

Abstract
This is the first comprehensive profile of cystic fibrosis transmembrane conductance regulator (CFTR) mutations and their corresponding haplotypes in the Iranian population. All of the 27 CFTR exons of 60 unrelated Iranian CF patients were sequenced to identify disease-causing mutations. Eleven core haplotypes of CFTR were identified by genotyping six high-frequency simple nucleotide polymorphisms. The carrier frequency of 2.5 in 100 (1 in 40) was estimated from the frequency of heterozygous patients and suggests that contrary to popular belief, cystic fibrosis may be a common, under-diagnosed disease in Iran. A heterogeneous mutation spectrum was observed at the CFTR locus in 60 cystic fibrosis (CF) patients from Iran. Twenty putative disease-causing mutations were identified on 64 (53%) of the 120 chromosomes. The five most common Iranian mutations together represented 37% of the expected mutated alleles. The most frequent mutation, ΔF508 (p.F508del), represented only 16% of the expected mutated alleles. The next most frequent mutations were c.1677del2 (p.515fs) at 7.5%, c.4041C>G (p.N1303K) at 5.6%, c.2183AA>G (p.684fs) at 5%, and c.3661A>T (p.K1177X) at 2.5%. Three of the five most frequent Iranian mutations are not included in a commonly used panel of CF mutations, underscoring the importance of identifying geographic-specific mutations in this population.
Cystic fibrosis (CF; MIM no. 219700) is caused by the inheritance of two nonfunctional copies of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. The protein product of the gene is a chloride channel and a member of the ATP-binding cassette membrane transporter superfamily. The CFTR channel is critical for the normal function of epithelial cells in the lungs, pancreas, intestine, gall bladder, and sweat glands. Excess mucus in the respiratory system of CF patients allows for chronic bacterial infections, leading to respiratory failure, which is the major cause of mortality. Most of the patients also fail to produce digestive enzymes in the pancreas, resulting in pancreatic insufficiency. Other clinical features are variably associated with the disease.1
CF is the most common life-threatening autosomal recessive disease in many Caucasian populations, including those of Europe and the United States. Approximately one in 2000 to 3000 newborns in populations of European ancestry are affected, and the average carrier frequency is about 1:25.2,3 The median survival age in economically developed countries is about 40 but is significantly less in other countries, such as Iran, where the incidence of CF has not been critically assessed.
Publication of the CFTR gene sequence in 1989 gave scientists the first opportunity to understand the molecular basis of cystic fibrosis (CFTR; MIM no. 60241). The p.F508del (ΔF508) allele of the gene is the most common mutation observed worldwide and is probably very old, dating to pre-Neolithic times.2,4 In Europe, its frequency exhibits a distinct northwest to southeast gradient, ranging from a high of 88% in Denmark to a low of 24% in Turkey.5,6,7,8,9 There are two competing theories about the origin of this gradient. The first theory suggests the location with highest frequency of ΔF508 is where it was first introduced into Europe. The second theory suggests the mutation may confer an advantage to heterozygous carriers who are less vulnerable to bacterial infections because the CFTR gene product is used as a receptor.10,11 In this scenario, the mutation would be propagated to different degrees depending on the amount of selective pressure. By analyzing the core haplotype background of the mutation in different geographic regions, it will be possible to distinguish between these two competing theories and dissect the origin and evolutionary history of a significant disease-causing mutation.
In addition to the ΔF508 mutation, more than 1000 other mutations in the CFTR gene have been identified (CF Genetic Analysis Consortium http://www.genet.sickkids.on.ca/cftr/). These mutations vary greatly in their frequency and distribution, but most are very rare. Only four (p.G542X, p.N1303K, p.G551D, and p.W1282X) have overall frequencies greater than 1%.12 Intriguingly, p.G542X and p.N1303K are found on the same haplotype background as ΔF508, suggesting that they arose in the same population.13
Two previous reports of CFTR mutations in Iran have been published. One reported the allele frequency of seven common mutations in exons 4 and 7 in 37 CF patients.14 The second one reported the allele frequency of the ΔF508 mutation in 24 CF patients.15 A relatively low frequency of 25% was reported, consistent with the frequencies of the mutation in neighboring countries. In the present study, all 27 of the CFTR exons of 60 unrelated Iranian CF patients (including the 24 previously studied) were sequenced with the aim of obtaining a profile of CFTR mutations in the Iranian population. In addition, the haplotypes associated with the mutations were assessed, and the carrier frequency was calculated based on the frequency of heterozygous patients.
Here, we describe 11 core haplotypes at the CFTR locus that are defined by 6 ancient bi-allelic polymorphisms. We believe these haplotypes will be useful for cross-comparison with other populations. We also describe the CFTR mutations found in the unique Iranian population. In ancient human history, Iran’s auspicious location made it a crossroads for travelers moving between Africa, Europe, India, and beyond. As such, it has a rich and valuable genetic legacy. By following disease-causing mutations in this population, we may come closer to tracking the origin of the most common disease-causing mutations outside of Africa. Finally, we hope that the knowledge of the unique mutation spectrum and the mutation frequency in this population will improve medical care CF patients in Iran.
Materials and Methods
The families of patients all consented to participate after being informed of the nature of the research. The 60 patients were diagnosed with cystic fibrosis based on elevated sweat chloride values (>60 mEq/L). Chloride ion concentrations are measured either by mercurimetric titration or a mercurimetric spectrophotometric method.16 Many patients had pulmonary complications and pancreatic insufficiency. The patients were recruited from four hospitals in Iran, but one-half were diagnosed at the Children’s Hospital Medical Center of Tehran University, which is the CF reference center in Iran, to which patients from all provinces are referred. The patients were from many different regions of Iran. DNA from the leukocytes of peripheral blood was prepared by the salting out protocol.17
Mutation Analysis
Each of the 27 exons of the CFTR gene, except exon 13 and its flanking intronic sequences, was amplified by the polymerase chain reaction (PCR). Multiplexing was not performed. Exon 13, because of its large size, was amplified in two overlapping segments. Sequences of most of the primers used have been reported.18,19 Newly designed primers were used for amplification of exons 1, 2, 23, and 24 (1F, 5′-AAGGAGGAGAGGAGGAAGGA-3′; 1R, 5′-ACCCACATTTTCTTTCAAAACA-3′; 2F, 5′-GCCTGTAAGAGATGAAGCCTG-3′; 2R, 5′-TCAAACTCCTGGTCTCAAGCA-3′; 23F, 5′-TTAGAGTCTACCCCATGGTTGA-3′; 23R, 5′-AAAGCTGGATGGCTGTATGA-3′; 24F, 5′-ATTTTCCTTTGAGCCTGTGC-3′; and 24R, 5′-CATCCTTGTTTTCTGAGGCA-3′). The primers were synthesized by Operon (Qiagen Co., Alameda, CA). All PCR products were sequenced in both the forward and reverse direction using the same primers used in the PCR reactions. Sequencing was done using the ABI Big Dye Terminator system and an ABI Prism 3700 and 3730 DNA sequencer (Applied Biosystems, Foster City, CA). Sequences were analyzed using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI).
Mutations and numbering were determined by comparison with the cDNA reference sequence for the CFTR gene (GenBank accession no. NM_000492), the protein sequence (NP_000483), and the genomic sequences AC000111 (first 18 exons) and AC000061 (last 9 exons). Every mutation was confirmed by sequencing at least two independent PCR amplification products. Predicted effects on splicing of mutations located in exons were determined by comparison with known exon splicing enhancer motifs (http://rulai.cshl.edu/tools/ESE/). Effects of intron mutations were determined by comparison with known canonical splice site motifs (http://www.fruitfly.org/seq_tools/splice.html).
Haplotype Analysis
Haplotypes were first defined by the eight most common intragenic polymorphisms noted among the 60 patients during the sequence analysis: c.867−33del4 (six or seven repeats of TTGA; rs4148700), c.1001+11 (C/T; rs1800503), c.1525−61 (A/G; rs number not designated), c.1540 (A/G; Met/Val; rs213950), c.3041−92 (G/A; rs number not designated), c.3601−65 (C/A; rs213989), c.4006−200 (G/A; rs214164), and c.4521 (G/A; Q1463Q; rs1800136). The frequencies of the minor alleles ranged from 14 to 45% of the CF chromosomes analyzed. Subsequently, six polymorphisms (all of the above except c.1525−61A>G and c.3041−92G>A) were shown to be sufficient to describe the haplotype diversity in this population and to compare it with a previously analyzed European cohort.
Four of the polymorphisms (c.1525−61A>G, c.1540A>G, 3601−65C>A, and c.4006−200A>G) were previously reported in the German population.20 Haplotype analysis did not include some common markers found in other populations, such as c.2694T>G found in Korea.21 The ancestral alleles of all markers were determined based on which allele was present in both the mouse and chimpanzee reference sequences (http://genome.ucsc.edu).
The phase of haplotypes in 85% of individuals was unambiguous because they were either homozygous or polymorphic at only one of the marker sites. The remaining haplotypes were deduced by maximum parsimony and the computer program Varia (Silicon Genetics, South San Francisco, CA). The haplotypes predicted by both methods were consistent with one another. Wherever possible, the phase of an ambiguous haplotype was assumed to be that remaining after subtracting one of the two most common haplotypes. In addition, the most likely haplotypes were assumed to be those with a minimum number of mutations or recombination events from a known unambiguous haplotype.
For analysis with the computer program Varia, text files containing unphased genotypes for eight markers were imported after conversion into internal Silicon Genetics format. All variations found in build 119 of the National Institutes of Health single nucleotide polymorphism (SNP) database (http://www.ncbi.nlm.nih.gov) were given their corresponding rs numbers; all newly discovered ones were assigned internal identifiers. After calculation of unambiguous phases, an EM-type algorithm was applied to estimate a unique haplotype for the ambiguous phases.22 Next, the frequencies of the deduced haplotypes were used to construct the haplotype map.23,24 Finally, a graphical representation of blocks along the chromosome was generated.
The haplotype blocks are the regions where the observed heterozygosity computed from haplotypes (HETobs) is less than the estimated heterozygosity computed from individual variations (HETest).25 The calculation of each block starts with a window (block) consisting of several consecutive variations. The score of the block is calculated with the equation S = HETobs/HETest. Next, the window slides along the chromosome and the score (S) is calculated for a new block, until the block with the best (the smallest) score is found. At last, to identify the final block, variations are added and removed at each end of the window—stopping when the best score is achieved. After the best block is found, the remaining variations are processed in the similar fashion, and more blocks are found.
The haplotype structure of the Iranian CF population was compared with a cohort of 88 unaffected individuals of European descent. The genotypes for the control group were downloaded from the National Center for Biotechnology Information Single Nucleotide Polymorphism database (http://www.ncbi.nlm.nih.gov/SNP) from the data sub-mitted by the International HapMap Project Consortium (http://www.hapmap.org) with the data ID designation “HapMap_chr7_CEU_BROAD_BEADARRAY”. Only four of the six polymorphisms used to define our six-marker haplotypes were represented in the dataset. These included c.1540 (A/G; Met/Val; rs213950), c.3601−65 (C/A; rs213989), c.4006−200 (G/A; rs214164), and c.4521 (G/A; Q1463Q; rs1800136).
Calculation of CF Carrier Frequency
The proportion of the patient population whose disease is not due to consanguinity can be used to calculate the frequency of CF-causing alleles in the population. The increase in incidence of disease due to consanguinity is expressed as X = (q + fp)/q, where q is the frequency of all mutated alleles, p is the frequency of normal alleles in the gene pool, and f is the average inbreeding coefficient.9 The maximum possible value for X is M/(h + H), where M is the number of patients in whom at least one mutation was found, h is the number of patients heterozygous for their mutation, and H is the number of patients homozygous for their mutation whose homozygosity is not due to consanguinity. Consanguinity in Iran is estimated at 25%.26 Assuming that one-half of consanguineous marriages are between first cousins and one-half are between second cousins, the average inbreeding coefficient was calculated to be
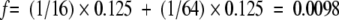 |
Hence, the CF allele frequency was determined by solving for the value of q, where p = 1− q, p is the CF normal allele frequency, and 2 pq is the carrier frequency.
Results
Putative disease-causing mutations were identified in 64 of the 120 CF chromosomes investigated, for a detection frequency of 53% (Table 1). Two mutations were detected in 27 patients (20 of which were homozygous), and only 1 was detected in 12. No mutation was found in the remaining 21 patients. Twenty putative disease-causing mutations and 21 putative polymorphisms were identified and categorized as such based on American College of Medical Genetics recommendations.27 One mutation altered the initiating methionine residue, eight mutations altered a single amino acid, two altered a canonical splice site, six were nonsense, and three were deletions. One of the deletions (ΔF508) was in frame and the other two caused a frameshift. One of the frameshift mutations (c.2183AA>G) also created an AG dinucleotide that could potentially act as a splice site. Two of the mutations were only recently reported, one affecting an amino acid alteration, c.3170C>T (p.P1013L), and another creating a stop codon, c.3661A>T (p.K1177X). They were reported in a Turkish and a Saudi Arabian population, respectively.28,29,30
Table 1.
CFTR Mutations Observed in Iranian CF Patients
| Gene location | Nucleotide mutation | Effect on protein | No. of Patients
|
Total alleles* | Associated haplotype | Global distribution | |
|---|---|---|---|---|---|---|---|
| Hom | Het | ||||||
| Exon 1 | c.134T>C | M1T | 1 | 1 | Rare | ||
| Exon 3 | c.386G>A | G85E | 1 | 1 | Global | ||
| Exon 4 | c.460G>C | D110H | 1 | 1 | H2 | Europe | |
| Exon 7 | c.1132C>T | R334W | 1 | 1 | H2 | Global | |
| Exon 7 | c.1145C>T | T338I | 1 | 1 | Europe | ||
| Intron 9 | c.1525−1G>A | Mis-splicing | 1 | 1 | H8 | Pakistan | |
| Exon 10 | c.1529C>G | S466X | 1 | 2 | H4 | Germany | |
| Exon 10 | c.1531C>T | L467F | 1 | 1 | Rare | ||
| Exon 10 | c.1649T>C | I506T | 1 | 2 | H8 | Lebanon | |
| Exon 10 | c.1652del3† | ΔF508 | 6 | 7 | 19 | H5 | Global |
| Exon 10 | c.1677delTA | 515fs | 4 | 1 | 9 | H1 | Europe |
| Exon 11 | c.1756G>T | G542X | 1 | 1 | H5 | Global | |
| Exon 12 | c.1821C>A | Y563X | 2 | 2 | Europe | ||
| Exon 13 | c.2183AA>G | 684fs | 3 | 6 | H3 | Europe | |
| Exon 17a | c.3170C>T | P1013L | 1 | 1 | Turkey | ||
| Exon 19 | c.3616C>T | R1162X | 2 | 2 | H2 | Germany | |
| Exon 19 | c.3661A>T | K1177X | 1 | 1 | 3 | H2 | Bahrain |
| Intron 20 | c.4005+1G>A | Mis-splicing | 1 | 2 | H2 | Europe | |
| Exon 21 | c.4041C>G | N1303K | 3 | 1 | 7 | H5 | Global |
| Exon 23 | c.4363C>T | Q1412X | 1 | 1 | Rare | ||
A total of 64 (53%) of the 120 expected alleles were observed.
The five most common mutations, shown in bold, accounted for 37% of the expected alleles.
Hom, homozygous; Het, heterozygous.
All of the eight missense mutations occurred at amino acids that were conserved in the CFTR orthologs of chimpanzee, mouse, rat, and chicken (http://genome.ucsc.edu). The mutation p.G85E has been shown to reduce gene activity by altering the tertiary structure of the CFTR channel;31 p.R334W leads to loss of conductivity of the large conductance channel of CFTR;32 p.T338I is a mutation in the sixth transmembrane segment of the MSD1 domain; p.L467F and p.I506T are mutations in the first nucleotide binding domain (NBD1); and p.N1303K is a mutation in the second nucleotide binding domain (NBD2) of the CFTR protein. Both domains are conserved in the ATP-binding cassette transporters.33 In addition, mutations p.D110H, p.R334W, p.T338I, p.L467F, and p.P1013L altered predicted exon splicing enhancer motifs in the CFTR gene, suggesting that they may have a deleterious effect on splicing.
ΔF508 was the most common mutation, representing 16% of the 120 CF alleles that were completely analyzed (19 of 120), but only 14% of the 140 CF alleles that were at least sequenced for exon 10 (19 of 140). This frequency is far lower than in populations of European descent (approximately 60%) but comparable with several countries of West Asia and North Africa (Algeria, 20%; Lebanon, 35%; Tunisia, 18%; Pakistanis in the United Kingdom, 19%; Turkey, 24%; and Saudi Arabia, 15%).6,9,28,30 The frequencies of the ΔF508 mutation in Iran and Saudi Arabia were the lowest yet reported for any country.
The next most frequent mutations were c.1677del2 (p.515fs) at 7.5%, c.4041C>G (p.N1303K) at 5.6%, c.2183AA>G (p.684fs) at 5%, and c.3661A>T (p.K1177X) at 2.5%. The two most frequent mutations were both in exon 10. These five most frequent mutations accounted for 37% of the mutated alleles in our patients. Five of the remaining detected mutant alleles were each found in two chromosomes and 10 were observed in only one chromosome.
Novel Polymorphisms
Twenty-one putative polymorphisms were detected in the CFTR gene of the Iranian patients (Table 2). The minor allele frequencies of eight polymorphisms were 14% or greater, and five were observed in only one chromosome. Eighteen were bi-allelic single-nucleotide polymorphisms, one (c.4006−90delC) was a single-basepair deletion, another (c.4005+121delTT) was a variation in the number of a single-nucleotide repeat (8T/6T), and a third (c.876−31delTTGA) was a variation in the number of a four-nucleotide repeat (7TTGA/6TTGA). Six were exonic polymorphisms that were either silent for an amino acid change or demonstrated to have little or no effect on protein function (p.E92E, p.I148T, p.M470V, p.E1194A, p.P1290P, and p.Q1463Q). Two of the variants that altered an amino acid (p.M470V and p.E1194A) were observed in at least one patient with two other mutations, adding credence to their assignment as neutral polymorphisms (however, see below). The CF chromosome carrying p.E1194A also carried the common p.515fs mutation, because the patient was homozygous for the latter. Furthermore, the amino acid at 1194 is not conserved in chimpanzee or macaque. A third variant (p.I148T) was confirmed as a neutral polymorphism by a recent report.34
Table 2.
Putative Polymorphism in the CFTR Gene of Iranian CF Patients
| Genomic position* | Polymorphism designation | Location in gene | Effect on protein | Minor allele frequency | DB-SNP number |
|---|---|---|---|---|---|
| 42296936 | c.185+45A>G | Intron 1 | 1% | ||
| 42325703 | c.297−75C>A | Intron 2 | 6% | ||
| 42347645 | c.408A>G | Exon 4 | E92E | 1% | |
| 42347812 | c.575T>C | Exon 4 | I148T | 2% | |
| 42352195 | c.875+40A>G | Intron 6a | 2% | ||
| 42353259 | c.876−33delTTGA | Intron 6a | 45% | rs4148700 | |
| 42353428 | c.1001+11C>T | Intron 6b | 30% | rs1800503 | |
| 42376147 | c.1525−61A>G | Intron 9 | 35% | ||
| 42376223 | c.1540A>G | Exon 10 | M470V | 45% | rs213950 |
| 42376407 | c.1716+8A>G | Intron 10 | 1% | ||
| 42423325 | c.3041−92G>A | Intron 15 | 15% | ||
| 42423346 | c.3041−71G>C | Intron 15 | 1% | ||
| 42423539 | c.3271+42A>T | Intron 16b | 1% | ||
| 42427170 | c.3272−93T>C | Intron 16b | 3% | ||
| 42444201 | c.3601−65C>A | Intron 18 | 14% | rs213989 | |
| 42444378 | c.3713A>C | Exon 19 | E1194A | 1% | |
| 42459334 | c.4002A>G | Exon 20 | P1290P | 3% | rs1800130 |
| 42459458 | c.4005+121delTT | Intron 20 | 2% | ||
| 42469386 | c.4006−200G>A | Intron 20 | 16% | rs214164 | |
| 42469496 | c.4006−90delC | Intron 20 | 2% | ||
| 42483798 | c.4521G>A | Exon 24 | Q1463Q | 19% | rs1800136 |
Genomic reference sequence NT_007933.
However, p.M470V may not be strictly neutral.21,35 It was found that the M470 protein matured more slowly and had increased intrinsic chloride channel activity compared with the V470 protein.35 In addition, the V470 allele was shown to reduce CFTR protein activity when present in combination with H1352.21 M470V may also affect exon 9 skipping during splicing processes.35,36 Its incidence in non-cystic fibrosis CFTR-related diseases also suggests that M470V has a role in those diseases.36,37 M470V in phase with the T5 allele of the polymorphic Tn locus was suggested to be a disease-causing allele in a Korean cystic fibrosis patient.38 The extent to which this allele may have contributed to the disease phenotype in our cohort is not clear and deserves investigation.
Four novel polymorphisms were observed in the Iranian CF patients, including c.297−75C>A, c.408A>G (p.E92E), c.1716+8A>G, and c.3713A>C (p.E1194A). The polymorphism c.1716+8A>G is located near a splice junction and was observed in only one patient in whom no other disease-causing mutation was found; however, it did not alter the canonical splice consensus sequence and so is unlikely to affect splicing.
Haplotype Analysis
Haplotype analysis with respect to putative polymorphisms found within the CFTR gene of our patients was done to determine the extent of variation in the genetic background of different individuals carrying the same mutation and also to obtain an assessment of identity by descent in patients homozygous for a mutation. Figure 1 illustrates the haplotype map of the major haplotypes observed.9
Figure 1.

Haplotype map of the major haplotypes identified by Varia. The bold horizontal line represents a segment of human chromosome 7 that contains the CFTR gene (bases 116,600,000 to 116,900,000 from GenBank build 34). The vertical lines along the chromosome represent the location of eight polymorphisms used to define core haplotypes in this study, specifically, c.867−33del4 (six or seven repeats of TTGA; rs4148700), c.1001+11 (C/T; rs1800503), c.1525−61 (A/G; rs number not designated), c.1540 (A/G; Met/Val; rs213950), c.3041−92 (G/A; rs number not designated), c.3601−65 (C/A; rs213989), c.4006−200 (G/A; rs214164), and c.4521 (G/A; Q1463Q; rs1800136). The first two polymorphisms coincide on the first vertical line. The horizontal segments connecting the polymorphic sites represent the calculated haplotype blocks. Haplotype blocks are nonoverlapping blocks of consecutive variations representing regions of strong linkage disequilibrium and low recombination rate within the CFTR gene. The corresponding haplotypes are listed below the haplotype blocks. The horizontal and diagonal lines connecting the haplotypes indicate those that co-transmit together. Note that rare haplotypes are not displayed, ie, each block lists only those haplotypes with an estimated frequency of at least 0.02, or if they occur in more than one instance.
Thirteen mutations were each found to be associated with a single six-marker haplotype, as summarized in Table 1. One mutation was found on H1, five on H2, one on H3, one on H4, three on H5, and two on H8. The association of each of the five most common mutations with a single haplotype may indicate that all of the chromosomes carrying the same mutation had a common origin. The three mutations on H5 are among the world’s five most common mutations.12
All patients homozygous for the ΔF508 mutation also carried six copies of the TTGA repeat in intron 6a on both their chromosomes, consistent with its association with haplotype H5. One patient heterozygous for ΔF508 had seven copies of the repeat on both his chromosomes, probably indicating a recombination event. One of the haplotypes associated with the mutation p.K1177X was also probably recombined.
All patients who were homozygous for their putative disease-causing mutation were also homozygous for a haplotype defined by the six polymorphic markers, consistent with their alleles being identical by descent. However, homozygosity for a mutation in three of the patients, because of heterozygosity in rare polymorphisms, should not be considered to be due to identity by descent. Therefore, of the 20 homozygous patients, probably only 17 were homozygous because of identity by descent.
The haplotypes listed in Table 3 were defined using six high-frequency bi-allelic markers, including one repeat polymorphism and five SNPs. All of the markers are widely distributed across human populations in different parts of the world and are believed to be very old. Consequently, they define the core haplotype framework in this gene. Because the Iranian samples had a high frequency of homozygosity, we were able to unambiguously define 11 SNP haplotypes at the CFTR gene, representing 85% of the chromosomes analyzed. Seven of the 11 haplotypes formed a step-wise parsimony network of single-mutation events originating from an ancestral haplotype (H0), and four were best explained by recombi-nation. The Varia program predicted only 9 of the 11 un-ambiguous haplotypes. Furthermore, the breakpoints predicted by the recombinant haplotypes did not correspond with the “haplotype blocks” as defined by the Varia program. Possibly, the discrepancy is due to the assumption of the EM algorithm that the examined population has had random mating and exhibits Hardy-Weinberg equilibrium, both of which were not valid assumptions in the Iranian CF samples.
Table 3.
Haplotypes Observed in Iranian CF Patients and a European Control Group
| Haplotype | c.876−33 (delTTGA) | c.1001+11 (C/T) | c.1540 (A/G) | c.3601−65 (C/A) | c.4006−200 (G/A) | c.4521 (G/A) | Iranian CF patients (%) | European controls (%) |
|---|---|---|---|---|---|---|---|---|
| H0 | 7 | C | A | C | G | A | 1 | 2 |
| H1 | 7 | C | G | C | G | A | 7 | 2 |
| H2 | 7 | C | G | C | G | G | 25 | 39 |
| H3 | 7 | C | A | C | G | G | 9 | – |
| H4 | 6 | C | A | C | G | G | 3 | – |
| H5 | 6 | T | A | C | G | G | 36 | – |
| H3, H4, H5 | – | – | A | C | G | G | (48) | 35 |
| H6 | 7 | C | A | A | G | G | 3 | 0 |
| H7 | 7 | C | G | A | G | G | 2 | 0 |
| H8 | 7 | C | A | C | A | G | 3 | 0 |
| H9 | 7 | C | A | A | A | A | 7 | 14 |
| H10 | 7 | C | G | A | A | A | 5 | 9 |
The last column in Table 3 lists the haplotype frequency of a European control cohort. Genotypes of 176 normal CFTR haplotypes generated from the Interna-tional HapMap project for four of the SNP markers de-fining our six-marker haplotypes permitted a limited comparison of haplotype structure across the two populations. The haplotype diversity was greater in the Iranian CF samples, with nine haplotypes compared with six in the European normals. Not surprisingly, the CF patient samples had a disproportionately high frequency of the haplotype associated with three common CF mutations (H5).
CF Carrier Frequency in Iran
The proportion of the patient population whose disease was not due to consanguinity was used to calculate the frequency of CF-causing alleles in the population. The increase in incidence of disease due to consanguinity in Iran was estimated to be f = 0.0098. This means q would equal 39/19 + 3 in our sample where 39 is the number of patients in whom at least one mutation was found, 19 the number of individuals heterozygous for their mutations, and 3 the number whose homozygosity was probably not due to consanguinity. Calculations yield a q value of 0.0125 and a carrier frequency (2pq) of 2.5 in 100, or 1 in 40. Even if all individuals homozygous for any mutation were considered to be from consanguineous marriages, the carrier frequency would only drop to 1.8 in 100, or 1 in 55.
Discussion
Considerable heterogeneity in the nature of CFTR mutations was observed among the Iranian cystic fibrosis patients of this study. Twenty putative disease-causing mutations were identified in this study. Four of the mutations have only been previously reported in countries that border Iran and therefore may be unique to West Asia. These are c.1525−1G>A found in Pakistan, c.1649T>C found in Lebanon, c.3170C>T found in Turkey, and c.3661A>T found in Bahrain.39,40,41,42 Two mutations that were common in the rest of the world, p.G551D and p.W1282X, were not found in this group of Iranian CF patients.43,44 This suggests that there has been some divergence in the CFTR mutation spectrum in recent human history and has implications for mutation screening in West Asia and North Africa. Twenty-one putative polymorphisms were also identified, four of which have not previously been reported.
The five most common mutations in Iran constituted 37% of the alleles of the patients studied. Each of these was found at a frequency of 2.5% or more. Assuming random mating, it is expected that 60% of Iranian CF patients would carry at least one of these mutations. Therefore a molecular assay that could identify these five may be an appropriate preliminary diagnostic tool for the Iranian patients. As in Iran, a large spectrum of CFTR mutations has also been found in Spain, Bulgaria, Greece, and Turkey, all of which served as “historic gateways” into Europe.6
Although the frequency of the ΔF508 among the Iranian patients (14 to 16%) was lower than its frequency in European countries, it was still the most frequent mutation. The ΔF508 mutation most likely originated outside of Europe before it was introduced by migration, possibly from the Middle East.4,45 As for patients of European, Bashkortostanian, and Turkish descent, the vast majority (18 of 19) of the ΔF508 alleles were associated with allele 6 of the TTGA repeat in intron 6b.9,46,47
All ΔF508 mutations were found on the same SNP haplotype background, consistent with a report that ΔF508 had extensive allele sharing of STR haplotypes in a French population48 and its complete linkage with a single variant of the intron 9 splice site polymorphism.49 All other CFTR mutations found on more than one chromosome, except p.Y563X, could be assigned to a single haplotype (Table 1). One of these (c.1677del2) was also associated with a single haplotype in Turkey, but another (c.2183AA>G) was associated with three haplotypes in that country.9 It is interesting that the c.1821C>A mutation was associated with two haplotypes. Two of the most common mutations among the Iranians were each associated with a single haplotype, suggesting a relatively recent introduction into the population followed by rapid expansion.
Because we defined haplotypes using SNP markers, we were only able to make limited comparisons with previously reported results. Simple tandem repeat (STR) haplotypes at the CFTR locus, based on analysis of microsatellite variation, have been defined in many different human populations.20,43 STR markers mutate at a rate 1000-fold greater than SNP markers50 and generate multiple mutations at a single site. Thus, STR polymorphisms have generally occurred more recently, and are more diverse than SNPs. Thus, the 11 SNP haplotypes described here can be used to visualize population substructure farther in the past than is possible using STR haplotypes, and we hope they will be used for future comparisons of CFTR haplotypes across different populations.
The mutation spectrum in Iran had a significant overlap with that observed in Turkey and was less similar to that observed in Europe. Specifically, the four most common Turkish mutations were found in Iran, including ΔF508, c.1677delTA, p.G542X, and c.2183AA>G.9,29 The p.G542X “Mediterranean mutation,” purported to be of Phoenician origin, was found on only one Iranian chromosome, whereas it was relatively frequent (3.6%) among the Turkish CF chromosomes.6,51 Another common mutations in Iran, p.K1177X, was not found in Turkey but was reported in Bahrain.39 In contrast, three mutations commonly found in Europe, including p.W1282X, p.G551D, and c.1717−1G>A,12 were not found in Iran. Two common mutations previously reported in Arab populations, including c.3601−111G>C52 and c.3120+1kbdel8.6kb,53 were not assayed in this study because they were outside the genomic regions amplified by PCR. Six additional mutations reported in at least two other Arab populations, including c.711+1G>A, p.R75X, c.1548delG, c.3120+1G>A, c.3199del6, and p.S549R,30,42,53,54,55,56 could have been detected in our assay but were not found in Iran.
Calculations of the frequency of carriers of disease-causing CFTR mutations in Iranians (1:40) is very similar to that of European populations (1:25), although there is greater allele diversity in Iran. Consequently, it is believed that the incidence of cystic fibrosis is also similarly high, and that the low incidence commonly believed to be associated with this non-European population is likely to be due to under-diagnosis. Furthermore, the frequency of CF may be higher in some isolated populations due to consanguinity, as has been previously reported in an isolated population in Israel.57 The head (A.K.) of the CF reference center in Iran, based on the history of patient referrals, corroborates that under-diagnosis of CF in Iran is notable. The same assessment has been proposed for populations of Turkey and Saudi Arabia.9,28,30 Therefore, in addition to Europe and the United States, CF is likely to be prevalent in many other countries as well, although the mutation spectrums may be different. We hope to ascertain CFTR mutation carrier frequencies and CF incidence among Iranians as soon as possible. A large cohort of patients is being collected for mutation analysis, and pilot trials for the screening of some variants in the general population has started.
Our mutation detection efficiency was a rather low 53%. We cannot be sure how much misdiagnosis contributed to this. Alternative methods of determining sweat chloride levels may be more accurate than those used in this study.58 Unfortunately, it is not possible to divide the patients into groups based on clinical and laboratory criteria because sufficient and uniform data on the patients was not available. Such a division may have identified groups that were responsible for overall low mutation detection. Some heterozygous point mutations may have gone undetected by both the Sequencher software and eye inspection of the sequence chromatograms. Use of denaturing high performance liquid chromatography has been shown to improve detection of CFTR mutations.59,60 However, large heterozygous deletions and mutations in intronic and control regions not amplified would have been missed by either method. Finally, it is possible that genes other than the CFTR gene may have caused CF in some of the patients. It is possible that the state of modifier genes in combination with the genotype of particular polymorphisms would result in the disease phenotype. In any case, even with the low detection efficiency, a very notable level of heterogeneity in the CFTR mutation profile was found. In the similarly heterogeneous population of Turkey, detection efficiency was also a relatively low 75%.9
We hope that the recognition of a probable high incidence of CF in Iran will attract increased attention to the diagnosis of this disease. We also hope that the identification of five relatively frequent mutations will assist in the development of a clinically appropriate assay for their detection as a preliminary test for diagnosis.
Acknowledgments
We thank all of the patients and their families for consenting to participate in this study. We also thank Sujatha Krishnakumar and Peter Oefner of the Stanford Genome Technology Center for technical discussions.
Footnotes
Supported by the Research Council of the Faculty of Sciences of Tehran University (Iran), The National Research Center for Genetic Engineering and Biotechnology (Iran), and National Institutes of Health grant 5P01HG00205.
References
- Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67:471–485. doi: 10.1046/j.1469-1809.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- Welsh M, Tsui L, Boat T, Beaudet A. Cystic Fibrosis. Scriver C, Beaudet A, Sly W, Valle D, editors. New York: McGraw-Hill,; The Metabolic and Molecular Bases of Inherited Disease. 1995:3799–3876. [Google Scholar]
- Farrell P. Improving the health of patients with cystic fibrosis through newborn screening. Adv Pediatr. 2000;49:79–115. [PubMed] [Google Scholar]
- Dawson K, Frossard P. The geographic distribution of cystic fibrosis mutations gives clues about population origins. Eur J Pediatr. 2000;159:496–499. doi: 10.1007/s004310051317. [DOI] [PubMed] [Google Scholar]
- Kerem B, Rommens J, Buchanan J, Markiewcz D, Cox T, Chakravarti A, Buchwald M, Tsui L. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–1080. doi: 10.1126/science.2570460. [DOI] [PubMed] [Google Scholar]
- Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- European Working Group on CF Genetics Gradient of distribution in Europe of the major CF mutation and its associated haplotypes. Hum Genet. 1990;85:436–445. doi: 10.1007/BF02428304. [DOI] [PubMed] [Google Scholar]
- Lucotte G, Hazout S, De Braekeleer M. Complete map of cystic fibrosis mutation DF508 frequencies in Western Europe and correlation between mutation frequencies and incidence of disease. Hum Biol. 1995;67:797–803. [PubMed] [Google Scholar]
- Kilinç M, Ninis V, Dagli E, Demirkol M, Özkinay F, Arikan Z, Çogulu Ö, Hüner G, Karakoç F, Tolun A. Highest heterogeneity for cystic fibrosis: 36 mutations account for 75% of all CF chromosomes in Turkish patients. Am J Hum Genet. 2002;113:250–257. doi: 10.1002/ajmg.10721. [DOI] [PubMed] [Google Scholar]
- Pier G. Role of the cystic fibrosis transmembrane conductance regulator in innate immunity to Pseudomonas aeruginosa infections. Proc Natl Acad Sci USA. 2000;97:8822–8828. doi: 10.1073/pnas.97.16.8822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak J, Pier G. Salmonella enterica serovar Typhi modulates cell surface expression of its receptor, the cystic fibrosis transmembrane regulator, on the intestinal epithelium. Infect Immun. 2002;70:6416–6423. doi: 10.1128/IAI.70.11.6416-6423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill X, Bancells C, Ramos C, the Biomed CF Mutation Analysis Consortium Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. Hum Mutat. 1997;10:135–154. doi: 10.1002/(SICI)1098-1004(1997)10:2<135::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Mateu E. Can a place of origin of the main cystic fibrosis mutations be identified? Am J Hum Genet. 2002;70:257–264. doi: 10.1086/338243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalalirad M, Houshmand M, Mirfakhraie R, Goharbari MH, Mirzajani F. First study of CF mutations in the CFTR gene of Iranian patients: detection of DF508, G542X, W1282X, A120T, R117H, and R347H mutations. J Trop Pediatr. 2004;50:359–361. doi: 10.1093/tropej/50.6.359. [DOI] [PubMed] [Google Scholar]
- Naghizadeh R, Elahi E. Determination of the frequency of the ΔF508 mutation amongst Iranian cystic fibrosis patients and the detection of carriers of this mutation using the ARMS-PCR protocol. Med J Islam Repub Iran. 1999;16:278–286. [Google Scholar]
- Schales O, Scales S. A simple and accurate method for the determination of chloride in biological fluids. J Biol Chem. 1941;140:879–884. [Google Scholar]
- Miller S, Dykes D, Polesky H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal C, Audrézet M, Quéré I, Raguénès O, Langonné S, Férec C. Complete and rapid scanning of the cystic fibrosis transmembrane regulator (CFTR) gene by denaturing high performance liquid chromatography (D-HPLC). Hum Genet. 2001;108:290–298. doi: 10.1007/s004390100490. [DOI] [PubMed] [Google Scholar]
- Zielenski J, Rozmahel R, Bozon D, Kerem B, Grzelczak Z, Riordan J, Rommens J, Tsui L. Genomic DNA sequencing of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10:214–228. doi: 10.1016/0888-7543(91)90503-7. [DOI] [PubMed] [Google Scholar]
- Dörk T, Neumann T, Wulbrand U, Wulf B, Kalin N, Maass G, Krawczak M, Guillermit H, Ferec C, Horn G. Intra- and extragenic marker haplotypes of CFTR mutations in cystic fibrosis families. Hum Genet. 1992;88:417–425. doi: 10.1007/BF00215676. [DOI] [PubMed] [Google Scholar]
- Lee JH, Choi JH, Namkung W, Hanrahan JW, Chang J, Song SY, Park SW, Kim DS, Yoon JH, Suh Y, Jang IJ, Nam JH, Kim SJ, Cho MO, Lee JE, Kim KH, Lee MG. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet. 2003;12:2321–2332. doi: 10.1093/hmg/ddg243. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Slatkin M. Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol. 1995;12:921–927. doi: 10.1093/oxfordjournals.molbev.a040269. [DOI] [PubMed] [Google Scholar]
- Reich D. Linkage disequilibrium in the human genome. Nature. 2001;411:199–204. doi: 10.1038/35075590. [DOI] [PubMed] [Google Scholar]
- Rioux J. Hierarchical linkage disequilibrium mapping of a susceptibility gene for Crohn’s disease to the cytokine cluster on chromosome 5. Nat Genet. 2001;29:223–228. [Google Scholar]
- Daly M, Rioux J, Schaffner S, Hudson T, Lander E. High-resolution haplotype structure in the human genome. Nat Genet. 2001;29:229–232. doi: 10.1038/ng1001-229. [DOI] [PubMed] [Google Scholar]
- Mani A, Meraji S-M, Houshyar R, Radhakrishnan J, Mani A, Ahangar M, Rezaie T, Taghavinejad M-A, Broumand B, Zhao H, Nelson-Williams C, Lifton R. Finding genetic contributions to sporadic disease: a recessive locus at 12q24 commonly contributes to patent ductus arteriousus. Proc Natl Acad Sci USA. 2002;99:15054–15059. doi: 10.1073/pnas.192582999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ACMG Laboratory Practice Committee Working Group ACMG recommendations for standards for interpretation of sequence variations. Genet Med. 2000;2:302–303. [Google Scholar]
- Banjar H, Kambouris M, Meyer BF, al-Mehaidib A, Mogarri I. Geographic distribution of cystic fibrosis transmembrane regulator gene mutations in Saudi Arabia. Ann Trop Paediatr. 1999;19:69–73. doi: 10.1080/02724939992671. [DOI] [PubMed] [Google Scholar]
- Onay T, Topaloglu O, Zielenski J, Gokgoz N, Kayserili H, Camcioglu Y, Cokugras H, Akcakaya N, Apak M, Tsui L, Kirdar B. Analysis of the CFTR gene in Turkish cystic fibrosis patients: identification of three novel mutations (3172delAC; P1013L; M1028I). Hum Genet. 1998;10:224–230. doi: 10.1007/s004390050683. [DOI] [PubMed] [Google Scholar]
- Banjar H. Geographic distribution of cystic fibrosis transmembrane regulator gene mutations in Saudi Arabia. East Mediterr Health J. 1999;5:1230–1235. [PubMed] [Google Scholar]
- Xiong X, Bragin A, Widdicombe JH, Cohn J, Skach WR. Structural cues involved in endoplasmic reticulum degradation of G85E and G91R mutant cystic fibrosis transmembrane conductance regulator. J Clin Invest. 1997;100:1079–1088. doi: 10.1172/JCI119618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue H, Devidas S, Guggino WB. The two halves of CFTR form a dual-pore ion channel. J Biol Chem. 2000;275:10030–10034. doi: 10.1074/jbc.275.14.10030. [DOI] [PubMed] [Google Scholar]
- Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- Claustres M, Altieri JP, Guittard C, Templin C, Chevalier-Porst F, Des Georges M. Are p.I148T, p.R74W and p.D1270N cystic fibrosis causing mutations? BMC Med Genet. 2004;5:19. doi: 10.1186/1471-2350-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuppens H, Lin W, Jaspers M, Costes B, Teng H, Vankeerberghen A, Jorissen M, Droogmans G, Reynaert I, Goossens M, Nilius B, Cassiman JJ. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes: the polymorphic (Tg)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J Clin Invest. 1998;101:487–496. doi: 10.1172/JCI639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Meeus A, Guittard C, Desgeorges M, Carles S, Demaille J, Claustres M. Linkage disequilibrium between the M470V variant and the IVS8 polyT alleles of the CFTR gene in CBAVD. J Med Genet. 1998;35:594–596. doi: 10.1136/jmg.35.7.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Moylan B, Leopold DA, Kim J, Rubenstein RC, Togias A, Proud D, Zeitlin PL, Cutting GR. Mutation in the gene responsible for cystic fibrosis and predisposition to chronic rhinosinusitis in the general population. JAMA. 2000;284:1814–1819. doi: 10.1001/jama.284.14.1814. [DOI] [PubMed] [Google Scholar]
- Ahn KM, Park HY, Lee JH, Lee MG, Kim JH, Kang IJ, Lee SI. Cystic fibrosis in Korean children: a case report identified by a quantitative pilocarpine iontophoresis sweat test and genetic analysis. J Korean Med Sci. 2005;20:153–157. doi: 10.3346/jkms.2005.20.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandarani HA. Cystic fibrosis transmembrane regulator gene mutations in Bahrain. J Trop Pediatr. 2002;48:348–350. doi: 10.1093/tropej/48.6.348. [DOI] [PubMed] [Google Scholar]
- Desgeorges M, Megarbane A, Guittard C, Carles S, Loiselet J, Demaille J, Claustres M. Cystic fibrosis in Lebanon: distribution of CFTR mutations among Arab communities. Hum Genet. 1997;100:279–283. doi: 10.1007/s004390050505. [DOI] [PubMed] [Google Scholar]
- Wahab A, Al Thani G, Dawod ST, Kambouris M, Al Hamed M. Rare CFTR mutation 1525–1G>A in a Pakistani patient. J Trop Pediatr. 2004;50:120–122. doi: 10.1093/tropej/50.2.120. [DOI] [PubMed] [Google Scholar]
- el-Harith EA, Dork T, Stuhrmann M, Abu-Srair H, al-Shahri A, Keller KM, Lentze MJ, Schmidtke J. Novel and characteristic CFTR mutations in Saudi Arab children with severe cystic fibrosis. J Med Genet. 1997;34:996–999. doi: 10.1136/jmg.34.12.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu E, Calafell F, Lao O, Bonne-Tamir B, Kidd JR, Pakstis A, Kidd KK, Bertranpetit J. Worldwide genetic analysis of the CFTR region. Am J Hum Genet. 2001;68:103–117. doi: 10.1086/316940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu E, Calafell F, Bonne-Tamir B, Kidd JR, Casals T, Kidd KK, Bertranpetit J. Allele frequencies in a worldwide survey of a CA repeat in the first intron of the CFTR gene. Hum Hered. 1999;49:15–20. doi: 10.1159/000022834. [DOI] [PubMed] [Google Scholar]
- Morral N, Bertranpetit J, Estivill X, Nunes V, Casals T, Gimenez J, Reis A, Varon-Mateeva R, Macek M, Kalaydjieva L, Angelicheva D, Dancheva R, Romeo G, Russo M, Garnerone S, Restagno G, Rerrari M, Magnani C, Claustres M, Desgeorges M, Schwartz M, Dallapiccola B, Novelli G, Ferec C, de Arce M, Kere J, Anvret M, Dahl N, Kadasi L. The origin of the major cystic fibrosis mutation (ΔF508) in European populations. Nat Genet. 1994;7:169–175. doi: 10.1038/ng0694-169. [DOI] [PubMed] [Google Scholar]
- Chehab F, Johnson J, Louie E, Goossens M, Kawasaki E, Erlich H. A dimorphic 4-bp repeat in the cystic fibrosis gene is in absolute linkage disequilibrium with the ΔF508 mutation: implications for parental diagnosis and mutation mutation origin. Am J Hum Genet. 1991;48:223–226. [PMC free article] [PubMed] [Google Scholar]
- Korytina G, Viktorova T, Baikova G, Khusnutdinova E. Analysis of the spectra of mutations and polymorphic loci of cystic fibrosis transmembrane conductance regulator in the population of Bashkortostan. Genetika. 2002;38:1270–1275. [PubMed] [Google Scholar]
- de Vries HG, van der Meulen MA, Rozen R, Halley DJ, Scheffer H, ten Kate LP, Buys CH, te Meerman GJ. Haplotype identity between individuals who share a CFTR mutation allele “identical by descent”: demonstration of the usefulness of the haplotype-sharing concept for gene mapping in real populations. Hum Genet. 1996;98:304–309. doi: 10.1007/s004390050211. [DOI] [PubMed] [Google Scholar]
- Dörk T, Fislage R, Neumann T, Wulf B, Tummler B. Exon 9 of the CFTR gene: splice site haplotypes and cystic fibrosis mutations. Hum Genet. 1994;93:67–73. doi: 10.1007/BF00218916. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Kimmel M, Stivers DN, Davison LJ, Deka R. Relative mutation rates at di-, tri-, and tetranucleotide microsatellite loci. Proc Natl Acad Sci USA. 1997;94:1041–1046. doi: 10.1073/pnas.94.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loirat F, Hazout S, Lucotte G. G542X as a probable Phoenician cystic fibrosis mutation. Hum Biol. 1997;69:419–425. [PubMed] [Google Scholar]
- Chillon M, Casals T, Gimenez J, Ramos MD, Palacio A, Morral N, Estivill X, Nunes V. Analysis of the CFTR gene confirms the high genetic heterogeneity of the Spanish population: 43 mutations account for only 78% of CF chromosomes. Hum Genet. 1994;93:447–451. doi: 10.1007/BF00201673. [DOI] [PubMed] [Google Scholar]
- Laufer-Cahana A, Lerer I, Sagi M, Rachmilewitz-Minei T, Zamir C, Rivlin JR, Abeliovich D. Cystic fibrosis mutations in Israeli Arab patients. Hum Mutat. 1999;14:543. doi: 10.1002/(SICI)1098-1004(199912)14:6<543::AID-HUMU16>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Frossard PM, Hertecant J, Bossaert Y, Dawson KP. Genotype-phenotype correlations in cystic fibrosis: clinical severity of mutation S549R(T→G). Eur Respir J. 1999;13:100–102. doi: 10.1183/09031936.99.13110099. [DOI] [PubMed] [Google Scholar]
- Messaoud T, Verlingue C, Denamur E, Pascaud O, Quere I, Fattoum S, Elion J, Ferec C. Distribution of CFTR mutations in cystic fibrosis patients of Tunisian origin: identification of two novel mutations. Eur J Hum Genet. 1996;4:20–24. doi: 10.1159/000472165. [DOI] [PubMed] [Google Scholar]
- Loumi O, Baghriche M, Delpech M, Kaplan JC, Bienvenu T. Analysis of the complete coding region of the CFTR gene in ten Algerian cystic fibrosis families. Hum Hered. 1999;49:81–84. doi: 10.1159/000022849. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Nissim-Rafinia M, Argaman Z, Genem A, Moran I, Kerem E, Kerem B. Screening of CFTR mutations in an isolated population: identification of carriers and patients. Eur J Hum Genet. 1998;6:181–184. doi: 10.1038/sj.ejhg.5200174. [DOI] [PubMed] [Google Scholar]
- Schrijver I, Ramalingam S, Sankaran R, Swanson S, Dunlop CLM, Keiles S, Moss RB, Oehlert J, Gardner P, Wassman ER, Kammesheidt A. Diagnostic testing by CFTR gene mutation analysis in a large group of Hispanics: novel mutations and assessment of a population-specific mutation spectrum. J Mol Diagn. 2005;7:289–299. doi: 10.1016/S1525-1578(10)60557-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Apice MR, Gambardella S, Bengala M, Russo S, Nardone AM, Lucidi V, Sangiuolo F, Novelli G. Molecular analysis using DHPLC of cystic fibrosis: increase of the mutation detection rate among the affected population in Central Italy. BMC Med Genet. 2004;5:8. doi: 10.1186/1471-2350-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnik-Glavac M, Atkinson A, Glavac D, Dean M. DHPLC screening of cystic fibrosis gene mutations. Hum Mutat. 2002;19:374–383. doi: 10.1002/humu.10065. [DOI] [PubMed] [Google Scholar]