 9 | 102 ± 11 | 155 ± 13 | 97 ± 7 | 115 ± 11 | 102 ± 7 | 189 ± 15 |
| O2 | 123 ± 10 | 119 ± 8 | 115 ± 12 | 121 ± 13 | 126 ± 11 | 118 ± 9 | 124 ± 11 | 207 ± 18 |
| Hsp70 | 14 ± 2 | 13 ± 3 | 11 ± 2 | 17 ± 4 | 14 ± 3 | 14 ± 4 | 13 ± 3 | 20 ± 4 |
| Ubiquitin | 53 ± 7 | 64 ± 8 | 49 ± 7 | 78 ± 6 | 50 ± 6 | 61 ± 5 | 54 ± 7 | 92 ± 9 |
| B. 1B6 | ||||||||
| NAO | 89 ± 7 | 93 ± 9 | 91 ± 7 | 97 ± 10 | 93 ± 7 | 91 ± 5 | 94 ± 8 | 90 ± 7 |
| H2O2 | 91 ± 8 | 122 ± 10 | 90 ± 8 | 155 ± 12 | 97 ± 6 | 145 ± 9 | 104 ± 8 | 193 ± 13 |
| O2 | 88 ± 8 | 93 ± 7 | 101 ± 9 | 126 ± 10 | 89 ± 7 | 101 ± 8 | 107 ± 12 | 248 ± 16 |
| Hsp70 | 2 ± 0 | 2 ± 0 | 2 ± 0 | 2 ± 0 | 2 ± 0 | 2 ± 0 | 2 ± 0 | 2 ± 0 |
| Ubiquitin | 12 ± 2 | 12 ± 3 | 12 ± 2 | 13 ± 4 | 13 ± 3 | 14 ± 5 | 12 ± 4 | 61 ± 7 |
Numbers in the tables represent median values of the fluorescence histograms after conversion of logarithmically amplified signals into values on a linear scale. Underlined values indicate a significant difference towards respective cisPt-untreated control cells.
ROS Production
To verify whether the mitochondrial hyperpolarization found in LCIFNSN cells was paralleled by alteration of the redox status of the cells, we evaluated ROS production. We used the fluorescent dye DHE, able to preferentially reveal superoxide anions, O2, and the oxidation-sensitive fluorescent probe DHR 123, capable of preferentially detecting hydrogen peroxide, H2O2. CisPt administration significantly (P < 0.01) increased ROS production in CIFN-producing cells and CIFN-treated cells [in both cell types 8863 (Table 1 ▶ A, second row) and 1B6 (Table 1 ▶ B, second row)]. This H2O2 hyperproduction appears paralleled by the elevation of ΔΨ observed at the same time (24 hours) as an early event in 1B6 and 8863 cells (see Figure 1C ▶ ). However, according to apoptosis data, this increase was more pronounced in LCIFNSN (CIFN-producing) cells with respect to cells treated with exogenous CIFN. Significant changes in the production of superoxide anion were detected (Table 1 ▶ , A and B, third row) in CIFN-producing cells only.
Stress Response
As shown in Table 1 ▶ , A and B (fourth row), Hsp70 protein expression was evaluated in both cell lines. This chaperone protein is increased in different stress conditions, ie, by ROS, and can protect cells from oxidative damage. 52 Our results showed a significant increase (P < 0.01) in the expression of this protein in LCIFNSN (CIFN-producing) 8863 cells after exposure to cisPt (Table 1 ▶ A). Notably, this increase was relevant only in these two 8863 cell lines (exogenous CIFN + cisPt versus exogenous CIFN, Δ = 34.7%; LCIFNSN + cisPt versus LCIFNSN, Δ = 33.4%) whereas it was absent in cisPt-treated parental and LXSN cells. Interestingly, 1B6 cells were almost negative for Hsp70 independently from cisPt administration (Table 1 ▶ B). This very low expression of Hsp70 could be of some relevance in explaining the high cisPt-induced apoptosis susceptibility observed in this cell line.
In the same vein, we evaluated the expression of ubiquitin, known to have a role in both shock response and apoptosis. 53 The results obtained clearly indicated that a marked increase of this protein was detectable in 8863 cells after cisPt administration and that this increase was highly significant in both CIFN-treated (CIFN versus CIFN + cisPt, Δ = 37.2%) and CIFN-producing (LCIFNSN versus LCIFNSN + cisPt, Δ = 40.8%) 8863 cells (Table 1 ▶ A, last row) cells. By contrast, in 1B6 cells, no significant variations were observed after cisPt treatment in parental cells, CIFN-treated cells and LXSN-transduced cells. In contrast, in CIFN-producing 1B6 cells the increase of ubiquitin expression after cisPt administration was dramatic and highly significant (P < 0.01, LCIFNSN versus LCIFNSN + cisPt, Δ = 80.0%).
Apoptotic Proneness Analysis
Finally, sensitivity of the 1B6 and 8863 cell lines to various apoptotic stimuli were considered: 1) STS (1 μmol/L 6 hours), known to induce mitochondrial-mediated apoptosis; 2) UVB radiation that induces cell death mainly involving ROS production and mitochondrial machinery; and 3) anti-Fas antibody, that triggers cell death via a receptor-mediated apoptotic cascade (typical type I stimulus). As a general rule, the results confirmed a major susceptibility to apoptosis of 1B6 cells (Figure 8A) ▶ with respect to 8863 cells (Figure 8B) ▶ . Furthermore, as observed for cisPt-induced apoptosis, CINF-producing (LCIFNSN) cells were significantly (P < 0.01) more susceptible to apoptosis with respect to parental and LXSN cells. This increased susceptibility was also detected in CIFN-treated cells although less markedly than in CIFN-producing cells. Importantly, this was clearly observed in both cell lines (1B6 and 8863; Figure 8, A and B ▶ ). By contrast, anti-Fas antibodies were not able to induce cell death in any 1B6 and 8863 cell lines. Interestingly, as demonstrated by cytofluorimetric analyses, CD95/Fas/APO1 was normally expressed on the surface of both 8863 and 1B6 cell lines (data not shown) and there were not significant (P > 0.05) differences in the expression of CD95/Fas between parental, CIFN-treated, control LXSN cells, and CIFN-producing cells (LCIFNSN). This is suggestive of a specific low susceptibility of both cell lines to type I apoptotic pathway. 8
Figure 8.
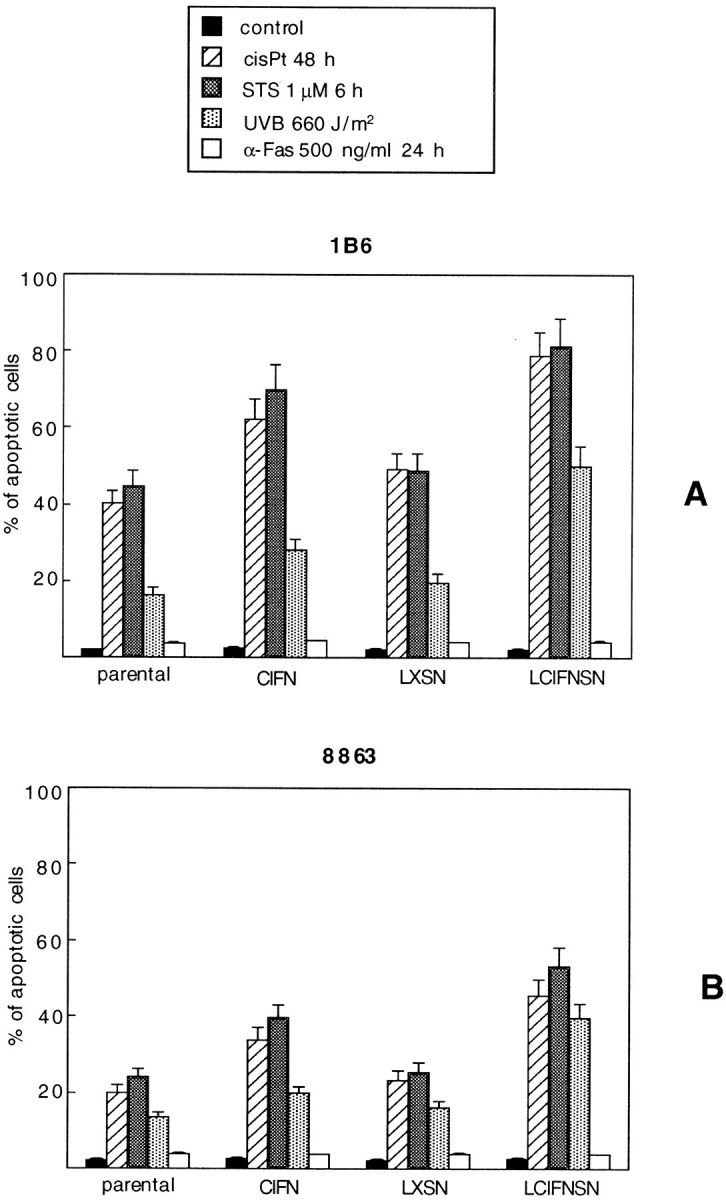
Apoptotic susceptibility. Cytofluorimetric quantitative evaluation of the percentage of apoptotic cells (as assessed by TUNEL reaction) in 1B6 (A) and 8863 (B) cell lines after induction with different apoptotic stimuli.
Discussion
Two different human melanoma cell lines, 8863 and 1B6, genetically modified to produce type I IFN, were demonstrated to undergo a powerful apoptotic response with suboptimal doses of cisPt. 22 In the present work we partially addressed the molecular mechanisms underlying this susceptibility. We found that irrespective of the apoptotic cascade used, caspase- or noncaspase-dependent, CIFN gene transfer confers an increased cisPt apoptotic susceptibility via a target effect on the major supervisor of type II apoptosis, ie, the mitochondrion. In fact, the two melanoma cell lines used here could be considered as different prototypes of different machineries for cell death: 1B6 cells underwent cisPt-induced apoptosis via caspase cascade whereas 8863 cells, lacking this machinery, use the alternative (less efficient) pathway represented by the cathepsin-mediated cell death. 54 In both situations, transfer of the human type I consensus IFN gene sensitized melanoma cells to cisPt-induced apoptosis. This sensitizing activity seemed to be significantly related to mitochondrial alterations observable in apoptotic process, ie, with changes of mitochondrial membrane potential. In fact, two different, and apparently conflicting, phenomena were described by the literature regarding mitochondria and apoptosis: 1) an increase of mitochondrial membrane potential (hyperpolarization) as well as 2) the loss of mitochondrial membrane potential (depolarization). It was postulated that, in some instances, the first phenomenon can represent a primitive event preceding the second. 42,44 Hyperpolarization can also represent a prerequisite for apoptotic susceptibility of cells administered with type II stimuli 35 and can be hypothesized to have a role in the cisPt sensitization of IFN-producing cells shown in the present work. In fact, increased ΔΨ found in our cell systems seems to be because of a specific target activity of type I IFN at the mitochondrial membrane level. In particular, shortly cisPt treatment (24 hours), ΔΨ change (hyperpolarization) was already appreciable in a high percentage of IFN-producing 1B6 and 8863 cells only. Subsequently, as expected, this change was followed, as a later event, by a depolarization of the mitochondrial membrane. These observations are in agreement with recent articles suggesting a direct activity of type I IFN in regulating mitochondrial function. 55
IFN-mediated enhancement of sensitivity to cisPt in CIFN-producing 1B6 melanoma cells was also associated with several events that paralleled the changes in the mitochondrial membrane described above. Namely, an early increase of caspase activity and an early intracellular redistribution of apoptosis-initiating key molecules such as cytochrome c and apaf-1 were found in IFN-producing 1B6 cells after cisPt addition. The first event was characterized by a fast and pronounced activation of caspase-9, the type II-associated caspase, which, in LCIFNSN better than in CIFN cells, was fully counteracted by its specific inhibitor, LEHD-CHO. Regarding the intracellular localization of cytochrome c and apaf-1, early changes were observed by analytical cytology studies in cisPt-treated IFN-producing 1B6 cells. In fact, both cytochrome c and apaf-1 molecules redistributed in the perinuclear region of IFN-producing cells only. Thus, as suggested by other authors 56,57 movements and trafficking of cell death-related molecules can play a key role in apoptosis proneness. Furthermore, parallel quantitative fluorescence-activated cell sorting analyses revealed that, unlike cytochrome c, the basal apaf-1 expression, ie, in the absence of cisPt treatments, was significantly higher in IFN-producing 1B6 cells with respect to control counterparts. Shortly after cisPt administration (24 hours), the highest absolute levels of apaf-1 expression were found in IFN-producing and IFN-treated 1B6 cells. These observations are in agreement with recent findings indicating that an increase in apaf-1 expression might represent a sensitizing factor toward apoptosis induction. 58
On the other hand, IFN-producing 8863 cells, also showing enhanced apoptotic proneness to cisPt, failed to display an active caspase cascade. These cells seemed to follow the alternative lysosome-associated pathway that seemed mainly mediated by cathepsins, a family of cysteine proteases associated with metastatic melanoma since 1986. 59 Although less efficiently, this enzymatic cascade was capable of apoptotic induction in 8863 cells. Importantly, IFN consensus gene transfection conferred to these cells the same behavior found in 1B6 melanoma cells, ie, apoptotic proneness. In fact, the earlier event in this cell line was represented by a significant increase of cathepsin B activity. Although parallel mechanisms cannot be ruled out, it can be hypothesized that this alternative route can play a role in the increased susceptibility of 8863 IFN-producing cells to cisPt induced apoptosis. As hypothesized above, this might be because of a specific activity of cathepsin B on mitochondrial homeostasis in LCIFNSN cells. Literature data are in fact suggestive for a target effect of cathepsin B on mitochondrial homeostasis and activation of a mitochondrially mediated apoptotic program. 50 Furthermore, in consideration of the possible implications of cathepsins in metastatic melanoma proliferation and invasiveness, 60 our results seem to be suggestive for a widespread reconsideration of pro-apoptotic drugs and their subcellular targets for metastatic melanoma. In fact, notably, regarding this point we can depict this paradox: 8863 cells that specifically use the cathepsin pathway to suicide are those with lower intracellular levels of this enzyme (with respect to 1B6 cells). This could be at least partially explained by the previously described release of cathepsins by metastatic cells. 61
The importance of mitochondria in the apoptotic cascade was assessed in a plethora of works describing the megapore opening and cytochrome c release as key factors in the initiation of type II apoptotic cascade. 8 Conversely, for type I stimuli, such as that represented by α-Fas monoclonal antibody (to which all cell lines considered here were refractory), this mitochondrial activity was described as subordinate (8,35). Notably, CyA and BA, specific inhibitors of mitochondrial pore opening, 47 significantly prevented apoptosis in cisPt-treated 1B6 and 8863 cells lines suggesting a key role for mitochondrial homeostasis in cisPt-induced apoptosis (Figure 2) ▶ . The difference in apoptotic sensitivity between 1B6 and 8863 cell lines can at least partially be explained by the different apoptotic machinery used. In fact, as already mentioned above, IFN-producing 8863 cells, although less efficiently, underwent significant cisPt-induced apoptosis by a mechanism involving the lysosomal cysteine protease cathepsin B and were impaired by a cathepsin-specific inhibitor, ie, cystatin C. 62 Albeit targeted to mitochondria 50 this cascade was still poorly described by literature and the 8863 cell line can thus represent an ideal cell model for future studies in this field.
A number of studies have demonstrated that DNA damage, caused by chemotherapeutic agents including cisPt, induces transient accumulation of p53 protein and results in programmed cell death. 63,64 The sensitivity to anti-cancer drugs has in fact been reported to be p53-dependent and the introduction of wild-type p53 into tumor cells has been reported to sensitize a variety of cancer cell lines to different anti-cancer drugs, including cisPt, both in vitro and in vivo. 65,66 In our previous study we showed that the extent and the kinetics of cisPt-induced apoptosis was correlated with the extent and the kinetics of IFN-dependent induction of p53. 22 This sensitization via IFN to p53-mediated apoptosis could be related to the results obtained on mitochondria presented in this study. In fact, it was hypothesized that mitochondria regulate p53 protein levels through a redox-dependent mechanism and that, in turn, p53 regulates mitochondrial membrane potential through ROS generation. 14 Moreover, a fraction of stress-induced p53 protein is known to localize at mitochondrial level at the onset of apoptosis. 67 Hence, in the present work we provide some additional link between the mechanisms underlying the control of cell fate via p53/mitochondrial function and we suggest that transfer of type I consensus IFN (CIFN) gene sensitizes human melanoma cells toward type II apoptotic triggering. 8,14
The last point to consider is represented by the role specifically played by oxidative stress, whose main cytoplasmic source is represented by mitochondria. In fact, apart from p53-associated mechanisms described above, the important role played by redox balance in ionic homeostasis of mitochondria and apoptosis is well established. 68 Strikingly, very few data are available in literature regarding a specific association of IFN and stress-associated molecules. 69 Here we show that only CIFN-producing cells significantly increased ROS production and stress-associated proteins (Hsp70 and ubiquitin) shortly after cisPt exposure. The first is well known to be associated with apoptosis induction and it was hypothesized to cause a marked hyperpolarization of mitochondria. 40 The second ones seem to be of relevance in the complex scenario provided by IFN-producing cells. In fact, the ubiquitin-proteasome pathway is the principal mechanism for the degradation of short-lived proteins in cells. This molecule, involved in apoptotic regulation, was found related to Hsp70 overexpression and it was hypothesized to be modulated by IFNs. 69 We can thus interpret these changes as counteracting mechanisms of cell resistance.
Although several aspects remain to be fully clarified, eg, different apoptotic susceptibility observed in CIFN-producing cells with respect to exogenous CIFN-treated cells, the results shown in the present work clearly indicate that type I IFN significantly sensitizes melanoma cells to type II apoptotic pathway, ie, mitochondrial pathway, induced by cisPt, radiation or STS. This booster effect led to an increased apoptotic proneness of major relevance in IFN-producing cells (LCIFNSN) rather than in exogenous IFN-administered cells (CIFN). Moreover, this proneness was detectable independently from the apoptotic cascade available in the cell (caspase-dependent or caspase-independent/cathepsin-dependent). This can suggest that the comprehension of mitochondrial role in drug-induced cell death program could also lead, in the long run, to the widespread use of highly active and mitochondrially targeted pro-apoptotic treatments in the control of tumor growth.
Footnotes
Address reprint requests to Prof. Walter Malorni, Department of Ultrastructures, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy. E-mail: malorni@iss.it.
Supported in part by grants from Ministero della Sanita’ and Ministero dell’ Universitiá e della Ricerca Scientifica Tecnologica (to W. M.) and by the Italian Association for Cancer Research (AIRC).
References
- 1.Reed CJ: Apoptosis and cancer: strategies for integrating programmed cell death. Semin Hematol 2000, 37:9-16 [DOI] [PubMed] [Google Scholar]
- 2.McCarthy NJ: Forever blebbing: a story of extended apoptosis. Trends Cell Biol 1997, 7:74. [DOI] [PubMed] [Google Scholar]
- 3.Villa P, Kaufman SH, Earnshaw WC: Caspases and caspase inhibitors. Trends Biochem Sci 1997, 22:388-393 [DOI] [PubMed] [Google Scholar]
- 4.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA: Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 1998, 94:325-337 [DOI] [PubMed] [Google Scholar]
- 5.Lesage S, Steff AM, Philippoussis F, Pagé M, Trop S, Mateo V, Hugo P: CD4+CD8+ thymocytes are preferentially induced to die following CD45 cross-linking, through a novel apoptotic pathway. J Immunol 1997, 159:4762-4771 [PubMed] [Google Scholar]
- 6.Green D, Kroemer G: The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol 1998, 8:267-271 [DOI] [PubMed] [Google Scholar]
- 7.Hengartner MO: The biochemistry of apoptosis. Nature 2000, 407:770-776 [DOI] [PubMed] [Google Scholar]
- 8.Schmitz I, Walczak H, Krammer PH, Peter ME: Differences between CD95 type I and type II cells detected with the CD95 ligand. Cell Death Differ 1999, 6:821-822 [DOI] [PubMed] [Google Scholar]
- 9.Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, Tsujimoto Y: Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc Natl Acad Sci USA 1998, 95:1455-1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green DR, Reed JC: Mitochondria and apoptosis. Science 1998, 281:1309-1312 [DOI] [PubMed] [Google Scholar]
- 11.Chakraborti T, Mondal M, Roychoudhury S, Chakraborti S: Oxidant, mitochondria and calcium: an overview. Cell Signal 1999, 11:77-85 [DOI] [PubMed] [Google Scholar]
- 12.Jurgensmeier J, Xie MZ, Deveraux Q, Ellerby L, Bredesen D, Reed JC: Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA 1998, 95:4997-5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vander Heiden MG, Chandel GHS, Williamson EK, Schumacker PT, Thompson CB: Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91:627-637 [DOI] [PubMed] [Google Scholar]
- 14.Li PF, Dietz R, von Harsdorf R: p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J 1999, 21:6027-6036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, Remy R, Xie ZH, Reed JC, Kroemer G: The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med 1998, 187:1261-1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G: Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 1998, 281:2027-2031 [DOI] [PubMed] [Google Scholar]
- 17.VanEijk M, deGroot C: Germinal center B cell apoptosis requires both caspase and cathepsin activity. J Immunol 1999, 163:2478-2482 [PubMed] [Google Scholar]
- 18.McClay EF, Albright KD, Jones JA, Eastman A, Christen R, Howell SB: Modulation of cisplatin resistance in human malignant melanoma cells. Cancer Res 1992, 52:6790-6796 [PubMed] [Google Scholar]
- 19.Simons JF, Mikhak B: Ex vivo gene therapy using cytokine-transduced tumor vaccines: molecular and clinical pharmacology. Semin Oncol 1998, 25:661-676 [PubMed] [Google Scholar]
- 20.Ferrantini M, Belardelli F: Gene therapy of cancer with interferon: lessons from tumor models and perspectives for clinical applications. Semin Cancer Biol 2000, 10:145-157 [DOI] [PubMed] [Google Scholar]
- 21.Pfeffer LM, Dinarello CA, Herberman RB, Williams BR, Borden EC, Bordens R, Walter MR, Nagabhushan TL, Trotta PP, Pestka S: Biological properties of recombinant alpha-interferons: 40th anniversary of the discovery of interferons. Cancer Res 1998, 58:2489-2499 [PubMed] [Google Scholar]
- 22.Mecchia M, Matarrese P, Malorni W, D’Agostino G, Sestili P, Santini SM, Gauzzi MC, Venditti M, Mazocchi A, Parmiani G, Belardelli F, Ferrantini M: Type I consensus interferon (CIFN) gene transfer into human melanoma cells up-regulates p53 and enhances cisplatin-induced apoptosis: implications for new therapeutic strategies with IFN-alpha. Gene Therapy 2000, 7:167-179 [DOI] [PubMed] [Google Scholar]
- 23.Davol PA, Goulette FA, Frackelton Jr AR, Darnowsky JW: Modulation of p53 expression by human recombinant interferon-2a correlates with abrogation of cisplatin resistance in a human melanoma cell line. Cancer Res 1996, 56:2522–2526 [PubMed]
- 24.Lotem J, Sachs L: Cytokine suppression of protease activation in wild-type p53-dependent and p53-independent apoptosis. Proc Natl Acad Sci USA 1997, 94:9349-9353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrantini M, Giovarelli M, Modesti A, Musiani P, Modica A, Venditti M, Peretti E, Lollini PL, Nanni P, Forni G: IFN-alpha1 gene expression into a metastatic murine adenocarcinoma (TS/A) results in CD8+ T cell-mediated tumor rejection and development of antitumor immunity. Comparative studies with IFN-gamma producing TS/A cells. J Immunol 1994, 153:4604-4615 [PubMed] [Google Scholar]
- 26.Belardelli F: Role of interferons and other cytokines in the regulation of the immune response. APMIS 1995, 103:161-179 [DOI] [PubMed] [Google Scholar]
- 27.Arienti F, Sulé-Suso J, Melani C, Maccalli C, Belli F, Illeni MT, Anichini A, Cascinelli N, Colombo MP, Parmiani G: Interleukin-2 gene-transduced human melanoma cells efficiently stimulate MHC-unrestricted and MHC-restricted autologous lymphocytes. Hum Gene Therapy 1994, 5:1139-1150 [DOI] [PubMed] [Google Scholar]
- 28.Sulé-Suso J, Arienti F, Melani C, Colombo MP, Parmiani G: A B7–1-transfected human melanoma line stimulates proliferation and cytotoxicity of autologous and allogeneic lymphocytes. Eur J Immunol 1995, 25:2737-2742 [DOI] [PubMed] [Google Scholar]
- 29.Miller AD, Rosman GJ: Improved retroviral vectors for gene transfer and expression. Biotechniques 1989, 7:980-984 [PMC free article] [PubMed] [Google Scholar]
- 30.Alton K, Stabinsky Y, Richards R, Ferguson B, Goldestein L, Altrock B, Miller L, Stebbings N: Production, Characterization and Biological Effects of Recombinant DNA-Derived Human IFN- and IFN- analogs. 1983:pp 119-128 H Shellenkens. Amsterdam, Elsevier Edited by E De Maeyer
- 31.Markowiitz D, Goff S, Bank A: A safe packaging line for gene transfer: separating viral gene on two different plasmid. J Virol 1988, 62:1120-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markowitz D, Goff S, Bank A: Construction and use of a safe and efficient amphotropic packaging cell line. Virology 1998, 167:400-406 [PubMed] [Google Scholar]
- 33.Zhang JF, Hu C, Geng Y, Blatt LM, Taylor MW: Gene therapy with an adeno-associated virus carrying an interferon gene results in tumor growth suppression and regression. Cancer Gene Ther 1996, 3:31-38 [PubMed] [Google Scholar]
- 34.Malorni W, Donelli G, Straface E, Santini MT, Paradisi S, Giacomoni PU: Both ultraviolet A and B induce cytoskeletal dependent surface blebbing in epidermoid cells. J Photochem Photobiol 1994, 26:265-270 [DOI] [PubMed] [Google Scholar]
- 35.Matarrese P, Testa U, Cauda R, Vella S, Gambardella L, Malorni W: Expression of P-170 glycoprotein sensitizes lymphoblastoid CEM cells to mitochondria-mediated apoptosis. Biochem J 2000, 355:587-595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malorni W, Rivabene R, Santini MT, Donelli G: N-acetylcysteine inhibits apoptosis and decreases viral particles in HIV-chronically infected U937 cells. FEBS Lett 1993, 327:75-78 [DOI] [PubMed] [Google Scholar]
- 37.Cossarizza A, Franceschi C, Monti D, Salvioli S, Bellesia E, Rivabene R, Biondo L, Rainaldi G, Tinari A, Malorni W: Protective effect of N-acetylcysteine in tumor necrosis factor alpha-induced apoptosis in U937 cells: the role of mitochondria. Exp Cell Res 1995, 220:232-240 [DOI] [PubMed] [Google Scholar]
- 38.Osborne BA: Apoptosis and the maintenance of homeostasis in the immune system. Curr Opin Immunol 1996, 8:245-254 [DOI] [PubMed] [Google Scholar]
- 39.Casciola-Rosen L, Nicholson DW, Chong T, Rowan KR, Thornberry NA, Miller D, Rosen A: Apopain/CPP32 cleaves proteins that are essential for cellular repair: a fundamental principle of apoptotic death. J Exp Med 1996, 183:1957-1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parlato S, Giammarioli AM, Logozzi M, Lozupone F, Matarrese P, Luciani F, Falchi M, Malorni W, Fais S: CD95 (APO-1/Fas) linkage to the actin cytoskeleton through ezrin in human T lymphocytes: a novel regulatory mechanism of the CD95 apoptotic pathway. EMBO J 2000, 19:5123-5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Such L, O’Connor JE, Saez GT, Gil F, Beltran JF, Moya A, Alberola A: Flow cytometric analysis of peroxidative activity in granulocytes from coronary and peripheral blood in acute methionine. Cytometry 1999, 37:140-146 [DOI] [PubMed] [Google Scholar]
- 42.Banki K, Hutter E, Gonchoroff NJ, Perl A: Elevation of mitochondrial transmembrane potential and reactive oxygen intermediate levels are early events and occur independently from activation of caspases in Fas signalling. J Immunol 1999, 162:466-470 [PMC free article] [PubMed] [Google Scholar]
- 43.Gross A, Pilcher K, Blachly-Dyson E, Basso E, Jockel J, Bassik MC, Korsmeyer SJ, Forte M: Biochemical and genetic analysis of the mitochondrial response of yeast to BAX and BCL-X(L). Mol Cell Biol 2000, 20:3125-3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsuyama S, Llopis J, Deveraux QL, Tsien RY, Reed JC: Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol 2000, 2:318-325 [DOI] [PubMed] [Google Scholar]
- 45.Kroemer G, Zamzami N, Susin SA: Mitochondrial control of apoptosis. Immunol Today 1997, 18:44-51 [DOI] [PubMed] [Google Scholar]
- 46.Kroemer G, Reed JC: Mitochondrial control of cell death. Nat Med 2000, 6:513-519 [DOI] [PubMed] [Google Scholar]
- 47.Desagher S, Martinou JC: Mitochondria as the central control point of apoptosis. Trends Cell Biol 2000, 10:369-377 [DOI] [PubMed] [Google Scholar]
- 48.Jiang X, Wang X: Cytochrome c promotes caspase-9 activation by inducing nucleotide binding to apaf-1. J Biol Chem 2000, 275:31199-31203 [DOI] [PubMed] [Google Scholar]
- 49.Perkins CL, Fang G, Kim CN, Bhalla KN: The role of Apaf-1, caspase-9, and bid proteins in etoposide- or paclitaxel-induced mitochondrial events during apoptosis. Cancer Res 2000, 60:1645-1653 [PubMed] [Google Scholar]
- 50.Guicciardi ME, Deussing J, Miyoshi H, Bronk S, Svingen PA, Peters C, Kaufmann SH, Gores GJ: Cathepsin B contributes to TNF-α-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome-c. J Clin Invest 2000, 106:1127-1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Um HD, Orenstein JM, Wahl SM: Fas mediated apoptosis in human monocytes by a reactive oxygen intermediate dependent pathway. J Immunol 1996, 156:3469-3477 [PubMed] [Google Scholar]
- 52.Polla BS, Kantengwa S, Francois D, Salvioli S, Marsac C, Cossarizza A: Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci USA 1996, 93:6458-6463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jentsch S, Pyrowolakis G: Ubiquitin and its kin: how close are the family ties? Trends Cell Biol 2000, 10:335-342 [DOI] [PubMed] [Google Scholar]
- 54.Johnson DE: Noncaspase proteases in apoptosis. Leukemia 2000, 14:1695-1703 [DOI] [PubMed] [Google Scholar]
- 55.Yanase N, Ohshima K, Ikegami H, Mizuguchi J: Cytochrome c release, mitochondrial membrane depolarization, caspase-3 activation, and BAX-alpha cleavage during IFN-alpha-induced apoptosis in Daudi B lymphoma cells. J Interferon Cytokine Res 2000, 20:1121-1129 [DOI] [PubMed] [Google Scholar]
- 56.Dinsdale D, Zhuang J, Chien GM: Redistribution of cytochrome c precedes the caspase-dependent formation of ultracondensed mitochondria, with a reduced inner membrane potential, in apoptotic monolayer. Am J Pathol 1999, 155:607-618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsu YT, Wolter KG, Youle RJ: Cytosol-to-membrane redistribution of Bax and Bclx(L) during apoptosis. Proc Natl Acad Sci USA 1997, 94:3668-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perkins C, Kim CN, Fang G, Bhalla KN: Overexpression of Apaf-1 promotes apoptosis of untreated and paclitaxel- or etoposide-treated HL-60 cells. Cancer Res 1998, 58:4561-4566 [PubMed] [Google Scholar]
- 59.Sloane BF, Rozhin J, Johnson K, Crissman JD, Honn KV: Cathepsin B: association with plasma membrane in metastatic tumors. Proc Natl Acad Sci USA 1986, 83:2483-2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldmann T, Suter L, Ribbert D, Otto F: The expression of proteolytic enzymes at the dermal invading front of primary cutaneous melanoma predicts metastasis. Pathol Res Pract 1999, 195:171-175 [DOI] [PubMed] [Google Scholar]
- 61.Honn KV, Timar J, Rozhin J, Sameni M, Ziegler G, Sloane BF: A lipoxygenase metabolite, 12-(S)-HETE, stimulates protein kinase C-mediated release of cathepsin B from malignant cells. Exp Cell Res 1994, 214:120-130 [DOI] [PubMed] [Google Scholar]
- 62.Kos J, Werle B, Lah T, Brunner N: Cysteine proteinases and their inhibitors in extracellular fluids: markers for diagnosis and prognosis in cancer. Int J Biol Markers 2000, 15:84-89 [DOI] [PubMed] [Google Scholar]
- 63.Soussi T: The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann NY Acad Sci 2000, 910:121-139 [DOI] [PubMed] [Google Scholar]
- 64.Weller M: Predicting response to cancer chemotherapy: the role of p53. Cell Tissue Res 1998, 292:435-445 [DOI] [PubMed] [Google Scholar]
- 65.Pestell KE, Hobbs SM, Titley JC, Kelland LR, Walton MI: Effect of P53 status on sensitivity to platinum complexes in a human ovarian cancer cell line. Mol Pharmacol 2000, 57:500-511 [DOI] [PubMed] [Google Scholar]
- 66.Lotem J, Sachs L: Differential suppression by protease inhibitors and cytokines of apoptosis induced by wild-type p53 and cytotoxic agents. Proc Natl Acad Sci USA 1996, 93:12507-12512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marchenco ND, Zaika A, Moll UM: Death signal-induced localization of p53 protein to mitochondria. J Biol Chem 2000, 257:16202-16212 [DOI] [PubMed] [Google Scholar]
- 68.Hall AG: The role of glutathione in the regulation of apoptosis. Eur J Clin Invest 1999, 29:238-245 [DOI] [PubMed] [Google Scholar]
- 69.Caraglia M, Abbruzzese A, Leardi A, Pepe S, Budillon A, Baldassarre G, Selleri C, Lorenzo SD, Fabbrocini A, Giuberti G, Vitale G, Lupoli G, Bianco AR, Tagliaferri P: Interferon-alpha induces apoptosis in human KB cells through a stress-dependent mitogen activated protein kinase pathway that is antagonized by epidermal growth factor. Cell Death Differ 1999, 6:773-780 [DOI] [PubMed] [Google Scholar]