

Abstract
The prognostic significance of somatic activating codon 816 c-kit mutations in pediatric urticaria pigmentosa has not yet been established in detail. Detection of such mutations in archival paraffin-embedded biopsies is usually hampered by an abundance of surrounding normal cells. Here we describe a method for the selective amplification and specific detection of c-kit mutation Asp816→Val in complete tissue sections cut from up to 24-year-old paraffin blocks. Peptide nucleic acid-mediated polymerase chain reaction clamping of the wild-type allele was combined with on-line mutation detection using oligonucleotide hybridization probes. In DNA extracted from HMC-1 cells heterozygously carrying the c-kit mutation Asp816→Val, the one-tube assay allowed specific detection of this mutation in a more than 1000-fold excess of normal background DNA within 1 hour and without the need for additional analytical steps. In a series of 38 cases with pediatric urticaria pigmentosa we detected c-kit codons 815 and 816 mutations in 16 cases. Mutation detection did not correlate with clinical outcome after a mean follow-up of 11.2 years. In conclusion, the procedure described may represent an ideal screening tool for all kinds of clinical applications, using point mutations as markers of, for example, early events in carcinogenesis, circulating metastatic tumor cells, and minimal residual disease.
Mastocytosis comprises a heterogeneous group of disorders characterized by accumulation of abnormal mast cells (MCs) in various tissues. An updated consensus classification system of MC diseases was recently published. 1 There are two main variants, cutaneous and systemic forms. 2,3 Cutaneous mastocytosis typically presents as urticaria pigmentosa (UP). 4 In systemic mastocytosis (SM) there is, by definition, involvement of at least two tissues/organs. Of the extracutaneous organs, the bone marrow is the most frequently affected. 2 SM is further subdivided into an indolent variant (ISM), SM with an associated clonal hematological non-MC lineage disease (AHNMD), and an aggressive subtype (ASM). 1
The c-kit proto-oncogene encodes for a transmembrane tyrosine kinase receptor (KIT) whose ligand is the stem cell factor, also known as MC growth factor (MGF). 5,6 c-kit is expressed on MCs 7 as well as on hemopoietic stem cells, 8-10 melanocytes, 11 and germ cells. 12 Activated c-kit mediates signals for proliferation and maturation in these cells. 13,14
Certain somatic gain-of-function point mutations cause stem cell factor-independent growth of MCs. 15,16 One of these mutations (Asp-816→Val) has been repeatedly detected in several subtypes of mastocytosis, e.g., mast cell leukemia, SM (with and without skin involvement, with and without AHNMD), and UP. 15,17-22 Mutations in other regions of the c-kit gene have been described in acute myeloid leukemia, gastrointestinal stromal tumors, and sinonasal T-cell lymphomas. 23-25
In pediatric mastocytosis, patients with activating codon 816 mutations tend to have more extensive and/or more persistent disease. 19 Such mutations can be detected in native tissue by amplification and further analysis of c-kit mRNA, which is expressed almost exclusively by MCs. In archival, formalin-fixed and paraffin-embedded tissue, however, the analysis of mRNA may be impossible because of age- and fixation-related degradation of nucleic acids. 26,27 Therefore, mutation screening has to focus on the analysis of genomic DNA. Tumor cells often represent only a minor fraction of the cells present in biopsy samples. Methods for the detection of sequence variants, such as single-strand conformation polymorphism, restriction fragment length polymorphism, DNA sequencing, allele-specific amplification, or hybridization with oligonucleotide probes, may allow detection of the extremely small tumor cell populations in such cases.
Peptide nucleic acid (PNA) oligomers have been used to enhance allele-specific amplification of minor background variants. 28 PNA-DNA hybrids have a higher thermal stability than DNA-DNA hybrids and PNA oligomers cannot be extended by DNA polymerases. 28 Assays using wild-type-specific PNA as competitors to mutation-specific primers have been described for the detection of point mutations in K-ras and p53. 29,30 However, a drawback of this set-up is that abundant readthrough from the suppressed wild type can still occur. The resulting products will contain the mutation making it indistinguishable from amplification products generated from the mutated alleles.
Hybridization probes consist of a pair of terminally fluorescent-labeled oligonucleotide probes designed to bind to a target strand in close proximity, enabling an energy transfer between both fluorophores. Measurement of the fluorescence energy is a sensitive monitor for base variations within the target region covered by the probes. 31 Hybridization probes are widely used for the analysis of mutations, for example in the HFE gene in hemochromatosis 32 or in the N-ras gene in cancer. 33 However, the analysis is restricted to see equivalent amounts of the variants or to detect the major variant only.
In this study, we combined the PNA-mediated PCR-clamping method with the on-line mutation detection. We used a wild-type-specific PNA and mutation-specific hybridization probes for the detection of activating c-kit Asp816→Val mutations. The samples we investigated were formalin-fixed and paraffin-embedded skin biopsy specimens from patients with pediatric cutaneous mastocytosis.
Materials and Methods
Patients
Thirty-eight consecutive patients with pediatric UP attending the Department of Dermatology of a Pediatric Hospital (Hospital del Niño Jesús, Madrid, Spain) were included in the study. UP was defined as skin infiltration by a large number of MCs in the form of multiple brown macules, papules, and nodules. Patients with other types of cutaneous mastocytosis (ie, mastocytoma, diffuse cutaneous mastocytosis, and telangiectasia macularis eruptiva perstans) were excluded from the study. The clinical diagnosis of UP was confirmed by skin biopsies stained with hematoxylin and eosin and Giemsa that exhibited large numbers of MCs in the upper dermis and hyperpigmentation of the basal layer of the epidermis. The age at which the lesions first appeared ranged from birth to 72 months, and the mean duration of follow-up was 11.2 ± 4.4 years (range, 3 to 20 years). With regard to outcome, patients were considered as having transient UP if all skin lesions had disappeared, and persistent UP if skin lesions were still present at the time of the last follow-up visit.
Tissue Samples
The presence of c-kit mutations was investigated in archival formalin-fixed, paraffin-embedded tissue: skin biopsies from 38 children with UP, bone marrow biopsies from 40 patients with acute myeloid leukemia, and 23 patients with (reactive) MC hyperplasia were investigated.
The cutaneous tissue was fixed in unbuffered formalin. All of the bone marrow biopsies were fixed in 5% neutral buffered formalin and decalcified in ethylenediaminetetraacetic acid. Paraffin sections (5 μm) were stained with Giemsa and immunostained with the antibody AA1 (DAKO Diagnostika, Hamburg, Germany) directed against tryptase, a very specific MC marker (Figure 1) ▶ . 34
Figure 1.

Photomicrographs of pediatric UP showing dense subepidermal MC infiltrates without any preferential attachment to pre-existing structures. Molecular analysis disclosed wild-type c-kit in A and mutation Asp816→Val in B. Results of melting curve analyses are shown in Figure 4, A and B ▶ . Paraffin sections, AA1 immunostain; original magnifications, ×200.
Genomic DNA extracted from HMC-1 cells served as a positive control for the detection of the c-kit mutation Asp816→Val. This cell line, which was established from a case of human mast cell leukemia, carries the mutation heterozygously. Human placental DNA served as a negative control for the mutation and as background DNA for dilution experiments undertaken to determine the sensitivity of the method.
DNA Extraction
For extraction of total genomic DNA, 10 8-μm-thick sections were cut from each paraffin block. The sections were dewaxed according to standard protocols. In brief, they were vortexed in 1 ml of xylene (100%) for 1 minute and centrifuged at 13,000 rpm for 5 minutes. This procedure was repeated three times. The samples were then washed twice in pure ethanol and vacuum-dried. Proteinase K digestion was performed in a final volume of 200 μl of buffer (50 mmol/L Tris, 1 mmol/L ethylenediaminetetraacetic acid, 0.5% Tween-20, pH 8.5) containing 0.2 mmol/L of proteinase K at 55°C (2 hours). Total DNA was extracted with phenol/chloroform/isoamyl alcohol (v/v/v, 25/24/1) and precipitated with ethanol (100%)/LiCl (8 mol/L) as described elsewhere. 35 Samples of 20 ng of the total extracted DNA were used for PCR amplification of the c-kit gene.
Primers and Hybridization Probes
The primers c-kit S (5′-CAG CCA GAA ATA TCC TCC TTA CT-3′) and c-kit B (5′-TTG CAG GAC TGT CAA GCA GAG-3′) were used to amplify a 138-bp fragment of the exon 17 (Figure 2) ▶ . The A7176T mutation site was covered by the 3′-fluorescein-labeled sensor probe (5′-AGC CAG AGT CAT CAA GAA TGA TTC TA-F 3′) being specific for the mutation (bold letter). We generally prefer to use the mutation-specific probe because the high melting temperature in the presence of the mutation is more significant than a decrease in the melting temperature that would be observed with a wild-type-specific probe. For our approach of a competition with a wild-type specific PNA this is even crucial. The anchor probe having a significantly higher (calculated) melting temperature was (5′-LC-ATG TGG TTA AAG GAA ACG TGA GTA CCC Ap-3′). The anchor probe was 5′-terminal labeled with LightCycler Red 640 (LC) and 3′-terminal blocked with a phosphate group (p). The LC dye was added using the respective NHS ester (Roche Diagnostics, Mannheim, Germany).
Figure 2.
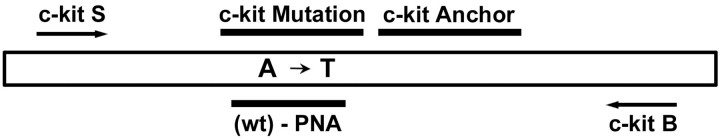
Positions of PCR primers (c-kit S, c-kit B), hybridization probes (c-kit mutation, c-kit anchor), and the PNA molecule targeting the wild-type sequence at codon 816 of the c-kit exon 17. Mutation Asp816→Val is caused by a A→T transition.
Primers, hybridization probes, and PNA were selected using the OLIGO 6.0 program (Molecular Biology Insights, Cascade, CO, USA) and BlueGene (Magic Works, Teltow, Germany). The probes were synthesized according to the manufacturer’s instructions (Roche Diagnostics).
PNA
PNAs are mimics of DNA oligonucleotides in which the phosphoribose backbone is replaced by a peptide-like repeat of (2-aminoethyl)-glycine units. Compared to DNA/DNA hybrids the corresponding PNA/DNA hybrids display greater thermal stability but are more easily destabilized by single bp mismatches. The PNA (5′-GCC AGA GAC ATC AAG AAT G-3′, site of mutation indicated in bold; Figure 2 ▶ ) corresponding to the wild-type sequence was synthesized using reagents from Applied Biosystems (Weiterstadt, Germany).
DNA Amplification and Mutation Detection
PCR amplification and mutation detection was performed with the LightCycler (software version 3.5, Roche Diagnostics) using the DNA Master Hybridization Probes kit as described by the manufacturer (Roche Diagnostics). The PCR mixture contained Taq polymerase, LightCycler buffer, dNTPs as supplied in the kit, 3 mmol/L MgCl2, 5 pmol (0.25 μmol/L) of each primer, 6 pmol (0.3 μmol/L) of both hybridization probes, and 15 pmol (0.75 μmol/L) of the PNA. Samples were amplified running 50 cycles of denaturation (10 seconds at 95°C), annealing of PNA (8 seconds at 68°C), annealing of primers (10 seconds at 56°C), and primer extension (10 seconds at 72°C). The first cycle was preceded by 10 minutes of denaturation at 95°C. Before melting point analysis the samples were denatured 5 minutes at 95°C. Melting curve analysis started at 40°C, increasing the temperature by 0.3°C/second up to 95°C.
Sequencing of PCR Products
For direct sequencing, PCR products were extracted from 2% Tris-acetate/ethylenediaminetetraacetic acid (TAE) agarose gels with a gel extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. PCR products were sequenced in both directions using primers c-kit S and c-kit B by the dye-deoxy terminator method on a ABI Prism Sequencer 377 (Applied Biosystems).
Results
Histology and Immunohistochemical Staining
MC infiltrates in the various tissues investigated were visualized by immunostaining for tryptase with the antibody AA1 (Figure 1) ▶ . MCs are the only cells that react with this antibody. Thus, this highly specific immunohistochemical reaction facilitated the identification of MC diseases of all subtypes and helped to assess semiquantitatively the numbers of infiltrating MCs. In UP, the dermis was infiltrated by strongly metachromatic MCs, typically located around small blood vessels and adnexae. MCs did not show signs of an epidermotropism. Control cases of MC hyperplasia (reactive states) showed loosely scattered also strongly metachromatic MCs but never compact (dense) clusters of MCs, diagnostic for mastocytosis.
Melting Analysis of PCR-Amplified Samples
We observed a melting temperature of 63.3°C for PCR products containing only the A7176T mutation (Asp-816→Val), and 57.7°C for wild-type samples. Heterozygous samples as in the case of DNA extracted from HMC-1 cells displayed both melting peaks (Figure 3A) ▶ .
Figure 3.

Effects of PNA-mediated PCR clamping. In melting curve analyses with Light Cycler hybridization probes wild-type (wt) c-kit and codon 17 mutation Asp816→Val (mut) can be discriminated through different melting peaks at ∼57°C (wt) and 63°C (mut) (A, continuous line). With only the use of hybridization probes, the method allows detection of the heterozygous mutation in an 10:1 excess of normal versus HMC-1 cells (A, broken line). Effects of PNA-mediated PCR clamping of the wild-type allele is demonstrated with various amounts of input PNA. In 100 ng of HMC-1 DNA, 0.75 μmol/L of PNA completely suppresses amplification of the wild-type allele (B, dotted line). Reduction of amplification efficiency of also the mutated allele by increasing amounts of PNA as a side effect must be taken into consideration when assessing the appropriate amount of input PNA (C). Compared to results in A, PNA-mediated PCR clamping of the wild-type allele results in a 100-fold increase of sensitivity yet allowing detection of the mutated allele in a 1000-fold excess of wild-type background DNA (D, three upper curves).
We determined the sensitivity of the melting analysis in dilution experiments of varying amounts of heterozygously mutated HMC-1 DNA in wild-type human placental DNA. The mutation-specific melting peaks were still detected in 10:1 mixtures hence containing only 5% of the mutated allele. However, the mutation was no longer detectable in a 100:1 mixture of 100 ng of wild-type DNA and 1 ng of HMC-1 DNA (0.5% mutated alleles, Figure 3A ▶ ). In direct sequencing of PCR products, the mutation was only detectable in the 1:1 mixture of wild-type and HMC-1 DNA (25% mutated alleles), but not in the 10:1 and 100:1 dilutions (data not shown).
Effects of PNA-Mediated PCR Clamping
The PNA was used to suppress the amplification of the wild-type target, possibly allowing detection of the mutant allele when the tumor cells represent only a minor fraction of all cells present and there is thus a large amount of wild-type background DNA.
First, the amount of PNA necessary to completely suppress amplification of the wild-type allele was determined. In 100 ng of HMC-1 DNA the addition of 0.25 μmol/L of PNA resulted in a marked decrease of the wild-type signal, 0.75 μmol/L of PNA resulted in a complete suppression of the wild-type melting peak (Figure 3B) ▶ . The suppression was also dependent on the amount of target DNA, indicating the importance of the ratio between PNA and target, more than the absolute concentration of PNA. Using 10 ng of HMC-1 DNA, 0.25 μmol/L of PNA suppressed the amplification of the wild type. Higher concentrated PNA (0.75 μmol/L and 1.25 μmol/L) lowered also the amplification of the mutant alleles (Figure 3C) ▶ .
To estimate the relative detection limit of our system mutation-specific c-kit hybridization probes and wild-type-specific c-kit PNA, dilution series of 1 μg of wild-type DNA and various amounts of HMC-1 DNA including 100 ng, 10 ng, 1 ng, 100 pg, and 10 pg were amplified with and without addition of PNA. Complete suppression of wild-type allele amplification again was achieved with 0.75 μmol/L of PNA. In repeated experiments, the mutation-specific melting peak at around 63°C reproducibly was detected in a 1000:1 excess of wild-type versus mutated cells (Figure 3D) ▶ . Detection of the mutation in 1 of 10,000 cells was not achieved reproducibly (data not shown).
c-kit Mutation Detection in Cutaneous Biopsies
To screen for c-kit exon 17 mutations in pediatric UP, cutaneous biopsies of 38 children were investigated. One sample failed in DNA amplification. From the residual 37 samples 16 displayed an aberrant melting curve indicating the presence of a sequence variation (Figure 4, B to E) ▶ . Ten samples had a melting point of 63°C, typical for the Asp816→Val mutation (patients 6, 9, 15, 18, 23, 25, 26, 31, 34, and 36), which was confirmed by DNA sequencing of the amplification products (Figure 4B) ▶ . The remaining six samples had other melting curves: the sample from patient 8 had a melting point of 53°C (Figure 4C) ▶ , samples 14 and 32 had 54°C (not shown), sample 22 had 55°C (Figure 4D) ▶ , and the samples from patients 7 and 16 had 59°C (Figure 4E) ▶ . Direct sequencing revealed mutations within the codons 815 and 816. We found two C/T transversions at nucleotide 7177, not resulting in amino acid exchanges in codon 816 (samples 14 and 32). One mutation in codon 815 (sample 8) was a G→A transversion at nucleotide 7173, resulting in a substitution of lysine for aspartate. Of the three alternative mutations in codon 816, two involved nucleotides 7175 and 7176 (GA→TT) resulting in a substitution of phenylalanine for aspartate (samples 7 and 16). The third mutation was a transversion at nucleotide 7175 (G→T) and resulted in a substitution of tyrosine for aspartate (sample 22). The results are summarized in Table 1 ▶ .
Figure 4.

Results of melting curve analyses and direct sequencing of PCR products in five cases of pediatric UP (Table 1 ▶ ; patients 8, 9, 16, 19, and 22). In figures on the left melting curves of PCR products amplified without PNA (PNA−, dotted line) and with PNA-mediated PCR clamping (PNA+, broken line) are shown. As controls, melting curves of PNA-amplified HMC-1 DNA, heterozygous for c-kit mutation Asp816→Val, are included in every figure (continuous line). Melting curve analysis was performed with hybridization probes c-kit mutation and c-kit anchor. Figures on the right display corresponding results of direct sequencing of PNA− and PNA+ amplification products. In A no exon 17 mutation was observed. B represents 1 of 10 cases with an A→T transition in codon 816. G→A and G→T transversions of codons 815 and 816 are shown in C and D. A combination of the mutations in B and D is shown in E. This form of mutation involving codon 816 has been detected in two patients (Table 1 ▶ , patients 7 and 16).
Table 1.
c-kit Mutations in Pediatric Urticaria Pigmentosa
| Patient no. | c-kit Mutation | Cutaneous involvement | Mutation detection | ||||
|---|---|---|---|---|---|---|---|
| Onset, months | Follow-up, years | Persistence | Seq. | PNA − | PNA + | ||
| 01 | wt | 0 | 8 | Yes | n.d. | − | − |
| 02 | wt | 4 | 5 | Yes | n.d. | − | − |
| 03 | wt | 3 | 5 | Yes | n.d. | − | − |
| 04 | wt | 72 | 3 | Yes | n.d. | − | − |
| 05 | wt | 9 | 5 | Yes | n.d. | − | − |
| 06 | GAC→GTC (Asp816→Val) | 4 | 6 | Yes | − | − | + |
| 07 | GAC→TTC (Asp816→Phe) | 3 | 7 | Yes | − | − | + |
| 08 | AGA→AAA (Arg815→Lys) | 0 | 11 | Yes | − | + | + |
| 09 | GAC→GTC (Asp816→Val) | 12 | 13 | Yes | − | (+) | + |
| 10 | wt | 2 | 12 | No | n.d. | − | − |
| 11 | wt | 0 | 12 | Yes | n.d. | − | − |
| 12 | wt | 24 | 14 | Yes | n.d. | − | − |
| 13 | wt | 2 | 14 | Yes* | n.d. | − | − |
| 14 | GAC→GAT (Asp816→Asp) | 0 | 15 | Yes | − | − | + |
| 15 | GAC→GTC (Asp816→Val) | 1 | 16 | Yes | − | (+) | + |
| 16 | GAC→TTC (Asp816→Phe) | 2 | 18 | Yes | − | − | + |
| 17 | No amplification | 3 | 4 | Yes | — | — | — |
| 18 | GAC→GTC (Asp816→Val) | 3 | 13 | Yes | + | (+) | + |
| 19 | wt | 12 | 6 | Yes | n.d. | − | − |
| 20 | wt | 1 | 6 | Yes | n.d. | − | − |
| 21 | wt | 0 | 8 | Yes | n.d. | − | − |
| 22 | GAC→TAC (Asp816→Tyr) | 3 | 11 | Yes | − | − | + |
| 23 | GAC→GTC (Asp816→Val) | 0 | 20 | No | − | (+) | + |
| 24 | wt | 1 | 16 | No | n.d. | − | − |
| 25 | GAC→GTC (Asp816→Val) | 1 | 14 | No | + | + | + |
| 26 | GAC→GTC (Asp816→Val) | 3 | 13 | No | − | (+) | + |
| 27 | wt | 24 | 12 | No | n.d. | − | − |
| 28 | wt | 1 | 17 | No | n.d. | − | − |
| 29 | wt | 0 | 17 | Yes | n.d. | − | − |
| 30 | wt | 12 | 17 | Yes | n.d. | − | − |
| 31 | GAC→GTC (Asp816→Val) | 4 | 6 | Yes | − | − | + |
| 32 | GAC→GAT (Asp816→Asp) | 2 | 5 | Yes | − | − | + |
| 33 | wt | 12 | 20 | Yes | n.d. | − | − |
| 34 | GAC→GTC (Asp816→Val) | 1 | 8 | Yes | + | + | + |
| 35 | wt | 12 | 18 | No | n.d. | − | − |
| 36 | GAC→GTC (Asp816→Val) | 0 | 15 | No | − | (+) | + |
| 37 | wt | 3 | 3 | Yes | n.d. | − | − |
| 38 | wt | 1 | 11 | Yes | n.d. | − | − |
| Σ | 16/37 | 2,5 (0–72) | 12 (3–20) | 29/38 | 3/16 | 3(9)/16 | 16/16 |
Seq., direct sequencing of PCR products; PNA−, PCR without PNA; PNA+, PNA-mediated PCR-clamping; Asp, aspartate; Val, valine; Phe, phenylalanine; Arg, arginine; Lys, lysine; Tyr, tyrosine; n.d., not done; (+), positive, but very weak signal.
*Symptoms greatly improved.
Without PNA only nine patients presented with aberrant melting curves more or less suspicious for the presence of a c-kit mutation. Highly suspicious were the curves of only three patients [patients 8 (Figure 4C ▶ , oblique arrow), 25, and 34 (Table 1) ▶ ]. In the remaining six patients, all of whom had the mutation Asp816→Val, besides the dominant wild-type peak at around 57°C there was only a very small additional mutation-specific peak at around 63°C [patients 9 (Figure 4B ▶ , oblique arrow), 15, 18, 23, 26, and 36 (Table 1) ▶ ].
Less sensitive is the identification of c-kit mutations by direct sequencing of the amplification products. Only 3 of 14 patients with mutations would have been identified by the presence of double peaks at the relevant positions (patients 18, 25, and 34 of Table 1 ▶ ). In one of these cases a double peak was observed only when the back primer c-kit B was used as the sequencing primer (patient 18, data not shown). In all other patients, mutation-specific sequencing signals were either indistinguishable from the background (data not shown), or totally absent. This can best be demonstrated in patient 9 (Figure 4B) ▶ for whom the melting curve displayed a small additional peak at around 63°C. In contrast, the sequencing result was completely normal. The findings in all 38 patients are summarized in Table 1 ▶ .
Clinical Outcome in Patients with UP
Of the 20 patients with 12 or more years of follow-up, 10 had persistent UP, 9 had transient UP, and 1 further patient experienced a great improvement in his skin condition. A cutoff at 12 years was arbitrarily chosen because no patient had experienced resolution of skin lesions before 12 years of follow-up. Of the nine patients whose skin lesions disappeared, four carried a recurrent GAC→GTC c-kit mutation, leading to the Asp816→Val substitution, whereas the other five patients carried the wild-type alleles. Of the 10 patients with persistent UP after more than 12 years of follow-up, 5 carried the wild-type alleles, 4 showed a c-kit mutation within codon 816, and 1 carried a silent codon mutation GAC→GAT, both coding for aspartate. The patient who experienced a great improvement after 14 years of follow-up carried the wild-type alleles. The data relating to clinical disease outcome are summarized in Table 2 ▶ .
Table 2.
Clinical Outcome in Patients with Pediatric Urticaria Pigmentosa (Only Patients with Follow-Up of 12 to 20 Years Included)
| n | Mean follow-up, years | Activating c-kit mutations | |
|---|---|---|---|
| Persistent UP | 11* | 15.4 | 4/11 (36.4%) |
| Transient UP | 9 | 15.2 | 4/9 (44.4%) |
| Total | 20 | 15.3 | 8/20 (40.0%) |
*Including one patient with marked improvement of the disease.
Screening for c-kit Mutations in Control Cases
Twenty-three cases with varying degrees of MC hyperplasia (no SM) and another 40 cases with various subtypes of acute myeloid leukemia (no associated MC disorders) were investigated as negative controls for exon 17 c-kit mutations. Using the same methods as in the 38 UP cases, no exon 17 c-kit mutations were detected in the 63 (negative) control cases.
Discussion
It has been suggested that the presence of somatic activating codon 816 mutations in MCs in children with UP might be a risk factor for persistence or progression of the disease, 19 and therefore might represent a valuable prognostic marker. The diagnosis of pediatric UP has to be established by histological examination of skin lesions. 36 The molecular analysis of paraffin-embedded tissue for long-term retrospective studies is hampered by fixation- and age-dependent degradation of both mRNA and DNA, especially when very old samples are used. 27,37 Because of the varying numbers of infiltrating mutated MCs, additional procedures may be required to enrich the number of these lesional cells against large amounts of unaffected cells. In nonsolid neoplasms such as UP, laser capture microdissection and amplification of pooled single cells have been used successfully. 38 However, laser capture microdissection is very laborious and time-consuming and the equipment is expensive and available only in some research centers.
In this report we develop a fast, highly sensitive, specific, and reproducible technique that facilitates the detection of a variety of codon 816 mutations of the c-kit proto-oncogene in complete tissue sections. The method was successfully applied with tissue obtained from paraffin blocks that were up to 24-years-old. The assay takes ∼1 hour to perform. Because of the single tube format the risk of contamination is low. The method combines the advantages of PNA-mediated PCR clamping and melting point analysis with two fluorescent-labeled oligonucleotide probes. 31,39
The latter technique, has found numerous applications, including quantitative PCR, identification and subtyping of infectious agents, and detection of point mutations in oncogenes and in hereditary metabolic diseases. 32,33,40,41 In contrast to many currently available methods for genotyping and mutation detection, including oligonucleotide ligation assay, 42 single-strand confirmation polymorphism, 43 allele-specific PCR, 44 and PCR-restriction fragment length polymorphism, 45 this technique overcomes the need for time-consuming analytical postamplification steps. Hybridization probes will also detect other sequence variations covered by the probes, which is not the case for restriction fragment length polymorphism analysis. In our case characteristic and reproducible melting peaks were found also for other mutations than Asp816→Val. However, without PNA the hybridization probes will detect only major components of gene variants. Usually sensitivity is limited and does not exceed 10%, ie, the mutation must be present in at least 10% of the gene copies. 33 Our results are consistent with this. In dilution experiments with DNA extracted from the c-kit mutation-bearing cell line HMC-1 and control DNA, we were unable to detect the mutation in dilutions containing a more than 10-fold excess of wild-type DNA using the hybridization probes alone.
The detection of mutations, present in an extremely small minority of nonmicrodissected (tumor) cells, ie, a needle-in-a-haystack situation, 46 requires techniques to specifically amplify the DNA of mutant alleles. A number of such techniques, including restriction endonuclease-mediated selective-PCR (REMS-PCR) 47 and selective stencil-aided mutation analysis (SAMA) 48 require certain restriction enzymes and/or restriction sites, limiting their general applicability. The method of allele-specific amplification 49 also in combination with PNA-mediated clamping 29,30,50-52 is generally limited to one specific mutation and will fail with related mutations. In addition, it bears the risk of amplification because of abundant readthrough: once the PCR primer has been extended from a wild-type allele, the amplification products are indistinguishable from those generated from the mutant allele.
In this study, as in investigations performed by a number of other groups, we used PNA molecules targeted against the wild-type sequence. However, unlike these other groups we did not use mutation-specific primers targeting the same region. Instead, we used outer PCR primers for a single round of amplifications. Because of the PNA-mediated PCR clamping, the mutant allele was selectively amplified and could easily be characterized by its specific melting peak. Although not directly addressed in this study, four other c-kit mutations were identified in addition to the most prevalent mutation Asp816→Val, each showing a characteristic melting peak in repeated experiments. All of the mutations were found in codons 815 and 816. Notably, besides two mutations described by Longley and colleagues, 19 two new mutations were identified, one of which represented a silent mutation at codon 816.
As far as sensitivity is concerned, most of the various methods can detect somewhere in the range of one mutant allele in the presence of approximately a 1000-fold excess of normal alleles. 30,47,51 The highest sensitivity was reported by Sun and colleagues, 52 who were able to detect as few as 3 mutant alleles in the presence of 10,000 normal alleles. Our results are within this range, the method consistently allowing detection of at least 1 mutant allele among 2000 normal alleles, in some cases we were able to identify 1 mutant allele among 20,000 normal alleles.
A comparison of the numbers of mutations unequivocally detected in our patients by the various methods showed that the combination of PNA-mediated PCR clamping and on-line mutation detection with hybridization probes was superior to conventional PCR with subsequent direct sequencing or melting point analysis, respectively.
When the findings of mutation analysis and long-term follow-up in patients with pediatric UP are compared on the basis of persistent or transient skin lesions, it is seen that codon 816 c-kit mutations were detected in both groups, patients with persistent disease as well as patients with transient disease, with similar frequencies. Especially the detection of activating c-kit mutation Asp816→Val in four of nine patients of the latter group is a remarkable finding, as it is in contrast to the data reported by Longley and colleagues 19 who did not find the mutation in any of the seven patients with transient pediatric UP in their series. False-positive results seem to be sufficiently excluded by the fact that no mutation has been detected in the 63 negative controls. According to our data, identification of activating c-kit mutations such as Asp816→Val does not serve as a independent prognostic marker for the clinical course of skin lesions in pediatric UP. In this regard it has to be emphasized that even though cutaneous lesions may eventually disappear, progression of the disease may not be halted. In some patients internal symptoms of MC disease may even reappear years later, thus underlining the importance of a prolonged follow-up of these patients. Probably, the identification of additional prognostic markers, eg, the abnormal expression of CD2/CD25 in MCs, may help to evaluate the prognosis for individual patients more reliably. 53
In conclusion, we describe the combined application of recently introduced methods for the detection of point mutations in the presence of excess amounts of normal DNA. Further clinical applications may be the detection of early events in carcinogenesis, of circulating metastatic tumor cells, and of minimal residual disease, as well as the identification of targets for drug therapies.
Footnotes
Address reprint requests to Karl Sotlar, M.D., Institute of Pathology, University of Tübingen, Liebermeisterstrasse 8, D-72076 Tübingen, Germany. E-mail: klsotlar@med.uni-tuebingen.de.
Supported in part by the Fondo de Investigaciones Sanitarias (grant FIS 01/0413) and the Comunidad de Madrid (grant CAM 08.1/0023/2000).
References
- 1.Valent P, Horny HP, Escribano L, Longley BJ, Li CY, Schwartz LB, Marone G, Nunez R, Akin C, Sotlar K, Sperr WR, Wolff K, Brunning RD, Parwaresch RM, Austen KF, Lennert K, Metcalfe DD, Vardiman JW, Bennett JM: Diagnostic criteria and classification of mastocytosis: a consensus proposal. Leuk Res 2001, 25:603-625 [DOI] [PubMed] [Google Scholar]
- 2.Lennert K, Parwaresch MR: Mast cells and mast cell neoplasia: a review. Histopathology 1979, 3:349-365 [DOI] [PubMed] [Google Scholar]
- 3.Horny HP, Parwaresch MR, Lennert K: Bone marrow findings in systemic mastocytosis. Hum Pathol 1985, 16:808-814 [DOI] [PubMed] [Google Scholar]
- 4.Stein DH: Mastocytosis: a review. Pediatr Dermatol 1986, 3:365-375 [DOI] [PubMed] [Google Scholar]
- 5.Yarden Y, Kuang WJ, Yang FT, Coussens L, Munemitsu S, Dull TJ, Chen E, Schlessinger J, Francke U, Ullrich A: Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 1987, 6:3341-3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu FH, Ray P, Brown K, Barker PE, Jhanwar S, Ruddle FH, Besmer P: Primary structure of c-kit: relationship with the CSF-1/PDGF receptor kinase family—oncogenic activation of v-kit involves deletion of extracellular domain and C terminus. EMBO J 1988, 7:1003-1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashman LK, Cambareri AC, To LB, Levinsky RJ, Juttner CA: Expression of the YB5.B8 antigen (c-kit proto-oncogene product) in normal human bone marrow. Blood 1991, 78:30-37 [PubMed] [Google Scholar]
- 8.Papayannopoulou T, Brice M, Broudy VC, Zsebo KM: Isolation of c-kit receptor-expressing cells from bone marrow, peripheral blood, and fetal liver: functional properties and composite antigenic profile. Blood 1991, 78:1403-1412 [PubMed] [Google Scholar]
- 9.Reisbach G, Bartke I, Kempkes B, Kostka G, Ellwart J, Birner A, Thalmeier K, Mailhammer R, Bornkamm GW, Ullrich A, Dörmer P: Characterization of hemopoietic cell populations from human cord blood expressing c-kit. Exp Hematol 1993, 21:74-79 [PubMed] [Google Scholar]
- 10.Ogawa M, Matsuzaki Y, Nishikawa S, Hayashi S, Kunisada T, Sudo T, Kina T, Nakauchi H: Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med 1991, 174:63-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orr UA, Avivi A, Zimmer Y, Givol D, Yarden Y, Lonai P: Developmental expression of c-kit, a proto-oncogene encoded by the W locus. Development 1990, 109:911-923 [DOI] [PubMed] [Google Scholar]
- 12.Manova K, Nocka K, Besmer P, Bachvarova RF: Gonadal expression of c-kit encoded at the W locus of the mouse. Development 1990, 110:1057-1069 [DOI] [PubMed] [Google Scholar]
- 13.Koch CA, Anderson D, Moran MF, Ellis C, Pawson T: SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 1991, 252:668-674 [DOI] [PubMed] [Google Scholar]
- 14.Pawson T: Protein-tyrosine kinases. Getting down to specifics. Nature 1995, 373:477-478 [DOI] [PubMed] [Google Scholar]
- 15.Furitsu T, Tsujimura T, Tono T, Ikeda H, Kitayama H, Koshimizu U, Sugahara H, Butterfield JH, Ashman LK, Kanayama Y, Matsuzawa Y, Kitamura Y, Kanamura Y: Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 1993, 92:1736-1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitayama H, Kanakura Y, Furitsu T, Tsujimura T, Oritani K, Ikeda H, Sugahara H, Mitsui H, Kanayama Y, Kitamura Y, Matsuzawa Y: Constitutively activating mutations of c-kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor-dependent hematopoietic cell lines. Blood 1995, 85:790-798 [PubMed] [Google Scholar]
- 17.Sperr WR, Walchshofer S, Horny HP, Fodinger M, Simonitsch I, Fritsche PR, Schwarzinger I, Tschachler E, Sillaber C, Hagen W, Geissler K, Chott A, Lechner K, Valent P: Systemic mastocytosis associated with acute myeloid leukaemia: report of two cases and detection of the c-kit mutation Asp-816 to Val. Br J Haematol 1998, 103:740-749 [DOI] [PubMed] [Google Scholar]
- 18.Longley BJ, Tyrrell L, Lu SZ, Ma YS, Langley K, Ding TG, Duffy T, Jacobs P, Tang LH, Modlin I: Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat Genet 1996, 12:312-314 [DOI] [PubMed] [Google Scholar]
- 19.Longley BJJ, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, Heitjan D, Ma Y: Activating and dominant inactivating c-KIT catalytic domain mutations in distinct clinical forms of human mastocytosis. Proc Natl Acad Sci USA 1999, 96:1609-1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagata H, Worobec AS, Oh CK, Chowdhury BA, Tannenbaum S, Suzuki Y, Metcalfe DD: Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA 1995, 92:10560-10564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akin C, Kirshenbaum AS, Semere T, Worobec AS, Scott LM, Metcalfe DD: Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp Hematol 2000, 28:140-147 [DOI] [PubMed] [Google Scholar]
- 22.Sotlar K, Marafioti T, Griesser H, Theil J, Aepinus C, Jaussi R, Stein H, Valent P, Horny HP: Detection of c-kit mutation Asp 816 to Val in microdissected bone marrow infiltrates in a case of systemic mastocytosis associated with chronic myelomonocytic leukaemia. Mol Pathol 2000, 53:188-193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, Vandenberghe E, Peake I, Reilly J: c-kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 1999, 105:894-900 [DOI] [PubMed] [Google Scholar]
- 24.Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad TG, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y: Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279:577-580 [DOI] [PubMed] [Google Scholar]
- 25.Hongyo T, Li T, Syaifudin M, Baskar R, Ikeda H, Kanakura Y, Aozasa K, Nomura T: Specific c-kit mutations in sinonasal natural killer/T-cell lymphoma in China and Japan. Cancer Res 2000, 60:2345-2347 [PubMed] [Google Scholar]
- 26.Rupp GM, Locker J: Purification and analysis of RNA from paraffin-embedded tissues. Biotechniques 1988, 6:56-60 [PubMed] [Google Scholar]
- 27.Mizuno T, Nagamura H, Iwamoto KS, Ito T, Fukuhara T, Tokunaga M, Tokuoka S, Mabuchi K, Seyama T: RNA from decades-old archival tissue blocks for retrospective studies. Diagn Mol Pathol 1998, 7:202-208 [DOI] [PubMed] [Google Scholar]
- 28.Orum H, Nielsen PE, Egholm M, Berg RH, Buchardt O, Stanley C: Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res 1993, 21:5332-5336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiede C, Bayerdorffer E, Blasczyk R, Wittig B, Neubauer A: Simple and sensitive detection of mutations in the ras proto-oncogenes using PNA-mediated PCR clamping. Nucleic Acids Res 1996, 24:983-984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Behn M, Thiede C, Neubauer A, Pankow W, Schuermann M: Facilitated detection of oncogene mutations from exfoliated tissue material by a PNA-mediated ‘enriched PCR’ protocol. J Pathol 2000, 190:69-75 [DOI] [PubMed] [Google Scholar]
- 31.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ: The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques 1997, 22:176-181 [DOI] [PubMed] [Google Scholar]
- 32.Mangasser-Stephan K, Tag C, Reiser A, Gressner AM: Rapid genotyping of hemochromatosis gene mutations on the LightCycler with fluorescent hybridization probes. Clin Chem 1999, 45:1875-1878 [PubMed] [Google Scholar]
- 33.Elenitoba-Johnson KS, Bohling SD, Wittwer CT, King TC: Multiplex PCR by multicolor fluorimetry and fluorescence melting curve analysis. Nat Med 2001, 7:249-253 [DOI] [PubMed] [Google Scholar]
- 34.Horny HP, Sillaber C, Menke D, Kaiserling E, Wehrmann M, Stehberger B, Chott A, Lechner K, Lennert K, Valent P: Diagnostic value of immunostaining for tryptase in patients with mastocytosis. Am J Surg Pathol 1998, 22:1132-1140 [DOI] [PubMed] [Google Scholar]
- 35.Sotlar K, Selinka HC, Menton M, Kandolf R, Bultmann B: Detection of human papillomavirus type 16 E6/E7 oncogene transcripts in dysplastic and nondysplastic cervical scrapes by nested RT-PCR. Gynecol Oncol 1998, 69:114-121 [DOI] [PubMed] [Google Scholar]
- 36.Wolff K, Komar M, Petzelbauer P: Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res 2001, 25:519-528 [DOI] [PubMed] [Google Scholar]
- 37.Goelz SE, Hamilton SR, Vogelstein B: Purification of DNA from formaldehyde fixed and paraffin embedded human tissue. Biochem Biophys Res Commun 1985, 130:118-126 [DOI] [PubMed] [Google Scholar]
- 38.Sotlar K, Fridrich C, Mall A, Jaussi R, Bültmann B, Valent P, Horny H-P: Detection of c-kit point mutation Asp-816Val in microdissected pooled single mast cells and leukemic cells in a patient with systemic mastocytosis and concomitant chronic myelomonocytic leukemia. Leuk Res 2002, 26:979-984 [DOI] [PubMed] [Google Scholar]
- 39.Orum H: PCR clamping. Curr Issues Mol Biol 2000, 2:27-30 [PubMed] [Google Scholar]
- 40.Barragan E, Bolufer P, Moreno I, Martin G, Nomdedeu J, Brunet S, Fernandez P, Rivas C, Sanz MA: Quantitative detection of AML1-ETO rearrangement by real-time RT-PCR using fluorescently labeled probes. Leuk Lymphoma 2001, 42:747-756 [DOI] [PubMed] [Google Scholar]
- 41.Schalasta G, Arents A, Schmid M, Braun RW, Enders G: Fast and type-specific analysis of herpes simplex virus types 1 and 2 by rapid PCR and fluorescence melting-curve-analysis. Infection 2000, 28:85-91 [DOI] [PubMed] [Google Scholar]
- 42.Porto G, Alves H, Rodrigues P, Cabeda JM, Portal C, Ruivo A, Justica B, Wolff R, De Sousa M: Major histocompatibility complex class I associations in iron overload: evidence for a new link between the HFE H63D mutation, HLA-A29, and non-classical forms of hemochromatosis. Immunogenetics 1998, 47:404-410 [DOI] [PubMed] [Google Scholar]
- 43.Hertzberg MS, McDonald D, Mirochnik O: Rapid diagnosis of hemochromatosis gene Cys282Tyr mutation by SSCP analysis. Am J Hematol 1998, 57:260-261 [DOI] [PubMed] [Google Scholar]
- 44.Smillie D: A PCR-SSP method for detecting the His63Asp mutation in the HFE gene associated with hereditary haemochromatosis. Mol Pathol 1998, 51:232-233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cesarman E, Inghirami G, Chadburn A, Knowles DM: High levels of p53 protein expression do not correlate with p53 gene mutations in anaplastic large cell lymphoma. Am J Pathol 1993, 143:845-856 [PMC free article] [PubMed] [Google Scholar]
- 46.Kwok PY: Finding a needle in a haystack: detection and quantification of rare mutant alleles are coming of age. Clin Chem 2000, 46:593-594 [PubMed] [Google Scholar]
- 47.Fuery CJ, Impey HL, Roberts NJ, Applegate TL, Ward RL, Hawkins NJ, Sheehan CA, O’Grady R, Todd AV: Detection of rare mutant alleles by restriction endonuclease-mediated selective-PCR: assay design and optimization. Clin Chem 2000, 46:620-624 [PubMed] [Google Scholar]
- 48.Lichtenstein AV, Serdjuk OI, Sukhova TI, Melkonyan HS, Umansky SR: Selective ‘stencil’-aided pre-PCR cleavage of wild-type sequences as a novel approach to detection of mutant K-RAS. Nucleic Acids Res 2001, 29:E90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Germer S, Holland MJ, Higuchi R: High-throughput SNP allele-frequency determination in pooled DNA samples by kinetic PCR. Genome Res 2000, 10:258-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kyger EM, Krevolin MD, Powell MJ: Detection of the hereditary hemochromatosis gene mutation by real-time fluorescence polymerase chain reaction and peptide nucleic acid clamping. Anal Biochem 1998, 260:142-148 [DOI] [PubMed] [Google Scholar]
- 51.Myal Y, Blanchard A, Watson P, Corrin M, Shiu R, Iwasiow B: Detection of genetic point mutations by peptide nucleic acid-mediated polymerase chain reaction clamping using paraffin-embedded specimens. Anal Biochem 2000, 285:169-172 [DOI] [PubMed] [Google Scholar]
- 52.Sun X, Hung K, Wu L, Sidransky D, Guo B: Detection of tumor mutations in the presence of excess amounts of normal DNA. Nature Biotechnol 2002, 20:186-189 [DOI] [PubMed] [Google Scholar]
- 53.Escribano L, Diaz-Agustin B, Bellas C, Navalon R, Nunez R, Sperr WR, Schernthaner GH, Valent P, Orfao A: Utility of flow cytometric analysis of mast cells in the diagnosis and classification of adult mastocytosis. Leuk Res 2001, 25:563-570 [DOI] [PubMed] [Google Scholar]