

Abstract
PTEN mutation and microsatellite instability are two of the most common genetic alterations in uterine endometrioid carcinoma. Furthermore, previous studies have suggested an association between the two alterations, however the basis and consequence of the association is not understood. Recently it has been shown that 100% of female Pten+/− mice develop complex atypical hyperplasia by 32 weeks of age that progresses to endometrial carcinoma in ∼20 to 25% of mice at 40 weeks. In an attempt to expand this mouse model of endometrial tumorigenesis and to further our understanding of the association betweenPten mutations and DNA mismatch repair deficiency, we generated Ptenheterozygous, Mlh1-null (mismatch repair deficient) mice. Significantly, the majority ofPten+/−/Mlh1−/−mice developed polypoid lesions in the endometrium at 6 to 9 weeks of age. By 14 to 18 weeks, all of the double-mutant mice had lesions histologically similar to those seen inPten+/− mice, and two of them exhibited invasive disease. Moreover, the frequency of loss of the wild-type Pten allele in the double-mutant mice at 14 to 18 weeks was similar to that seen in lesions from 40-week-old Pten+/− mice. Taken together, our results indicate that DNA mismatch repair deficiency can accelerate endometrial tumorigenesis inPten heterozygous mice and suggests that loss of the wild-type Pten allele is involved in the development/progression of tumors in this setting.
To date, the most common genetic alterations identified in uterine endometrioid carcinoma (UEC), the most prevalent type of endometrial cancer, are mutations in the PTEN tumor suppressor gene and the presence of a molecular phenotype called microsatellite instability (MI). PTEN mutations have been detected in 35 to 83% and MI in 20 to 45% of UECs. 1-7 In addition, both alterations have also been detected in complex atypical hyperplasia (CAH), the direct precursor of UEC. 5,8,9 Together these findings implicate an important and early role for PTEN mutations and DNA mismatch repair (MMR) deficiency, detected as MI, in the pathogenesis of UEC. Moreover, several studies have shown a statistically significant association between PTEN mutations and MI, 1-3,10,11 however the basis and consequence of this association remains unknown.
In this and previous studies it has been shown that 100% of 32-week-old female mice lacking one wild-type copy of Pten spontaneously develop lesions that closely resemble CAH in humans. Of note, ∼20 to 25% of mice at 40 weeks of age develop invasive carcinoma with morphological similarities to UEC. 12,13 To exploit this mouse model of endometrial tumorigenesis and to further our understanding of the association between Pten mutations and DNA MMR deficiency, we developed Pten+/−/Mlh1−/− double-mutant mice. Here we present data showing that DNA mismatch repair deficiency can accelerate endometrial tumorigenesis in Pten heterozygous mice and that it is associated with loss of the wild-type Pten allele.
Materials and Methods
Mice
Male Pten heterozygous C57BL6/129SvJ mice were obtained from R. Parsons (Institute of Cancer Genetics, College of Physicians and Surgeons, Columbia University, New York, NY) 12 and bred to C57BL6 wild-type female mice (The Jackson Laboratory, Bar Harbor, ME). Mlh1 heterozygous C57BL6 mice were provided by R. Kucherlapati (Department of Molecular Genetics, Albert Einstein College of Medicine, Bronx, NY) 14 and interbred to increase the colony size. Mlh1+/− and Pten+/− mice were bred to generate double heterozygotes that were then interbred to produce Pten+/−/Mlh1−/− mice and appropriate littermate controls.
Genotyping
Animals were genotyped using multiplex polymerase chain reaction (PCR) on genomic DNA prepared from tail samples. Mlh1 genotyping was performed as described previously. 14 For Pten genotyping the following primers were used: mPTEN EX/I, GGGATTATCTTTTTGCAACAGT; mPTEN R, GGGCCTCTTGTGCCTTTA; and PGK6: AGAAAGCGAAGGAGCAAAG. The reaction conditions were: 5 minutes at 95°C, 1 minute at 95°C, 1 minute at 60°C, and 1 minute at 72°C for 40 cycles followed by 5 minutes at 72°C. The amplified product from the wild-type allele is 222 bp (mPTEN Ex/I and mPTEN R) and the product from mutant allele is 467 bp (mPTEN Ex/I and PGK6).
Analysis of Tumors
Animals were sacrificed according to guidelines and the vagina was flushed with saline, sampled with a cotton swab and the exfoliated cells were spread on glass slides, stained, and examined by light microscopy to determine the phase of the estrus cycle. The entire female genital tract was removed, representative sections of each horn were fixed in 10% buffered formalin and paraffin embedded. Routine hematoxylin and eosin (H&E)-stained sections were reviewed by the same gynecological pathologist (LHE). The remaining representative sections of uteri were frozen in liquid nitrogen and stored at −80°C.
Microdissection and DNA Extraction
Tissue sections were deparaffinized and lightly stained with hematoxylin. Distinct areas of CAH and endometrial carcinoma were microdissected using a 26-gauge needle under light microscopic guidance to at least 90% purity. Normal tissue was microdissected for each mouse in a similar manner for isolation of control DNA for both loss of heterozygosity (LOH) and MI analysis. DNA extraction was performed as previously described. 1
LOH Analysis
PCR-based LOH analysis of Pten was performed using a common 5′ primer within the intron of exon 5 and two 3′ primers. Wild-type primers: GGGATTATCTTTTTGCAACAGT and GGGCCTCTTGTGCCTTTA; mutant primers: GGGATTATCTTTTTGCAACAGT and TTCCTGACTAGGGGAGGAGT. DNA was prepared from either microdissected normal or lesional tissue or from tails of Pten+/− and wild-type mice. PCR was performed in 50-μl reactions containing 10 mmol/L Tris-HCl (pH 9.2), 1.5 mmol/L MgCl2, 75 mmol/L KCl, 0.4 μmol/L of each 3′ primer, 0.8 μmol/L 5′ primer, 160 μmol/L each dNTP, and 2.5 U of Taq polymerase (Life Technologies, Inc., Gaithersburg, MD). Forty cycles of PCR were performed, each cycle consisted of 1 minute at 95°C, 1 minute at 57°C, and 1 minute at 72°C, followed by a single 5-minute extension at 72°C. The products were separated on 2% agarose gel, stained with ethidium bromide, and the relative intensities of the bands were visually scored. A mutant to wild-type ratio greater than 2:1 was scored as a LOH of the wild-type Pten allele. Each sample was repeated and scored separately in a blinded manner by two individuals.
MI Analysis
Six microsatellite loci were amplified with incorporation of [α-32P]dCTP (10 mCi/ml; Amersham Pharmacia Biotech Inc., Piscataway, NJ) as described previously 15 using dinucleotide repeats (Research Genetics, Huntsville, AL) D1Mit36, D7Mit91, and D10Mit2; and mononucleotide repeats U12235, MBAT25, and MBAT37. 16,17 Amplified PCR products were separated on denaturing polyacrylamide gels and visualized by autoradiography.
Immunohistochemistry
Five-μm sections on glass slides were prepared from formalin-fixed, paraffin-embedded tissue. The slides were baked at 60°C for 1 hour, and the tissue was rehydrated. Antigen retrieval was performed in 1 mmol/L of ethylenediaminetetraacetic acid (pH 7.5) solution and heated in a microwave oven for 10 minutes. Tissue was incubated in 10% normal goat serum at room temperature, followed by incubation in a 1:200 dilution of Pten Ab-2 (Lab Vision Corporation, Fremont, CA) in Tris-buffered saline (TBS) for 1 hour at room temperature, or phospho-Akt (Ser473) (Cell Signaling Technology, Beverly, MA) at a 1:50 dilution overnight at room temperature. The sections were washed in TBS-T (0.01% Tween 20 in TBS) and then incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA). Endogenous peroxidase was blocked by incubation in 0.3% hydrogen peroxide for 12 minutes. Immunoenzymatic reaction for Pten staining was developed using avidin-biotinylated horseradish-peroxidase complex and 3,3′-diaminobenzidine as substrate (Vector Laboratories). The reactions for phospho-Akt staining were performed using avidin peroxidase and 3-amino-9-ethyl-carbazole (Sigma Chemical Co., St. Louis, MO). The slides were counterstained with Methyl Green or hematoxylin, respectively. Negative controls lacking primary antibody were performed on each section. Staining for Pten was scored as following: 0 for negative staining, 1+ for weak staining, 3+ for strong staining, and 2+ for those staining with an intensity between 1+ and 3+. Only membrane staining for phosphorylated Akt was scored as positive.
Results
Evaluation of Endometrial Pathology in Pten Heterozygous Mice
We first analyzed the uteri in a series of female Pten+/− mice at 16, 24, 32, and 40 weeks. Light microscopic evaluation of H&E-stained sections revealed endometrial lesions characterized by increased architectural complexity and cytological atypia of endometrial glands and surface luminal epithelium. The lesions were multifocal and closely resembled CAH in humans (Figure 1b) ▶ . The number and size of individual lesions were determined in each mouse. The incidence, number, and size of endometrial lesions increased with age in Pten+/− mice (Table 1) ▶ . All of the Pten+/− female mice that were 32 weeks of age had extensive CAH. Two of eight (25%) mice at 40 weeks of age had endometrial carcinoma with stromal invasion. These lesions, similar to well-differentiated UEC in humans, consisted of cribriform, confluent glands without intervening stroma (Figure 1c) ▶ . Notably, age-matched wild-type mice did not develop lesions in any of the age groups (Figure 1a) ▶ .
Figure 1.
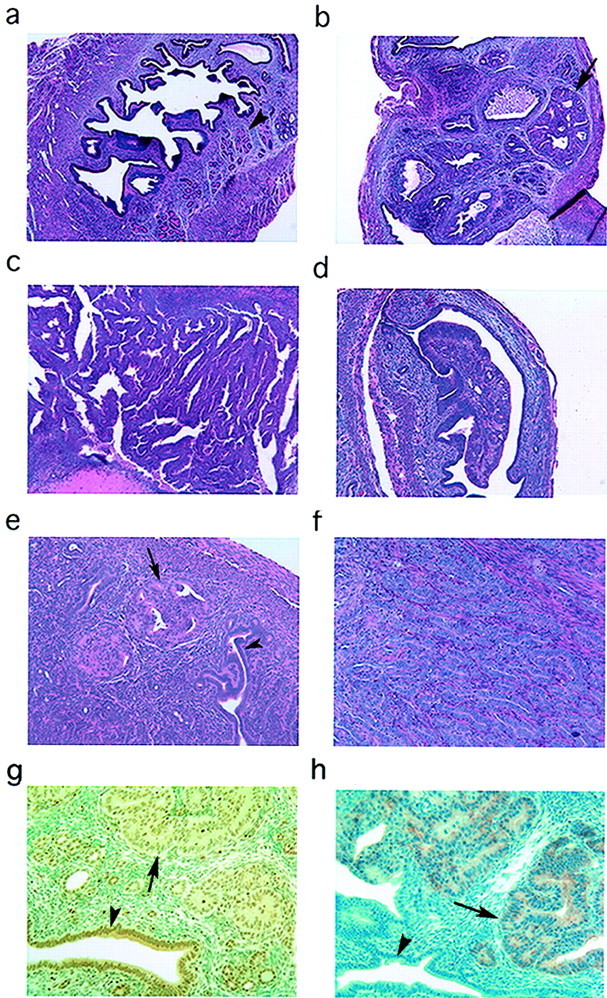
Light microscopic evaluation and immunohistochemical analysis of Pten+/− and Pten+/−/Mlh1−/− uteri. a: Pten+/+ at 32 weeks showing normal glands (arrowhead) and central lumen. b: Pten+/− at 32 weeks with multifocal CAH (arrow). c: Pten+/− at 40 weeks with invasive adenocarcinoma confined to the stroma. d:Pten+/−/Mlh1−/− at 6 weeks with polypoid lesion arising from the central luminal epithelium. e:Pten+/−/Mlh1−/− at 16 weeks showing multifocal CAH (arrow) with adjacent normal glands (arrowhead). f:Pten+/−/Mlh1−/− at 14 weeks showing well-differentiated carcinoma with myometrial invasion. g: Immunohistochemical analysis of Pten in 32-week-old Pten+/− mouse showing decreased staining in CAH (arrow). Normal gland (arrowhead) with strong cytoplasmic staining. h: Increased P-Akt expression in lesions (arrow) of an adjacent serial section from the same 32-week-old Pten+/− mouse. Normal gland (arrowhead) lacks staining for P-Akt. H&E; original magnifications: ×40 (a, b, d); ×100 (c, e, f ).
Table 1.
Incidence, Number, and Size of Neoplastic Endometrial Lesions in Pten+/− and Pten+/−/Mlh1−/− Mice
| Pten genotype | Mlh1 genotype | Age (weeks) | n | No. (%) of mice with lesions | No. (%) of mice with CA* | No. of lesions per mouse (mean ± SD) | Size of lesion (mm2) | Range of size (mm2) | LOH (%) |
|---|---|---|---|---|---|---|---|---|---|
| +/− | + /+ | 16 | 7 | 5 (71.4) | 0 | 1.14 ± 1.07 | 0.09± 0.10 | 0.04→0.32 | NA† |
| +/− | + /+ | 24 | 9 | 8 (88.9) | 0 | 9.78± 5.91 | 0.19 ± 0.16 | 0.04→0.64 | 30 |
| +/− | + /+ | 32 | 9 | 9 (100) | 0 | 18.56± 8.57 | 0.22± 0.12 | 0.04→0.75 | 30 |
| +/− | + /+ | 40 | 8 | 8 (100) | 2 (25) | 28.75± 15.34 | 0.84± 5.61 | 0.04→80.75 | 60 |
| +/− | − /− | 6–9 | 7 | 6 (85.7) | 0 | 3.43± 2.99 | NA | NA | NA |
| +/+ | − /− | 6–9 | 4 | 0 | 0 | 0 | |||
| +/− | + /+ | 6–9 | 5 | 0 | 0 | 0 | |||
| +/− | − /− | 14–18 | 5 | 5 (100) | 2 (40) | 12.20± 9.09 | 0.98± 2.39 | 0.04→12 | 60 |
| +/+ | − /− | 14–18 | 5 | 0 | 0 | 0 | 0 |
*CA, Carcinoma.
†NA, Not analyzed.
Accelerated Endometrial Tumorigenesis in Pten+/−/Mlh1−/− Mice
To assess the role of DNA mismatch repair in endometrial tumorigenesis in the setting of Pten mutations, we developed Pten+/−/Mlh1−/− double-mutant mice. MLH1 encodes a protein involved in DNA mismatch repair and the absence of its expression is associated with MI in the vast majority of MI-positive sporadic human UECs. 18 Pten+/−/Mlh1−/− mice were sacrificed at two time points, 6 to 9 weeks and 14 to 18 weeks. In the 6- to 9-week-old age group, six of seven (85.7%) mice developed polypoid lesions that protruded into the endometrial cavity and appeared to arise from the luminal surface epithelium (Table 1) ▶ . Although the lesions were architecturally distinct from the hyperplastic lesions, the cells showed an increase in size and nuclear atypia, similar to those of CAH (Figure 1d) ▶ . None of the Pten+/− or Mlh1−/− littermates developed any lesions at this early age. By 14 to 18 weeks all of the double-mutant mice had lesions histologically similar to the CAH seen in Pten+/− mice (Figure 1e) ▶ , and two of them exhibited invasive disease (Table 1) ▶ . One of the invasive tumors maintained its glandular differentiation, mimicking infiltrating well-differentiated carcinomas in humans, and extensively invaded the myometrium with extension onto the serosal surface of the uterus (Figure 1f) ▶ . The other carcinoma exhibited only stromal invasion, as seen in the Pten+/− mice at 40 weeks old. Compared to the Pten+/− mice of the same age, the number and the size of lesions in Pten+/−/Mlh1−/− mice were increased ∼10-fold. None of the age-matched Mlh1−/− littermates developed any endometrial lesions (Table 1) ▶ . Thus, Pten+/−/Mlh1−/− mice showed a decrease in the age of onset of disease and an increase in the severity of the disease.
The MI phenotype of the Pten+/−/Mlh1−/− mice was confirmed in both tail DNA (diluted to 1 to 3 genomes) and DNA prepared from microdissected endometrial lesions (invasive and noninvasive) (Figure 2, a and b) ▶ . MI was detected in 4 of 10 (40%) microdissected lesions from Pten+/−/Mlh1−/− mice, whereas it was detected in 1 of 7 (14.3%) lesions in Pten+/− mice. In Pten+/−/Mlh1−/− mice each MI-positive endometrial lesion showed instability at one to two mononucleotide repeat tracts (U12235 and MBAT37) yet instability was not detected in any of the dinucleotide repeats. Only one lesion from a Pten+/− mice displayed instability where it was seen in two dinucleotide repeats (D7Mit91 and D10Mit2). These results indicate that mismatch repair deficiency, because of lack of Mlh1, accelerates endometrial tumorigenesis in Pten heterozygous mice.
Figure 2.

MI and LOH analysis in Pten+/−/Mlh1−/− mouse. a: MI analysis on genomic DNA with U12235 locus. Lane 1: Undiluted tail DNA. Lanes 2 and 3: Two diluted DNA samples from the same mouse showing instability. b: MI analysis on uterine lesions with U12235. Lane 1: Microdissected DNA from normal myometrium. Lanes 2 and 3: DNA from two microdissected CAH lesions from the same mouse showing a shift in lane 3 but not definitively in lane 2. c: PCR-based analysis of LOH of Pten in endometrial lesions from Pten+/−/Mlh1−/− mice. Lane M: One-kb ladder. Lane 1: Amplification of tail DNA from Pten+/− mouse showing both mutant and wild-type alleles. Lane 2: Amplification of tail DNA from Pten+/+ mouse. Lane 3: Amplification of DNA from a microdissected lesion without LOH. Lanes 4 and 5: Amplification of DNA from two microdissected lesions showing LOH of the wild-type allele.
Immunohistochemical Analysis
Immunohistochemical analysis of uterine sections demonstrated reduced Pten expression in most of the endometrial lesions. Overall, we found that histologically normal epithelium in uteri from Pten heterozygous mice stained less strongly (2+) than age-matched wild-type controls (3+). Histologically normal endometrial glands never demonstrated a complete lack of Pten expression. In both Pten+/− mice and Pten+/−/Mlh1−/− mice at all age groups, the lesions showed a spectrum of staining patterns with the majority of the lesions showing reduced (1+) or negative (0) staining (Figure 1g) ▶ . However, occasional endometrial lesions showed 2+ to 3+ staining that may be secondary to alterations in the remaining allele of Pten that result in elevated levels of abnormal Pten protein. On the other hand, phosphorylated Akt (P-Akt) was expressed in every lesion that was detected on H&E-stained sections (Figure 1h) ▶ and was absent in intervening normal endometrium of Pten+/−, Pten+/−/Mlh1−/− mice and in normal endometrium of wild-type mice. The immunohistochemical studies support an inverse relationship between Pten expression and phosphorylated Akt levels in endometrial tumorigenesis. In addition, we have analyzed expression of p27, cyclin D1, and cyclin D3 by immunohistochemistry in Pten+/− mice and have failed to detect any correlation with morphological changes and Pten or P-Akt expression (data not shown). Clearly, additional studies are required to determine in vivo, at the molecular level, the consequence of aberrations in Pten alone or in conjunction with MI that result in endometrial lesions.
LOH Analysis
To determine the status of the wild-type Pten allele we performed LOH analysis at the Pten locus on DNA from microdissected endometrial lesions. In Pten+/−/Mlh1−/− double mutants at 14 to 18 weeks of age, 60% of microdissected CAH and endometrial carcinoma showed LOH of the wild-type Pten allele (Figure 2c) ▶ , whereas 30%, 30%, and 60% of lesions in the Pten+/− mice at 24, 32, and 40 weeks showed LOH, respectively (Table 1) ▶ . Thus, the frequency of loss of the wild-type allele increased with age in Pten+/− mice and was increased at 14 to 18 weeks in the double-mutant mice. This finding suggests that tumor development/progression in both backgrounds involves loss of the wild-type Pten allele and that this is accelerated in the Pten+/−/Mlh1−/− mice. All of the lesions with LOH had either markedly decreased or absent staining for Pten. In addition, the majority of lesions without detectable LOH also showed decreased or absent staining, however some lesions without LOH maintained normal or even elevated levels of expression. The lesions with decreased Pten expression but without LOH suggest that Pten may be inactivated in these lesions by other mechanisms, such as intragenic mutations, epigenetic alterations (eg, by methylation), and posttranscriptional or posttranslational regulation. We are in the process of sequencing Pten in those lesions that lack LOH.
Discussion
Mutations in the PTEN tumor suppressor gene have been detected in ∼35 to 50% of sporadic human UECs 1-4 in several independent studies and in 83% in one study. 5 Moreover, mutations have been identified in 20% of noninvasive precursor lesions called hyperplasias 8,9 and in 55% in one study. 5 Here, and in previous studies, it has been shown that mice carrying a germline mutation in one allele of Pten spontaneously develop preinvasive and invasive endometrial neoplastic lesions that are morphologically similar to the human disease. These findings support a fundamental role for PTEN in the control of endometrial epithelial growth/proliferation.
An additional observation from studies on primary human tumors is the significant association of PTEN mutations and MI. 1-3,10,11 In studies from our laboratory and others, PTEN mutations were detected in 75 to 85% of MI-positive UECs, compared to only 30 to 35% in MI-negative UECs. 1,2 Furthermore, the majority of MI-positive sporadic UECs demonstrates hypermethylation of the hMLH1 promoter and lack expression of the protein product. 18-22 In this study we have shown that Pten+/−/Mlh1−/− mice demonstrate an accelerated onset and increased severity of endometrial tumorigenesis compared to Pten+/− mice. Importantly, aged matched Mlh1−/− littermates did not develop any endometrial lesions. 14,23-26 Our results, in conjunction with the association of these genetic alterations in human UECs, indicate an important relationship between aberrations in the PTEN pathway and mismatch repair deficiency in the pathogenesis of endometrial tumorigenesis. Although the causal mechanism of this relationship and its contribution to the development and/or progression of endometrial cancer are not completely elucidated by the present studies they do provide some preliminary insights. In mice with both mutations, LOH of the wild-type Pten allele was seen in 60% of the hyperplastic lesions at 14 to 18 weeks of age. In Pten+/− mice this frequency of LOH was not seen until 40 weeks of age. These findings suggest that inactivation of the remaining Pten allele promotes the progression of hyperplastic lesions and that mismatch repair deficiency may hasten this event.
Previous studies showed that the spectrum of mutations in PTEN was similar in both MI+ and MI− human UECs. 2,7 Moreover, 100% of Pten mutant mice develop CAH, but MMR-deficient mice lack any endometrial lesions. These observations suggest that PTEN mutations are not directly attributable to MI in endometrial carcinoma. In contrast, in human colorectal tumors the mutational spectrum of APC, a gene thought to initiate colorectal tumorigenesis, is substantially different in MI+ tumors when compared to MI− tumors. 27 Furthermore, in Apc mutant mice loss of Apc gene function is mediated through loss of wild-type Apc, whereas tumors in Apc+/−/Mlh1−/− mice reveal an increase in intragenic mutations in the wild-type copy of Apc. 26 These findings suggest that Apc is a target of MMR deficiency in colorectal tumorigenesis in both humans and mice. Our data show that the majority of endometrial lesions from Pten+/−/Mlh1−/− mice at 14 to 18 weeks of age display LOH of wild-type Pten allele at a frequency similar to that in Pten+/− mice at 40 weeks. This result, along with the studies on the human tumors, implies that the mechanism involved in acceleration of endometrial tumorigenesis in Pten+/−/Mlh1−/− mice may be distinct from that described for colorectal carcinoma. Although the coding region of Pten contains two [A]6 tracts that represent potential mutational targets in tumors with defective mismatch repair, repeat tract mutation in PTEN is an uncommon event in MI+ endometrial cancers. 7,28 Based on the above observations, PTEN may not be a direct target of MI and mutations may not be attributable to MI in endometrial carcinoma.
In addition, numerous studies have demonstrated the ability of Pten to negatively influence cellular-survival signaling via regulation of PKB/Akt and implicated PKB/Akt activation by phosphorylation in the development of Pten-related tumors. 13,29-31 Our studies also support a critical role for elevated levels of P-Akt in the development of endometrial lesions. In every lesion detectable by light microscopic evaluation, irrespective of size or architecture, P-Akt was detected in both Pten+/− and Pten+/−/Mlh1−/− mice by immunohistochemical analysis. Although other downstream targets in the Pten pathway may play a role in the development/progression of endometrial tumorigenesis these studies suggest a central role for P-Akt.
Genetic mouse models of human disease are often limited by the fact that the phenotype produced in the mouse does not recapitulate the human disease. In the studies presented here we have shown that mice with germline mutations in two of the most commonly altered genes in human UEC spontaneously develop neoplastic endometrial lesions that morphological resemble the human disease. Moreover, the mice reveal a spectrum of changes from preinvasive to invasive disease mimicking the progression model that has been developed through studies on the human disease. Future studies of this mouse model should lead to an understanding of this disease process at the molecular level. In addition, it offers a potential experimental system for the development of clinically relevant diagnostic and treatment modalities for this common disease of women.
Footnotes
Address reprint requests to Dr. Lora Hedrick Ellenson, Department of Pathology, Weill Medical College of Cornell University, 1300 York Ave., New York, NY 10021. E-mail: lhellens@med.cornell.edu.
Supported by grants from The Mary Kay Ash Charitable Foundation, the Dorothy Rodbell Cohen Foundation for Sarcoma Research (to L. H. E.), and the National Cancer Institute (CA84301 to R. K.).
References
- 1.Tashiro H, Blazes MS, Wu R, Cho KR, Bose S, Wang SI, Li J, Parsons R, Ellenson LH: Mutations in PTEN are frequent in endometrial carcinoma but rare in other common gynecological malignancies. Cancer Res 1997, 57:3935-3939 [PubMed] [Google Scholar]
- 2.Kong D, Suzuki A, Zou TT, Sakurada A, Kemp LW, Wakatsuki S, Yokoyama T, Yamakawa T, Sato M, Ohuchi N, Sato S, Yin J, Wang S, Abraham JM, Souza RF, Smolinski KN, Meltzer SJ, Horii A: PTEN is frequently mutated in primary endometrial carcinomas. Nat Genet 1997, 17:143-144 [DOI] [PubMed] [Google Scholar]
- 3.Risinger JI, Hayes K, Maxwell GL, Carney ME, Dodge RK, Barrett JC, Berchuck A: PTEN mutation in endometrial cancers is associated with favorable clinical and pathologic characteristics. Clin Cancer Res 1998, 4:3005-3010 [PubMed] [Google Scholar]
- 4.Simpkins SB, Schneider SP, Mutch DG, Gersell D, Goodfellow PJ: PTEN mutations in endometrial cancers with 10qLOH: additional evidence for the involvement of multiple tumor suppressors. Gynecol Oncol 1998, 71:391-395 [DOI] [PubMed] [Google Scholar]
- 5.Mutter GL, Lin MC, Fitzgerald JT, Kum JB, Baak JP, Lees JA, Weng LP, Eng C: Altered PTEN expression as a diagnostic marker for the earliest endometrial. J Natl Cancer Inst 2000, 92:924-930 [DOI] [PubMed] [Google Scholar]
- 6.MacDonald ND, Salvesen HB, Ryan A, Iversen OE, Akslen LA, Jacobs IJ: Frequency and prognostic impact of microsatellite instability in a larger population-based study of endometrial carcinomas. Cancer Res 2000, 60:1750-1752 [PubMed] [Google Scholar]
- 7.Gurin CC, Federici MG, Kang L, Boyd J: Cause and consequences of microsatellite instability in endometrial carcinoma. Cancer Res 1999, 59:462-466 [PubMed] [Google Scholar]
- 8.Levine RL, Cargile CB, Blazes MS, van Rees B, Kurman RJ, Ellenson LH: PTEN mutations and microsatellite instability in complex atypical hyperplasia, a precursor lesion to uterine endometrioid carcinoma. Cancer Res 1998, 58:3254-3258 [PubMed] [Google Scholar]
- 9.Maxwell GL, Risinger JI, Gumbs C, Shaw H, Bentley RC, Barrett JC, Berchuck A, Futreal PA: Mutation of the PTEN tumor suppressor gene in endometrial hyperplasias. Cancer Res 1998, 58:2500-2503 [PubMed] [Google Scholar]
- 10.Maxwell GL, Risinger JI, Alvarez AA, Barrett JC, Berchuck A: Favorable survival associated with microsatellite instability in endometrioid endometrial cancers. Obstet Gynecol 2001, 97:417-422 [DOI] [PubMed] [Google Scholar]
- 11.Bussaglia E, del Rio E, Matias-Guiu X, Prat J: PTEN mutations in endometrial carcinomas: a molecular and clinicopathologic analysis of 38 cases. Hum Pathol 2000, 31:312-317 [DOI] [PubMed] [Google Scholar]
- 12.Podsypanina K, Ellenson LH, Nemes A, Gu J, Tamura M, Yamada KM, Cordon-Cardo C, Catoretti G, Fisher PE, Parsons R: Mutation of Pten/Mmac1 in mice causes neoplasia in multiple organ systems. Proc Natl Acad Sci USA 1999, 96:1563-1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stambolic V, Tsao MS, Macpherson D, Suzuki A, Chapman WB, Mak TW: High incidence of breast and endometrial neoplasia resembling human Cowden syndrome in Pten+/− mice. Cancer Res 2000, 60:3605-3611 [PubMed] [Google Scholar]
- 14.Edelmann W, Cohen PE, Kane M, Lau K, Morrow B, Bennett S, Umar A, Kunkel T, Cattoretti G, Chaganti R, Pollard JW, Kolodner RD, Kucherlapati R: Meiotic pachytene arrest in MLH1-deficient mice. Cell 1996, 85:1125-1134 [DOI] [PubMed] [Google Scholar]
- 15.Tashiro H, Lax SF, Gaudin PB, Isacson C, Cho KR, Hedrick L: Microsatellite instability is uncommon in uterine serous carcinoma. Am J Pathol 1997, 150:75-79 [PMC free article] [PubMed] [Google Scholar]
- 16.Yao X, Buermeyer AB, Narayanan L, Tran D, Baker SM, Prolla TA, Glazer PM, Liskay RM, Arnheim N: Different mutator phenotypes in Mlh1-versus Pms2-deficient mice. Proc Natl Acad Sci USA 1999, 96:6850-6855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blake C, Tsao JL, Wu A, Shibata D: Stepwise deletions of polyA sequences in mismatch repair-deficient colorectal cancers. Am J Pathol 2001, 158:1867-1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Staebler A, Lax SF, Ellenson LH: Altered expression of hMLH1 and hMSH2 protein in endometrial carcinomas with microsatellite instability. Hum Pathol 2000, 31:354-358 [DOI] [PubMed] [Google Scholar]
- 19.Esteller M, Levine R, Baylin SB, Ellenson LH, Herman JG: MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 1998, 17:2413-2417 [DOI] [PubMed] [Google Scholar]
- 20.Esteller M, Catasus L, Matias-Guiu X, Mutter GL, Prat J, Baylin SB, Herman JG: hMLH1 promoter hypermethylation is an early event in human endometrial tumorigenesis. Am J Pathol 1999, 155:1767-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salvesen HB, MacDonald N, Ryan A, Iversen OE, Jacobs IJ, Akslen LA, Das S: Methylation of hMLH1 in a population-based series of endometrial carcinomas. Clin Cancer Res 2000, 6:3607-3613 [PubMed] [Google Scholar]
- 22.Parc YR, Halling KC, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN, Halling AC: Microsatellite instability and hMLH1/hMSH2 expression in young endometrial carcinoma patients: associations with family history and histopathology. Int J Cancer 2000, 86:60-66 [DOI] [PubMed] [Google Scholar]
- 23.Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM: Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms2 and Pms2 DNA mismatch repair. Nat Genet 1998, 18:276-279 [DOI] [PubMed] [Google Scholar]
- 24.Heyer J, Yang K, Lipkin M, Edelmann W, Kucherlapati R: Mouse models for colorectal cancer. Oncogene 1999, 18:5325-5333 [DOI] [PubMed] [Google Scholar]
- 25.Prolla TA, Abuin A, Bradley A: DNA mismatch repair deficient mice in cancer research. Semin Cancer Biol 1996, 7:241-247 [DOI] [PubMed] [Google Scholar]
- 26.Edelmann W, Yang K, Kuraguchi M, Heyer J, Lia M, Kneitz B, Fan K, Brown AM, Lipkin M, Kucherlapati R: Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res 1999, 59:1301-1307 [PubMed] [Google Scholar]
- 27.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, Kinzler KW, Vogelstein B, Dunlop MG: APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA 1996, 93:9049-9054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cohn DE, Basil JB, Venegoni AR, Mutch DG, Rader JS, Herzog TJ, Gersell DJ, Goodfellow PJ: Absence of PTEN repeat tract mutation in endometrial cancers with microsatellite instability. Gynecol Oncol 2000, 79:101-106 [DOI] [PubMed] [Google Scholar]
- 29.Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW: Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95:29-39 [DOI] [PubMed] [Google Scholar]
- 30.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H: PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci USA 1999, 96:6199-6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahia PL, Aguiar RC, Alberta J, Kum JB, Caron S, Sill H, Marsh DJ, Ritz J, Freedman A, Stiles C, Eng C: PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanisms in hematological malignancies. Hum Mol Genet 1999, 8:185-193 [DOI] [PubMed] [Google Scholar]