

Abstract
Tumors express more than a single angiogenic growth factor. To investigate the relative impact of fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF) on tumor growth and neovascularization, we generated tumor cell transfectants differing for VEGF and/or FGF-2 expression. Human endometrial adenocarcinoma HEC-1-B-derived Tet-FGF-2 cells that express FGF-2 under the control of the tetracycline-responsive promoter (Tet-off system) were further transfected with a VEGF121 anti-sense (AS-VEGF) cDNA. Next, Tet-FGF-2 and AS-VEGF/Tet-FGF-2 cells were transplanted subcutaneously in nude mice that received tetracycline or not in the drinking water. Simultaneous expression of FGF-2 and VEGF in Tet-FGF-2 cells resulted in fast-growing lesions characterized by high blood vessel density, patency and permeability, and limited necrosis. Blood vessels were highly heterogeneous in size and frequently associated with pericytes. Inhibition of FGF-2 production by tetracycline caused a significant decrease in tumor burden paralleled by a decrease in blood vessel density and size. AS-VEGF expression resulted in a similar reduction in blood vessel density associated with a significant decrease in pericyte organization, vascular patency, and permeability. The consequent decrease in tumor burden was paralleled by increased tumor hypoxia and necrosis. A limited additional inhibitory effect was exerted by simultaneous down-regulation of FGF-2 and VEGF expression. These findings demonstrate that FGF-2 and VEGF stimulate vascularization synergistically but with distinctive effects on vessel functionality and tumor survival. Blockade of either one of the two growth factors results in a decrease in blood vessel density and, consequently, in tumor burden. However, inhibition of the expression of VEGF, but not of FGF-2, affects also vessel maturation and functionality, leading to tumor hypoxia and necrosis. Our experimental model represents an unique tool to investigate anti-neoplastic therapies in different angiogenic environments.
New blood vessel formation and differentiation are important steps in tumor progression. 1 Tumor angiogenesis is controlled by positive and negative modulators produced by neoplastic, stromal, and tumor-infiltrating cells. 2 Individual tumors express a variety of angiogenic factors whose relative production can change throughout time. 3 Among them, fibroblast growth factor-2 (FGF-2) was one of the first identified angiogenic growth factors. 4,5 FGF-2 is a heparin-binding protein that shows angiogenic activity in different experimental models. 6 In situ hybridization and immunolocalization experiments have shown the presence of FGF-2 mRNA and/or protein in neoplastic cells within human tumors. 7-10 Anti-sense cDNAs for FGF-2 and FGF receptor (FGF-R)-1 inhibit neovascularization and growth of human melanomas in nude mice. 11 A significant correlation between the presence of FGF-2 in cancer cells and advanced tumor stage has been reported. 12-14 Moreover, FGF-2 is detectable in urine of patients with a wide spectrum of cancers 15,16 and in cerebrospinal fluid of children with brain tumors. 17 Also, the anti-angiogenic activity of interferon-α/β appears to be related, at least in part, to the capacity to down-regulate FGF-2 expression. 18 These data suggest that FGF-2 production and release may occur in vivo and may influence solid tumor growth and neovascularization. 19-22 Relevant to this point is the observation that a secreted FGF-binding protein that mobilizes stored extracellular FGF-2 can serve as an angiogenic switch for different tumor cell lines, including squamous cell carcinoma and colon cancer cells. 23
Vascular endothelial growth factor (VEGF), also known as vascular permeability factor, appears to play a major role in tumor neovascularization. 24,25 VEGF acts through its tyrosine kinase receptors VEGF-R-1/Flt-1 and VEGF-R-2/KDR/Flk-1 to modulate motility and proliferation of endothelial cells and vascular permeability. 26,27 The VEGF gene encodes five alternative spliced isoforms 26,27 that show distinct biochemical features and exert unique functions in tumor vascularization. 28,29 VEGF and VEGF-R antagonists, including neutralizing antibodies, 30,31 anti-sense cDNA, 32 dominant-negative receptor mutants, 33 and VEGF-R tyrosine-kinase inhibitors 34 affect tumor growth and vascularization in different experimental models. Also, VEGF levels in tumor biopsies correlate with blood vessel density of the neoplastic tissue and may be of prognostic significance. 35,36 Furthermore, VEGF has been described in the biological fluids of patients with malignant neoplasia. 37
The capacity of tumor cells to express various angiogenic factors has profound implications for the understanding of tumor angiogenesis per se and for the design of efficacious anti-angiogenic therapies. However, few studies have investigated the impact of the expression of multiple angiogenic factors on tumor vascularization and response to anti-angiogenic intervention. As far as FGF-2 and VEGF are concerned, targeting FGF-binding protein with specific ribozymes inhibits the growth and vascularization of xenografted tumors in mice 23 despite the high levels of VEGF produced by these cells. 38 Recently, we have shown that constitutive 39,40 or tetracycline-regulated 41 FGF-2 overexpression causes a significant increase in the angiogenic activity and tumorigenic capacity of the VEGF-producing human endometrial adenocarcinoma HEC-1-B cell line. 41 These data suggest that modulation of FGF-2 expression may allow a fine tuning of the angiogenesis process even in the presence of VEGF. 38
In the present study, we generated stable HEC-1-B cell transfectants differing for VEGF and/or FGF-2 expression to investigate the relative impact of the two growth factors on tumor growth and neovascularization. HEC-1-B-derived Tet-FGF-2 cells 41 that express FGF-2 under the control of the tetracycline-responsive promoter (Tet-off system) were further transfected with a VEGF121 anti-sense (AS) cDNA. Transfectants (AS-VEGF/Tet-FGF-2 cells) constitutively express reduced levels of VEGF and high levels of FGF-2 when maintained in the absence of tetracycline. Tet-FGF-2 and AS-VEGF/Tet-FGF-2 cells were evaluated for their angiogenic and tumorigenic capacity in the absence or in the presence of tetracycline. We describe that FGF-2 and VEGF affect tumor growth and angiogenesis synergistically. Inhibition of either one of the two growth factors results in a significant reduction in tumor burden and vascularization. However, FGF-2 and VEGF affect vessel functionality and maturation differently. This has implications for the development of therapeutic interventions based on anti-angiogenic approaches.
Materials and Methods
Cell Cultures and Transfection
Human endometrial adenocarcinoma HEC-1-B cells were obtained from American Type Culture Collection (Rockville, MD). Tet-FGF-2 15H cells, originated by transfection of HEC-1-B cells with the human FGF-2 cDNA under the control of the tetracycline-responsive promoter (Tet-off system), produce and release significant amounts of FGF-2 whose expression is hampered by tetracycline treatment. 41
To generate stable AS-VEGF transfectants (AS-VEGF/Tet-FGF-2 cells), Tet-FGF-2 15H cells (3.0 × 10 5 cells/100-mm dish) were transfected with 80 μg of lipofectamine added with 4 μg of pcDNA-3 expression vector harboring the VEGF121 anti-sense cDNA 42 (kindly provided by G. Viglietto, University of Naples, Naples, Italy). After 20 hours, G418 sulfate antibiotic was added to the culture medium at 500 μg/ml. G418-resistant AS-VEGF/Tet-FGF-2 clones were isolated, expanded, and tested for AS-VEGF mRNA expression. Mock cells (Tet-FGF-2 cells) were transfected with the empty pcDNA-3 vector and selected as above. Cells were grown in Eagle’s minimal essential medium supplemented with 1% nonessential amino acids, 1% sodium pyruvate, and 10% fetal bovine serum.
To evaluate their growth rate in vitro, clones were seeded at 25,000 cells/well in 24-well plates in complete cell culture medium. At the indicated time points, cells were trypsinized and counted.
AS-VEGF mRNA Expression
Northern blot analysis of total RNA isolated from Tet-FGF-2 and AS-VEGF/Tet-FGF-2 transfectants and from tumor xenografts was performed according to standard procedures. Briefly, total RNA samples (20 μg/lane) were run on formaldehyde/agarose gel, blotted overnight to a nylon membrane, and fixed under ultraviolet light. Uniform loading of the gels was assessed by methylene blue staining of the filter. Blots were hybridized with riboprobes to AS-VEGF mRNA labeled with digoxigenin-UTP by in vitro transcription with T7 RNA polymerase DIG RNA labeling kit (Roche Applied Science, Basel, Switzerland). Hybridized filters were stained with anti-digoxigenin alkaline phosphatase-conjugated antibody followed by enzyme-catalyzed color reaction with 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium salt (Nucleic Acid Detection Kit, Roche Applied Science).
VEGF and FGF-2 Production by Tet-FGF-2 and AS-VEGF/Tet-FGF-2 Transfectants
For the determination of VEGF production, Tet-FGF-2 and AS-VEGF/Tet-FGF-2 cells were incubated in fresh medium for 2 days. Then, conditioned media were collected and VEGF content was quantified using the Quantikine VEGF Immunoassay (R&D Systems, Minneapolis, MN). Data were normalized for the protein content of the cell extracts. Also, conditioned media were concentrated on Centricon YM-10 filters (Millipore, Billerica, MA), 60 μg of proteins were run on sodium dodecyl sulfate-15% polyacrylamide gel electrophoresis, electrophoretically transferred to nitrocellulose membranes, and probed with anti-VEGF antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Immunocomplexes were visualized by chemiluminescence using the ECL Western blotting kit (Amersham Biosciences, Amersham, UK) according to the manufacturer’s instructions.
For the determination of FGF-2 production, cells were incubated in fresh medium in the absence or in the presence of 10 ng/ml of tetracycline (Sigma Chemical Co., St. Louis, MO). After 3 days, cells were washed with phosphate-buffered saline (PBS), scraped with a rubber policeman, sonicated on ice at 50 W for 20 seconds in PBS, and centrifuged at 10,000 × g for 10 minutes. Next, 500-μg aliquots of the cell supernatants were loaded onto 0.1-ml heparin-Sepharose columns. After a 0.5-mol/L NaCl wash, resin beads were boiled in reducing sample buffer, samples were run on sodium dodecyl sulfate-15% polyacrylamide gel electrophoresis and analyzed by Western blotting with a rabbit polyclonal anti-FGF-2 antibody (Santa Cruz Biotechnology).
Endothelial Cell Growth Assay
Cultures of the different clones were grown in the absence or in the presence of tetracycline for 3 days. Then, all cultures were incubated for 3 more days in serum-free medium in the absence of tetracycline. 41 Conditioned media were prepared as described above. Fetal bovine aortic endothelial GM 7373 cells (obtained from the Institute for Medical Research, Camden, NJ) were maintained in Eagle’s minimal essential medium containing 10% fetal bovine serum, vitamins, and essential and nonessential amino acids. Cell proliferation assay was performed as described. 41 Briefly, GM 7373 cells were seeded at 75,000 cells/cm2 in 24-well dishes. After overnight incubation, cells were incubated for 24 hours in fresh medium containing 0.4% fetal bovine serum in the absence or in the presence of aliquots (from 12 to 100 μg/ml) of the different conditioned media. At the end of incubation, cells were trypsinized and counted in a Burker chamber. Cell proliferation was expressed as percentage of the proliferation measured in cells grown in 10% fetal bovine serum, equal to 1.0 cell population doublings.
Tumor Growth Assay
Female NCr-nu/nu mice were obtained from the animal production colony of the National Cancer Institute, Frederick Cancer Research and Development Center (Frederick, MD) and used when 6 to 8 weeks old. Tumor cells were harvested by brief exposure to 0.25% trypsin/0.02% ethylenediaminetetraacetic acid, washed twice, and suspended in Hanks’ balanced salt solution. Mice were given a subcutaneous injection in the dorsal scapular region of 1 × 106 cells suspended in 0.2 ml of Hanks’ balanced salt solution. The tumor mass was measured twice a week with calipers, and tumor volume (mm3) was estimated by the formula for ellipsoid: (length × width2/2. When indicated, animals received tetracycline in the drinking water (2.0 mg/ml) with a change every other day throughout the whole experimental period starting 4 days before tumor cell transplantation. 41 No significant differences were observed in response to tetracycline when the antibiotic was administered starting on day of tumor cell implantation (data not shown). At sacrifice, tumors were harvested and processed for mRNA expression analysis and immunohistological analysis (see below). When indicated, tumor-bearing mice were treated with the monoclonal neutralizing antibody (DC101) targeting the murine VEGF receptor-2/Flk-1 (a gift from D. J. Hicklin, ImClone Systems Inc., NY). Mice were given doses of DC101 (800 μg/injection) or vehicle once every 4 days throughout the whole experimental period starting 1 day after cell implantation.
Tumor Histology
Tumor Necrosis and Mononuclear Cell Infiltrate
Xenograft specimens were embedded in OCT-compound and immediately frozen. Five-μm sections were obtained with a cryostat microtome and stained with H&E. The percentage of necrotic parenchyma was quantified by computerized image analysis of digitized whole tissue sections captured with a stereomicroscope at ×2.5 magnification (two slides per tumor, five animals per group) using the Image-Pro Plus software (Media Cybernetics, Silver Spring, MD). For evaluation of mononuclear phagocyte infiltrate, sections were fixed with ice-cold acetone for 10 minutes, rinsed in PBS, and incubated for 10 minutes with 0.006% H2O2 in absolute methanol to block endogenous peroxidase. Then, a 30-minute preincubation with diluted normal serum was followed by 90 minutes of incubation with a 1:100 dilution of goat polyclonal anti-CD11b antibody (M-19, Santa Cruz Biotechnology) in a humidified chamber. Sections were then exposed to biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) and to avidin-biotin-peroxidase complex (ABComplex HRP; DAKO Corp., Carpinteria, CA) for 30 minutes. Peroxidase color reaction was developed with 3-amino-9-ethyl-carbazole (Sigma) and the sections were lightly counterstained with Mayer’s hematoxylin. CD11b+ cells were counted in the most vascularized areas (×400 field, three fields per tumor) (see below).
CD31 Staining and Computer-Assisted Analysis of Tumor Vessels
To evaluate microvessel density, frozen sections from each tumor were immunostained with the rat monoclonal anti-mouse CD31 antibody MEC 13.3 (kindly provided by A. Vecchi, Istituto Mario Negri, Milano, Italy) for the detection of blood vessels, accordingly to the procedure described above. Sections were examined at low-power magnification to identify the areas with the highest density of CD31+ vessels. In each case, the most vascularized area was selected, and microvessels in a ×400 field were counted. Because part of the xenografts were characterized by a poorly vascularized central zone of necrosis, CD31+ vessels were counted within the tumor parenchyma at the periphery of the lesion where angiogenesis is more robust. 43 No significant differences in microvessel counts were observed between paired sections of individual tumors.
For computer-assisted analysis of tumor vessels, CD31-stained sections were captured with a digital camera at ×20 magnification and morphometric analysis was performed on the Image-Pro Plus software. At least three different 0.4-mm2 fields in each section were examined for vessel caliber and for relative area occupied by tumor blood vessels.
CD31 and α-SMA Immunostaining
Tumor sections were double-labeling immunostained to visualize CD31+ endothelial cells and α-smooth muscle actin (α-SMA)+ cells. To this purpose, frozen sections were blocked with 5.0% goat serum in PBS containing 1.0% bovine serum albumin and 0.1% Triton X-100. Then, sections were incubated for 1 hour with a 1:50 dilution of R-phycoerythin (RPE)-conjugated anti-CD31 antibody (BD PharMingen, San Diego, CA) plus a 1:100 dilution of murine monoclonal anti-α-SMA antibody (clone 1A4, Sigma) followed by a 1-hour incubation with a 1:500 dilution of fluorescein isothiocyanate-conjugated anti-mouse secondary antibody (Molecular Probes, Eugene, OR). Confocal microscopy was performed using a three laser confocal microscope (Radiance 2100; BioRad, Hercules, CA). Fluorescent signals from single optical sections were sequentially acquired and analyzed by Paint Shop Pro software (JascSoftware, Eden Prairie, MN).
Blood Vessel Permeability
To assess tumor blood vessel permeability, tumor-bearing mice were injected in the tail vein with fluorophore Hoechst 33342 (40 mg/kg body wt, Sigma) and killed 2 minutes later. 44 Tumor frozen sections were serially incubated with anti-CD31 antibody, biotinylated secondary antibody, and Texas Red Avidin D (Vector Laboratories). CD31 immunofluorescence and Hoechst 33342 fluorescence images of the same microscopic field were captured with a digital camera under a fluorescence microscope at ×20 magnification. Then, the two images were overlaid using Paint Shop Pro software.
HIF-1α Staining
Frozen sections were incubated with a 1:8000 dilution of monoclonal anti-hypoxia-inducible factor-α (HIF-1α) antibody (ab8366; Abcam Limited, Cambridge, UK), processed according to the Animal Research Peroxidase Kit (DAKO Corp.) manufacturer’s instructions, and lightly counterstained with Mayer’s hematoxylin. Semiquantitative scoring of HIF-1α immunostaining was performed on the whole tumor sections (five tumors per group) using an arbitrary scale from 0 (negative) to 4+ (strongly positive).
Results
Anti-Sense VEGF cDNA Transfection of Tet-FGF-2 Cells
The single expression vector version of the Tet-off system was used to modulate FGF-2 production in HEC-1-B cells. 41 We generated stable Tet-FGF-2 cells in which FGF-2 expression is under the control of the tetracycline-responsive promoter (Tet-off system). Also, Tet-FGF-2 cells secrete significant amounts of VEGF whose levels are not affected by FGF-2 expression. 41 On this basis, Tet-FGF-2 cells were further transfected with a AS-VEGF cDNA (see Materials and Methods). Stable transfectants were obtained and AS-VEGF mRNA expression was verified by Northern blotting. As shown in Figure 1A ▶ , representative AS-VEGF transfected cells (AS-VEGF/Tet-FGF-2 A5 and C1 clones), but not mock-transfected cells (Tet-FGF-2 A4 and C7 clones), express significant steady-state levels of AS-VEGF mRNA. Western blot analysis (Figure 1C) ▶ and enzyme-linked immunosorbent assay of the conditioned media confirmed that anti-sense expression was paralleled by an ∼40 to 50% decrease in the levels of VEGF121 and VEGF165 isoforms in AS-VEGF/Tet-FGF-2 cells (enzyme-linked immunosorbent assay values ranging between 1.8 and 2.7 ng total VEGF/48 hours/106 cells) when compared to Tet-FGF-2 cells (ranging between 2.9 and 3.4 ng total VEGF/48 hours/106 cells). Tetracycline administration did not affect VEGF levels in all of the clones examined (data not shown). In contrast, all of the clones expressed similar levels of FGF-2 protein that were significantly reduced after a 3-day incubation with 10 ng/ml of tetracycline (Figure 1D) ▶ . Proliferation of fetal bovine aortic endothelial GM 7373 cells was differently enhanced by incubation with the conditioned medium from Tet-FGF-2 A4 or AS-VEGF/Tet-FGF-2 C1 clones, the former one being the most effective. For both clones, down-regulation of FGF-2 expression by tetracycline caused a significant reduction of the mitogenic activity of their conditioned media (Figure 1E) ▶ . Tetracycline had no direct effect on endothelial cell proliferation (data not shown).
Figure 1.

VEGF and FGF-2 expression by Tet-FGF-2 and AS-VEGF/Tet-FGF-2 transfectants. Tet-FGF-2 15H cells were transfected with the pcDNA-3 expression vector harboring the VEGF121 anti-sense cDNA. A: Stable transfectants (AS-VEGF/Tet-FGF-2 A5 and C1 clones) and mock-transfected cells (Tet-FGF-2 A4 and C7 clones) were analyzed for AS-VEGF mRNA steady-state levels by Northern blotting of total RNA (20 μg/lane) using a digoxigenin-UTP-labeled AS-VEGF riboprobe. B: Uniform loading of the gel was assessed by methylene blue staining of the filter. C: Serum-free conditioned media from the different clones (60 μg of protein) were probed with anti-VEGF antibodies by Western blotting showing decreased levels of the secreted forms of VEGF in AS-VEGF/Tet-FGF-2 cells. D: Lysates (500 μg) from cells grown in the absence (−) or in the presence (+) of tetracycline were loaded onto 0.1-ml heparin-Sepharose columns and bound material was probed with anti-FGF-2 antibodies in a Western blot. All of the clones express the 24-, 22-, and 18-kd FGF-2 isoforms whose expression is hampered by tetracycline treatment. E: Endothelial GM 7373 cells were incubated for 24 hours with increasing concentrations of the conditioned media from untreated and tetracycline-treated (+tet) Tet-FGF-2 A4 and AS-VEGF/Tet-FGF-2 C1 clones. At the end of incubation cells were trypsinized and counted. Data are the mean ± SE of four determinations.
Mock and AS-VEGF transfectants showed a similar replication rate in vitro under standard culture conditions (Figure 2A) ▶ and their proliferation was not affected by tetracycline (data not shown). Nevertheless, AS-VEGF/Tet-FGF-2 clones grew poorly after transplantation in the flank of nude mice when compared to Tet-FGF-2 clones (Figure 2B) ▶ . Accordingly, all AS-VEGF/Tet-FGF-2 xenografts express significant levels of AS-VEGF mRNA that was instead undetectable in Tet-FGF-2 lesions (Figure 2B ▶ , inset). On this basis, the terms “AS-VEGF/Tet-FGF-2 cells” and “Tet-FGF-2 cells” will refer to AS-VEGF/Tet-FGF-2 C1 and Tet-FGF-2 A4 clones, respectively, throughout the article unless specified otherwise.
Figure 2.

In vitro and in vivo growth properties of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 transfectants. A: AS-VEGF/Tet-FGF-2 A5 and C1 clones (▵, ▴) and mock Tet-FGF-2 C7 and A4 clones (○, •) were seeded at 25,000 cells/well in 24-well plates in complete cell culture medium. At the indicated time points, cells were trypsinized and counted. B: Cells were transplanted subcutaneously in nude mice (1 × 106 cells/animal) and tumor growth was reported. Each point is the mean ± SE of five tumors. Statistical analysis of tumor volume by orthogonal comparison analysis of variance at day 45 showed a significant difference (P < 0.001) between Tet-FGF-2 and AS-VEGF/Tet-FGF-2 clones (**). At sacrifice, three Tet-FGF-2 A4 tumors and three AS-VEGF/Tet-FGF-2 C1 tumors were analyzed for AS-VEGF mRNA expression (arrow) by Northern blotting (inset). Uniform loading of the gel was assessed by methylene blue staining of the filter.
Growth of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 Xenografts
Tetracycline administration in the drinking water completely suppresses FGF-2 expression in Tet-FGF-2 cells transplanted in nude mice. 41 On this basis, we have now assessed the impact of the inhibition of FGF-2 and/or VEGF expression on the growth of the Tet-FGF-2 model. Nude mice were randomized to receive tetracycline or not in the drinking water. Four days later each group was further randomized to receive a subcutaneous transplant of Tet-FGF-2 or AS-VEGF/Tet-FGF-2 cells and tumor growth was monitored.
Tetracycline administration caused a significant decrease (P < 0.01) in the growth rate of Tet-FGF-2 xenografts (Figure 3) ▶ . The growth rate of AS-VEGF/Tet-FGF-2 lesions was even slower than that of tetracycline-treated Tet-FGF-2 tumors (P < 0.01) and it was only marginally affected by the addition of tetracycline (Figure 3) ▶ . Thus, both FGF-2 down-regulation by tetracycline administration and VEGF down-regulation by AS-VEGF expression inhibit tumor growth. This is further slightly suppressed by the combination of the two inhibitory stimuli. Similar results were obtained in three independent experiments performed on the same clones or when AS-VEGF/Tet-FGF-2 A5 and Tet-FGF-2 C7 clones were used (not shown).
Figure 3.

Effect of tetracycline on Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumor growth. Nude mice were transplanted subcutaneously with 1 × 106 Tet-FGF-2 A4 cells (○, •) or AS-VEGF/Tet-FGF-2 C1 cells (▵, ▴). Animals were left untreated (•, ▴) or received 2 mg/ml of tetracycline in the drinking water throughout the whole experimental period starting 4 days before cell transplantation (○, ▵). Each point is the mean ± SE of five tumors. The results are representative of three independent experiments. Statistical analysis of tumor volumes by analysis of variance followed by Tukey-Kramer posthoc test showed a significant difference (P < 0.01) between the untreated Tet-FGF-2 group and all of the other groups (day 43, **) and between tetracycline-treated Tet-FGF-2 group and the two AS-VEGF/Tet-FGF-2 groups (day 43 and day 64, *). No significant difference was observed between untreated and tetracycline-treated AS-VEGF/Tet-FGF-2 groups at any time point.
No morphological differences were observed in H&E-stained sections between Tet-FGF-2 and AS-VEGF/ Tet-FGF-2 tumors. They showed the features of poorly differentiated adenocarcinomas with papillary and adenomatous pattern 39 both in the absence and in the presence of tetracycline treatment (data not shown). However, AS-VEGF/Tet-FGF-2 lesions were characterized by large areas of necrotic parenchyma when compared to Tet-FGF-2 tumors (Figure 4 ▶ ; A to C). FGF-2 down-regulation by tetracycline administration had no significant effect on tumor necrosis in both tumor types, thus indicating that VEGF but not FGF-2 suppression affects tumor survival in vivo.
Figure 4.
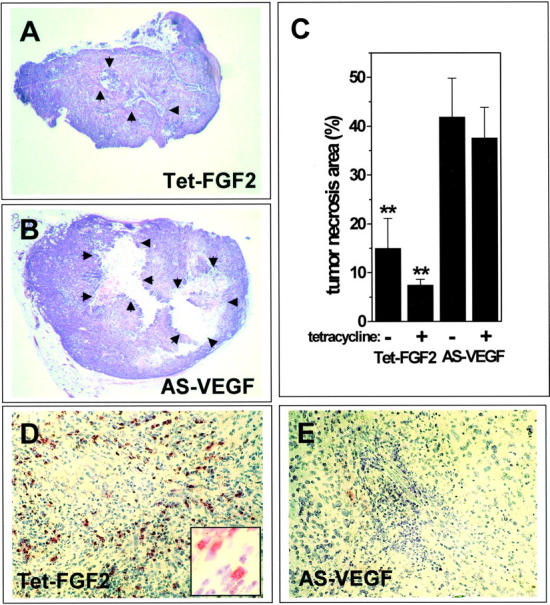
Tumor necrosis and mononuclear cell infiltrate in Tet-FGF-2 and AS-VEGF/Tet-FGF-2 xenografts. At the end of the experimentation (shown in Figure 3 ▶ ), animals were sacrificed and tumor sections were stained with H&E (A, B). Note the large necrotic area at the center of AS-VEGF/Tet-FGF-2 lesions (arrows in B) compared to the limited necrosis observed in Tet-FGF-2 tumors (A). In C, the percentage of necrotic tumor parenchyma was quantified by computerized image analysis of tissue sections (two slides per tumor, five animals per group) from Tet-FGF-2 and AS-VEGF/Tet-FGF-2 xenografts treated (+) or not (−) with tetracycline. **, Statistically different from AS-VEGF (P < 0.05 or better). D and E: CD11b immunostaining of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors. Large areas of CD11b+ monocyte/macrophage infiltrates are observed in Tet-FGF-2 xenografts (D) but not in AS-VEGF/Tet-FGF-2 lesions (E). A similar immunostaining was observed in all tumors examined (five tumors per group). Original magnifications: ×2.5 (A, B); ×20 (D, E); ×63 (inset in D).
Interestingly, Tet-FGF-2 xenografts showed a higher mononuclear phagocyte content than AS-VEGF/Tet-FGF-2 lesions, as indicated by CD11b immunostaining of tumor sections. Indeed, large areas of CD11b+ cell infiltrate were observed within the stroma of Tet-FGF-2 xenografts but not of AS-VEGF/Tet-FGF-2 lesions (Figure 4, D and E) ▶ . FGF-2 down-regulation by tetracycline administration had no effect on mononuclear phagocyte infiltrates in both tumor types. Accordingly, counting of the monocytes infiltrating the vascular hot spots (see below) showed the presence of 67 ± 20 and 52 ± 7 CD11b+ cells per field in control and tetracycline-treated Tet-FGF-2 lesions and of 5 ± 1 and 6 ± 1 CD11b+ cells per field in control and tetracycline-treated AS-VEGF/Tet-FGF-2 lesions, respectively (three tumors per group).
Neovascularization of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 Xenografts
The above data prompted us to assess the impact of the modulation of FGF-2 and/or VEGF expression on tumor vascularization. To this purpose, vascular density was evaluated in CD31-immunostained tumor sections (Figure 6, A and B) ▶ . Because part of the tumors were characterized by a poorly vascularized central zone of necrosis (see above), CD31+ vessels were counted within areas of viable tumor parenchyma at the periphery of the lesion where angiogenesis is more robust. 43 As shown in Figure 5A ▶ , tetracycline administration causes a significant decrease in blood vessel density of Tet-FGF-2 xenografts to values similar to those observed in AS-VEGF/Tet-FGF-2 lesions given tetracycline or not. Also, striking differences in the morphology of CD31+ microvessels were observed in Tet-FGF-2 lesions of control group when compared to the other experimental groups (Figure 6, A and B) ▶ . Indeed, computer-assisted image analysis of CD31+ vessels indicated that the microvasculature of Tet-FGF-2 control lesions was characterized by a remarkable heterogeneity in lumen diameter with numerous large-caliber vessels (Table 1) ▶ . In contrast, all tumors originated by Tet-FGF-2 cells in tetracycline-treated animals or by AS-VEGF/Tet-FGF-2 cells in control and tetracycline-treated animals were characterized by more homogenous small-caliber vessels. This resulted in a significant decrease in the mean value of blood vessel diameter (Table 1) ▶ with 90% reduction of the total vascular area in tetracycline-treated Tet-FGF-2 lesions and in untreated AS-VEGF/Tet-FGF-2 lesions when compared to control Tet-FGF-2 tumors (Figure 5B) ▶ . The simultaneous inhibition of FGF-2 and VEGF expression (AS-VEGF/Tet-FGF-2 + tetracycline) caused a further decrease in total vascular area to values 95% lower than those measured in Tet-FGF-2 tumors (Figure 5B) ▶ . Similar results were obtained when tumors originated by AS-VEGF/Tet-FGF-2 A5 and Tet-FGF-2 C7 clones were analyzed (data not shown).
Figure 6.
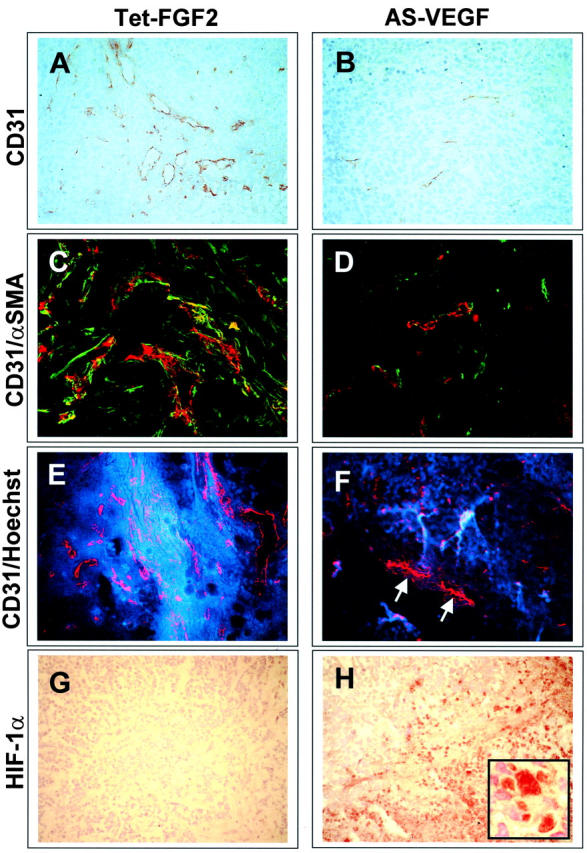
Immunohistochemical characterization of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors from tetracycline-untreated mice. Representative images from Tet-FGF-2 (A, C, E, G) and AS-VEGF/Tet-FGF-2 (B, D, F, H) lesions are shown. See Figure 5 ▶ for quantitative analysis performed on both tumor types in the absence and in the presence of tetracycline treatment. A and B: Tumor sections were processed for CD31 immunostaining. Note the large-sized vessels in Tet-FGF-2 tumors (A) when compared to small-caliber microvessels in AS-VEGF/Tet-FGF-2 lesions (B). Small-caliber microvessels were observed also in all of the Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors from tetracycline-treated animals (not shown). C and D: Tumor sections were double-labeling immunostained to visualize CD31+ endothelial cells (in red) and α-SMA+ cells (in green) by confocal microscopy. Note the numerous α-SMA+ cells frequently associated with CD31+ blood vessels in Tet-FGF-2 lesions (C) when compared to AS-VEGF/Tet-FGF-2 tumors (D). E and F: Animals were injected intravenously 2 minutes before sacrifice with fluorescent Hoechst 33342. A remarkable extravasation of the dye (in blue) from patent CD31+ vessels (in red) was observed in Tet-FGF-2 lesions (E) when compared to AS-VEGF/Tet-FGF-2 tumors (F). Note the nonpatent Hoechst 33342-negative/CD31+ vessels frequently detected in AS-VEGF/Tet-FGF-2 lesions (arrows in F). G and H: HIF-1α immunostaining of Tet-FGF-2 (G) and AS-VEGF/Tet-FGF-2 (H) tumors showing a strong, mainly nuclear (inset), HIF-1α expression in AS-VEGF-transfected lesions. Tetracycline administration had no significant effect on α-SMA and HIF-1α immunostaining and on Hoechst 33342 extravasation in both Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors when compared to untreated lesions (data not shown). Original magnifications: ×20 (A–H); ×63 (inset in H).
Figure 5.

Vascularization of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors. Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumor sections from control (−) and tetracycline-treated (+) nude mice (see Figure 3 ▶ ) were processed for CD31 immunostaining. For each section, the most vascularized area within the tumor parenchyma at the periphery of the lesion was selected and microvessels in a ×400 field were counted (A). Total CD31+ blood vessel area (B) and total pericyte/myofibroblast α-SMA+ area (C) in 0.4-mm2 fields of viable tumor parenchyma (three fields per tumor) were quantified by computerized image analysis. D: Semiquantitative scoring of HIF-1α immunostaining was performed on the whole tumor sections using an arbitrary scale from 0 (negative) to 4+ (strongly positive). n = 5 mice per group. **, P < 0.05 or better.
Table 1.
Quantitative Computer-Assisted Analysis of Blood Vessel Size in Tet-FGF-2 and AS-VEGF/Tet-FGF-2 Xenografts
| Tetracycline | Number of measurements | Blood vessel diameter, μm | Large-sized vessels, area > 1000 μm2 (% of total) | ||
|---|---|---|---|---|---|
| Range | Mean ± SE | ||||
| Tet-FGF-2 cells | − | 766 | 5.1–172.8 | 23.5 ± 8.4 | 20.0 ± 3.0 |
| + | 950 | 4.9–63.4 | 13.1 ± 4.2 | 3.0 ± 1.0 | |
| AS-VEGF/Tet-FGF-2 cells | − | 622 | 4.8–47.8 | 11.6 ± 3.6 | 3.0 ± 1.5 |
| + | 587 | 5.2–29.4 | 9.5 ± 3.2 | 0.8 ± 0.4 | |
Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors from mice treated with tetracycline or not (five tumors per group) were immunostained with anti-CD31 antibody. Then, quantitative computer-assisted image analysis was performed on digitized images of the indicated numbers of CD31+ tumor blood vessels. The percentage of large caliber vessels (vessel area > 1,000 μm2) was also calculated.
Functional Features of Tumor Blood Vessels in Tet-FGF-2 and AS-VEGF/Tet-FGF-2 Xenografts
The data indicate that both AS-VEGF expression and FGF-2 down-regulation reduce the tumor burden and have a similar impact on tumor blood vessel density, heterogeneity, and size. However, only AS-VEGF expression was accompanied by a significant increase in tumor necrosis. This observation prompted us to assess whether inhibition of VEGF and/or FGF-2 expression differently affects blood vessel maturation and functionality.
To assess the maturation of blood vessels in Tet-FGF-2 and AS-VEGF/Tet-FGF-2 xenografts, we performed a double-labeling immunostaining of tumor sections to visualize CD31+ endothelial cells and α-SMA+ cells (pericytes/myofibroblasts) by confocal microscopy. 45 As shown in Figure 6C ▶ , the majority of CD31+ vessels in Tet-FGF-2 lesions were associated with α-SMA+ pericytes. Also, numerous α-SMA+ cells with no apparent association with blood vessels, possibly representing myofibroblasts, 45 were detectable in these lesions. In contrast, very few α-SMA+ cells could be found in AS-VEGF/Tet-FGF-2 xenografts (Figure 6D) ▶ where they appeared as isolated cells scattered within the tumor parenchyma with no or very limited association with tumor blood vessels. Tetracycline administration did not affect the distinguishing features of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 xenografts (data not shown). This was confirmed by the quantification of α-SMA+ cells by computerized image analysis that demonstrated a fourfold to fivefold decrease of the total α-SMA+ area in both control and tetracycline-treated AS-VEGF/Tet-FGF-2 lesions when compared to their Tet-FGF-2 counterparts (Figure 5C) ▶ . Similar results were obtained when tumor sections were stained for desmin immunoreactivity (data not shown), a different tumor pericyte marker. 45
As an index of blood vessel functionality, vascular patency was measured by intratumor detection of the cell-permeable fluorophore Hoechst 33342 injected into the tail vein. Tet-FGF-2 tumors contained patent vessels (Figure 6E) ▶ . In contrast, nonpatent Hoechst 33342-negative/CD31+ vessels were frequently detected in AS-VEGF/Tet-FGF-2 lesions (Figure 6F ▶ , arrows). Also, a remarkable increase in the extravasation of the fluorescent dye from patent vessels was observed in Tet-FGF-2 lesions when compared to AS-VEGF/Tet-FGF-2 tumors (Figure 6, E and F) ▶ . This occurred independently of tetracycline administration in both types of tumors (data not shown). This suggests that AS-VEGF expression, but not FGF-2 down-regulation, strongly affects blood vessel permeability. Indeed, 5 minutes after intravenous injection, we observed an extensive interstitial diffusion of high molecular weight fluorescein isothiocyanate-labeled dextran into the parenchyma of Tet-FGF-2 tumors but not of AS-VEGF/Tet-FGF-2 tumors. Again, this occurred independently of tetracycline administration in both types of tumors (data not shown).
A decrease in blood vessel patency and permeability may lead to tumor hypoxia. 46 On this basis, we evaluated the expression of the hypoxia-inducible HIF-1α-transactivating factor 47,48 as an indirect index of oxygenation of Tet-FGF-2 and AS-VEGF/Tet-FGF-2 tumors (Figure 5D) ▶ . As expected, AS-VEGF/Tet-FGF-2 lesions were characterized by an intense HIF-1α immunostaining (Figure 6H) ▶ . In agreement with previous findings, 49,50 HIF-1α expression was primarily nuclear (Figure 6H ▶ , inset) and detectable throughout the tumor area and in perinecrotic regions. In contrast, only few scattered HIF-1α+ cells were observed in Tet-FGF-2 lesions (Figure 6G) ▶ . In keeping with the observed blood vessel patency and permeability, HIF-1α expression was not affected by FGF-2 down-regulation after tetracycline administration in both tumor types (Figure 5D) ▶ .
Effect of Neutralizing Anti-VEGF-R-2/Flk-1 Antibody on Tet-FGF-2 Tumor Growth and Vascularization
AS-VEGF mRNA expression leads to an ∼50% decrease in the production of both VEGF121 and VEGF165 isoforms in our transfectants. To confirm that the described effects on tumor growth and vascularization in AS-VEGF/Tet-FGF-2 lesions were indeed related to VEGF down-regulation, we used a complementary approach in which monoclonal neutralizing anti-murine VEGF-R-2/Flk-1 antibodies (DC101) were given to nude mice bearing Tet-FGF-2 cells. These antibodies had been shown to inhibit VEGF-induced angiogenesis and tumor growth in various experimental settings. 31,51,52 Animals were injected subcutaneously with Tet-FGF-2 cells and received an intraperitoneal administration of DC101 antibody or vehicle starting from day 1 in the absence or in the presence of tetracycline in the drinking water. At day 44, mice were sacrificed and tumors harvested and analyzed. As observed with the AS-VEGF approach, DC101 administration causes a significant decrease in tumor burden together with an increase in the percentage of necrotic tumor parenchyma (Table 2) ▶ . This was paralleled by a significant decrease in blood vessel density, mean vessel diameter, and total vascular area. DC101 appeared to be slightly more efficacious that tetracycline whereas DC101 and tetracycline co-administration exerted a partially additional effect (Table 2) ▶ .
Table 2.
Effect of DC101 Antibody on Tet-FGF-2 Tumor Growth and Neovascularization
| Tumor volume, mm3 | Necrosis, % | Vessel density, no. vessels/field | Vessel diameter, μm | Vascular area, mm2/field × 10−3 | |
|---|---|---|---|---|---|
| Vehicle | 438 ± 128 | 21 ± 5 | 60 ± 1 | 25.7 ± 1.0 | 31.3 ± 2.6 |
| Tetracycline + vehicle | 255 ± 75* | 26 ± 8 | 30 ± 6* | 16.9 ± 0.2* | 6.6 ± 0.6* |
| DC101 | 182 ± 24* | 46 ± 1* | 20 ± 2* | 13.5 ± 0.5* | 2.9 ± 0.3* |
| Tetracycline + DC101 | 103 ± 30* | 44 ± 5* | 14 ± 2* | 14.0 ± 1.7* | 2.3 ± 0.5* |
Tet-FGF2 cells were transplanted subcutaneously in nude mice (1 × 106 cells/animal). Tetracycline treatment (2.0 mg/ml in the drinking water) started 4 days before cell transplantation whereas intraperitoneal DC101 antibody administration (800 μg/animal every four days) started at day 1. At day 44, mice were sacrificed and tumors harvested and analyzed. The percentage of necrotic tumor parenchyma was calculated by computerized image analysis of the whole tissue sections. Analysis of CD31+ tumor vessels was performed at ×20 magnification on digitized images of 0.4-mm2 microscopic fields. Data are the mean ± SE of five animals.
*Statistically different from vehicle (Student’s t-test, P < 0.05 or better).
Discussion
Multiple angiogenic growth factors are expressed in tumors. For many of them, the individual role in vasculogenesis and angiogenesis has been extensively investigated. Nevertheless, little is known how these factors coordinately regulate tumor neovascularization. The present study sought to determine the relative impact of FGF-2 and/or VEGF expression on tumor growth and angiogenesis. To this purpose, the human endometrial adenocarcinoma HEC-1-B-derived Tet-FGF-2 cells that express FGF-2 under the control of the tetracycline-responsive promoter 41 were further transfected with a VEGF121 anti-sense cDNA, thus generating AS-VEGF/Tet-FGF-2 cells. This allowed the comparison of the in vivo properties of tumors originating from the same cell line but differing for FGF-2 and/or VEGF expression. Indeed, Tet-FGF-2 cells express high levels of both FGF-2 and VEGF; tetracycline-treated Tet-FGF-2 cells express high levels of VEGF only; AS-VEGF/Tet-FGF-2 cells express high levels of FGF-2 and reduced levels of VEGF; tetracycline-treated AS-VEGF/Tet-FGF-2 cells express reduced levels of VEGF only.
The findings demonstrate profound differences in tumor growth and vascularization as a function of the relative expression of the two angiogenic factors. The expression of both FGF-2 and VEGF resulted in fast-growing, highly vascularized Tet-FGF-2 tumors. Tumor blood vessels are highly heterogeneous in size with numerous large caliber vessels. They are frequently associated with α-SMA+ pericytes and surrounded by numerous α-SMA+ myofibroblasts. The patency and permeability of blood vessels in these lesions allow a sufficient oxygenation of tumor parenchyma, as indicated by the low levels of HIF-1α expression and limited necrosis.
FGF-2 down-regulation after tetracycline administration causes a significant decrease in Tet-FGF-2 tumor burden. Because of the in vitro proliferation of Tet-FGF-2 cells unaltered by FGF-2 production and/or tetracycline administration, 41 the reduced rate of growth of tetracycline-treated Tet-FGF-2 tumors is the likely consequence of the observed decrease in blood vessel density and size that results in a dramatic reduction in the total vascular area of these lesions. Thus, FGF-2 can deeply affect tumor growth and vascularization even in the presence of a high background of VEGF whose production is unchanged by tetracycline treatment. However, even though FGF-2 down-regulation clearly reduces tumor vascular density, the scarce blood vessels are still patent and highly permeable to both low-molecular weight fluorophore Hoechst 33342 and to high-molecular weight fluorescein isothiocyanate-labeled dextran. The functionality of the reduced tumor vasculature is sufficient to sustain a slow rate of tumor growth and to avoid massive necrosis and hypoxia.
In contrast, the growth delay and reduced vascularization caused by AS-VEGF transfection is paralleled by a drastic reduction in blood vessel patency and permeability, in keeping with the dual role of VEGF as an angiogenic and vascular permeability factor. 53 Vessel leakiness is an important feature of tumor vasculature. 54 It correlates with tumor histological grade and malignant potential. 55 Also, it may favor the dissemination of tumor metastases via the bloodstream and the extravasation of macromolecules into the tumor. 56 As a consequence of the reduced blood vessel patency and permeability, AS-VEGF/Tet-FGF-2 tumors are highly necrotic and hypoxic, as indicated by the intense HIF-1α immunoreactivity of tumor parenchyma. Interestingly, a significant correlation between tumor necrosis and nuclear expression of HIF-1α has been observed in human head and neck cancer. 57 The modifications of tumor vasculature functionality caused by AS-VEGF expression, as well as by anti-VEGF-R-2/Flk-1 DC101 antibody treatment (data not shown), are unique features that distinguish the down-regulation of VEGF activity. In fact this phenomenon occurred despite the presence of high levels of FGF-2. On the other hand, the preservation of vascular functionality observed in those tumors in which only FGF-2 is down-regulated might be because of the action of background VEGF.
Thus, inhibition of FGF-2 or VEGF expression results in a similar decrease in blood vessel density. However, the two growth factors differently affect tumor blood vessel maturation and functionality, with different consequences on tumor oxygenation and viability. Indeed, microvessel density does not necessarily reflect tumor perfusion and blood supply. 58 Even though quantification of the number of CD31+ microvessels in vascular hot spots represents a widely used procedure to assess tumor vascularization in experimental and clinical protocols, microvessel density may not represent a measure of the angiogenic dependence of the tumor and it may not be an indicator of anti-angiogenic response to therapy. 58
Inhibition of the expression of both FGF-2 and VEGF, as it occurs in tetracycline-treated AS-VEGF/Tet-FGF-2 lesions, caused only a marginal additional decrease in tumor growth and vascularization when compared to lesions in which only one of the two growth factors is down-regulated. Similarly, administration of both tetracycline and anti-VEGF-R-2/Flk-1 DC101 antibody to Tet-FGF-2 tumor-bearing mice resulted in a limited further suppression of tumor growth and vascularization when compared to tumors treated with DC101 or tetracycline only. Taken together, the data indicate that each growth factor individually exerts a limited effect on tumor burden and blood vessel density whereas the expression of both FGF-2 and VEGF results in a synergistic effect. This extends previous observations about the synergistic action exerted by the two growth factors in stimulating angiogenesis in vitro or in an animal model of limb ischemia. 59,60 Also, the synergistic effect of VEGF and of a signal peptide-FGF-2 chimera on tumor growth and vascularization has been recently reported in a murine hepatocellular carcinoma model. 61
Modulation of FGF-2 and/or VEGF activity deeply affect tumor blood vessel morphology. Tet-FGF-2 tumors are characterized by a striking heterogeneity in blood vessel diameter with numerous large caliber vessels. Large caliber vessels with abrupt changes in diameter are frequently observed in human tumors, 62 even though they do not represent a specific feature in tumor angiogenesis, being found in various forms of secondary angiogenesis as in wound healing. 63 Remarkably, FGF-2 down-regulation by tetracycline administration has a profound impact on microvessel morphology, causing a significant decrease in diameter heterogeneity and the disappearance of large caliber vessels. Accordingly, tridimensional morphometric analysis of microvascular corrosion casts indicated that blood vessels of FGF-2-overexpressing tumors are characterized by a wider average vascular diameter when compared to the microvasculature of parental lesions and by an extreme variability of the diameter of each individual vessel. 40 AS-VEGF mRNA expression as well as DC101 treatment cause a decrease in microvessel size similar to that observed in tetracycline-treated Tet-FGF-2 lesions. A decrease in microvessel density and diameter has been shown also in tumors treated with anti-VEGF antibody. 53 Conversely, VEGF overexpression induces an increase in tumor vessel density and size 28 and exogenous VEGF induces alterations in vascular patterning and regulation of vessel size during embryonic development. 64 Thus, the unbalanced production of one or more angiogenic factors (ie, FGF-2 and/or VEGF) may result in profound alterations of blood vessel morphology.
At variance with normal blood capillaries and postcapillary venules, tumor blood vessels are characterized by an abnormally loose association with α-SMA+ pericytes. 45 Also, pericyte coverage on microvessels varies extensively in different human tumors. 65 Here, VEGF expression, but not FGF-2 expression, is associated with the recruitment of α-SMA+ cells in Tet-FGF-2 xenografts. Also, we have observed that a significant fraction of these cells are not apparently associated with tumor microvessels and may represent stromal myofibroblasts. 45 These observations are in keeping with the ability of VEGF to mediate a chemotactic response in smooth muscle cells 66 and with the hypothesis that pericytes and myofibroblasts share a common mesenchymal origin and may be interconvertible. 45 Both the platelet-derived growth factor-B/platelet-derived growth factor receptor-β and the angiopoietin/Tie2 receptor systems play an important role in blood vessel maturation. 67,68 The cross-talk between these systems and tumor-derived VEGF and/or FGF-2 in pericyte recruitment during tumor vascularization is at present under investigation in our laboratory.
AS-VEGF cDNA transfection results also in a dramatic decrease in monocyte recruitment within the tumor. These data parallel clinical studies demonstrating a positive correlation between macrophage infiltration and VEGF expression levels or vascular grade in human breast cancer. 69,70 Accordingly, VEGF overexpression stimulates angiogenesis and macrophage recruitment in human ovarian cancer xenografts. 71 VEGF has been reported to be a chemotactic agent for monocytes. 72 Also, VEGF causes the up-regulation of macrophage chemoattractant protein-1 in endothelial cells. 73 Tumor-associated macrophages may play an important role in tumor growth and vascularization driven by VEGF. 74
In our experimental model, tetracycline treatment and AS-VEGF cDNA transfection or DC101 treatment recapitulate the action of selective inhibitors of the FGF-2/FGF-R and VEGF/VEGF-R systems, respectively. Our results demonstrate that inhibition of the FGF-2/FGF-R system results in a significant reduction of tumor size and vascularization that is slightly less efficacious than that caused by the inhibition of the VEGF/VEGF-R system. Inhibition of both systems caused a further, albeit limited decrease in the rate of tumor growth and angiogenesis. However, none of the treatments was able to fully suppress the growth of our transfectants. Other growth factors produced during tumor progression may partially rescue tumorigenicity in the absence of sufficient FGF-2 and VEGF. Alternatively, the possibility exists that our approach may cause only a partial inhibition of either one of the two systems. Indeed, AS-VEGF mRNA expression led to an ∼50% decrease in the production of both VEGF121 and VEGF165 isoforms in our transfectants. On the other hand, previous observations on tumor cell lines of different origin had shown that partial inhibition of VEGF expression by AS-VEGF transfection was sufficient to cause a significant effect on tumor growth and vascularization. 42,75,76 Also, the loss of a single allele after targeted inactivation of the VEGF gene is lethal in mouse embryo in which angiogenesis and blood-island formation are impaired, resulting in several developmental anomalies. 77
Inhibition of either the FGF-2/FGF-R system or the VEGF/VEGF-R system results in a significant decrease in the rate of tumor growth. Nevertheless, the high percentage of necrotic tumor parenchyma in AS-VEGF transfected or DC101-treated lesions indicates that VEGF/VEGF-R blockade may represent a more efficacious anti-neoplastic approach compared to FGF-2/FGF-R blockade. It is however possible that the reduction in vessel permeability and tumor oxygenation consequent to this approach may limit the access of therapeutic molecules to tumor cells. Also, a low intratumoral pO2 predicts a poor response to radiotherapy in different solid tumors. 78,79 On the other hand, inhibition of tumor angiogenesis might result in an increased efficacy of conventional therapies 80 and recent studies have shown that anti-angiogenesis treatment normalizes tumor vasculature, leading to an increased drug transport into the tumor. 81 Whatever the best combination/schedule might be, experimental evidence points to the combination of anti-angiogenic compounds with other therapeutic interventions for the treatment of solid tumors. 82-84 The model here described provides an unique system to design combination-based therapeutic approaches in different angiogenic environments.
Acknowledgments
We thank G. Bertolino for his help in statistical analysis and L. Naldini and M. De Palma (University of Turin, Italy) for their assistance in confocal microscopy studies.
Footnotes
Address reprint requests to Marco Presta, Department of Biomedical Sciences and Biotechnology, School of Medicine, University of Brescia, viale Europa 11, 25123 Brescia, Italy. E-mail: presta@med.unibs.it.
Supported in part by grants from the Fondazione Italiana per la Ricerca sul Cancro and Progetti Strategici Oncologia (to R. G.), the Associazione Italiana per la Ricerca sul Cancro (to M. P. and R. G.), and the National Research Council (Target Project on Biotechnology), Ministero dell’Istruzione, dell’Università e della Ricerca (Cofin 2002, Centro di Eccellenza IDET, FIRB “Novel Biotechnological and Postgenomic Tools for Tissue Revascularization”) (to M. P.).
R. G. and B. S. contributed equally to this work.
References
- 1.Folkman J: Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995, 1:27-31 [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK: Angiogenesis in cancer and other diseases. Nature 2000, 407:249-257 [DOI] [PubMed] [Google Scholar]
- 3.Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL: Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 1997, 57:963-969 [PubMed] [Google Scholar]
- 4.Moscatelli D, Presta M, Rifkin DB: Purification of a factor from human placenta that stimulates capillary endothelial cell protease production, DNA synthesis, and migration. Proc Natl Acad Sci USA 1986, 83:2091-2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shing Y, Folkman J, Sullivan R, Butterfield C, Murray J, Klagsbrun M: Heparin affinity: purification of a tumor-derived capillary endothelial cell growth factor. Science 1984, 223:1296-1299 [DOI] [PubMed] [Google Scholar]
- 6.Folkman J, Klagsbrun M: Angiogenic factors. Science 1987, 235:442-447 [DOI] [PubMed] [Google Scholar]
- 7.Zagzag D, Miller DC, Sato Y, Rifkin DB, Burstein DE: Immunohistochemical localization of basic fibroblast growth factor in astrocytomas. Cancer Res 1990, 50:7393-7398 [PubMed] [Google Scholar]
- 8.Ohtani H, Nakamura S, Watanabe Y, Mizoi T, Saku T, Nagura H: Immunocytochemical localization of basic fibroblast growth factor in carcinomas and inflammatory lesions of the human digestive tract. Lab Invest 1993, 68:520-527 [PubMed] [Google Scholar]
- 9.Takahashi JA, Mori H, Fukumoto M, Igarashi K, Jaye M, Oda Y, Kikuchi H, Hatanaka M: Gene expression of fibroblast growth factors in human gliomas and meningiomas: demonstration of cellular source of basic fibroblast growth factor mRNA and peptide in tumor tissues. Proc Natl Acad Sci USA 1990, 87:5710-5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Statuto M, Ennas MG, Zamboni G, Bonetti F, Pea M, Bernardello F, Pozzi A, Rusnati M, Gualandris A, Presta M: Basic fibroblast growth factor in human pheochromocytoma: a biochemical and immunohistochemical study. Int J Cancer 1993, 53:5-10 [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Becker D: Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor-1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med 1997, 3:887-893 [DOI] [PubMed] [Google Scholar]
- 12.Yamanaka Y, Friess H, Buchler M, Beger HG, Uchida E, Onda M, Kobrin MS, Korc M: Overexpression of acidic and basic fibroblast growth factors in human pancreatic cancer correlates with advanced tumor stage. Cancer Res 1993, 53:5289-5296 [PubMed] [Google Scholar]
- 13.Azuma M, Yuki T, Motegi K, Sato M: Enhancement of bFGF-export associated with malignant progression of human salivary gland cell clones. Int J Cancer 1997, 71:891-896 [DOI] [PubMed] [Google Scholar]
- 14.Kitadai Y, Ellis LM, Tucker SL, Greene GF, Bucana CD, Cleary KR, Takahashi Y, Tahara E, Fidler IJ: Multiparametric in situ mRNA hybridization analysis to predict disease recurrence in patients with colon carcinoma. Am J Pathol 1996, 149:1541-1551 [PMC free article] [PubMed] [Google Scholar]
- 15.Chodak GW, Hospelhorn V, Judge SM, Mayforth R, Koeppen H, Sasse J: Increased levels of fibroblast growth factor-like activity in urine from patients with bladder or kidney cancer. Cancer Res 1988, 48:2083-2088 [PubMed] [Google Scholar]
- 16.Nguyen M, Watanabe H, Budson AE, Richie JP, Hayes DF, Folkman J: Elevated levels of an angiogenic peptide, basic fibroblast growth factor, in the urine of patients with a wide spectrum of cancers. J Natl Cancer Inst 1994, 86:356-361 [DOI] [PubMed] [Google Scholar]
- 17.Li VW, Folkerth RD, Watanabe H, Yu C, Rupnick M, Barnes P, Scott RM, Black PM, Sallan SE, Folkman J: Microvessel count and cerebrospinal fluid basic fibroblast growth factor in children with brain tumours. Lancet 1994, 344:82-86 [DOI] [PubMed] [Google Scholar]
- 18.Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ: Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci USA 1995, 92:4562-4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baird A, Mormede P, Bohlen P: Immunoreactive fibroblast growth factor (FGF) in a transplantable chondrosarcoma: inhibition of tumor growth by antibodies to FGF. J Cell Biochem 1986, 30:79-85 [DOI] [PubMed] [Google Scholar]
- 20.Gross JL, Herblin WF, Dusak BA, Czerniak P, Diamond MD, Sun T, Eidsvoog K, Dexter DL, Yayon A: Effects of modulation of basic fibroblast growth factor on tumor growth in vivo. J Natl Cancer Inst 1993, 85:121-131 [DOI] [PubMed] [Google Scholar]
- 21.Hori A, Sasada R, Matsutani E, Naito K, Sakura Y, Fujita T, Kozai Y: Suppression of solid tumor growth by immunoneutralizing monoclonal antibody against human basic fibroblast growth factor. Cancer Res 1991, 51:6180-6184 [PubMed] [Google Scholar]
- 22.Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G: Fibroblast growth factors are required for efficient tumor angiogenesis. Cancer Res 2000, 60:7163-7169 [PubMed] [Google Scholar]
- 23.Czubayko F, Liaudet-Coopman ED, Aigner A, Tuveson AT, Berchem GJ, Wellstein A: A secreted FGF-binding protein can serve as the angiogenic switch in human cancer. Nat Med 1997, 3:1137-1140 [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N: Vascular endothelial growth factor and the regulation of angiogenesis. Recent Prog Horm Res 2000, 55:15-35 [PubMed] [Google Scholar]
- 25.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 27.Thomas KA: Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem 1996, 271:603-606 [DOI] [PubMed] [Google Scholar]
- 28.Yu JL, Rak JW, Klement G, Kerbel RS: Vascular endothelial growth factor isoform expression as a determinant of blood vessel patterning in human melanoma xenografts. Cancer Res 2002, 62:1838-1846 [PubMed] [Google Scholar]
- 29.Grunstein J, Masbad JJ, Hickey R, Giordano F, Johnson RS: Isoforms of vascular endothelial growth factor act in a coordinate fashion to recruit and expand tumor vasculature. Mol Cell Biol 2000, 20:7282-7291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N: Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362:841-844 [DOI] [PubMed] [Google Scholar]
- 31.Prewett M, Huber J, Li Y, Santiago A, O’Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ: Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res 1999, 59:5209-5218 [PubMed] [Google Scholar]
- 32.Saleh M, Stacker SA, Wilks AF: Inhibition of growth of C6 glioma cells in vivo by expression of antisense vascular endothelial growth factor sequence. Cancer Res 1996, 56:393-401 [PubMed] [Google Scholar]
- 33.Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A: Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 1994, 367:576-579 [DOI] [PubMed] [Google Scholar]
- 34.Mendel DB, Schreck RE, West DC, Li G, Strawn LM, Tanciongco SS, Vasile S, Shawver LK, Cherrington JM: The angiogenesis inhibitor SU5416 has long-lasting effects on vascular endothelial growth factor receptor phosphorylation and function. Clin Cancer Res 2000, 6:4848-4858 [PubMed] [Google Scholar]
- 35.Samoto K, Ikezaki K, Ono M, Shono T, Kohno K, Kuwano M, Fukui M: Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res 1995, 55:1189-1193 [PubMed] [Google Scholar]
- 36.Takahashi Y, Cleary KR, Mai M, Kitadai Y, Bucana CD, Ellis LM: Significance of vessel count and vascular endothelial growth factor and its receptor (KDR) in intestinal-type gastric cancer. Clin Cancer Res 1996, 2:1679-1684 [PubMed] [Google Scholar]
- 37.Poon RT, Fan ST, Wong J: Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol 2001, 19:1207-1225 [DOI] [PubMed] [Google Scholar]
- 38.Rak J, Kerbel RS: bFGF and tumor angiogenesis—back in the limelight? Nat Med 1997, 3:1083-1084 [DOI] [PubMed] [Google Scholar]
- 39.Coltrini D, Gualandris A, Nelli EE, Parolini S, Molinari-Tosatti MP, Quarto N, Ziche M, Giavazzi R, Presta M: Growth advantage and vascularization induced by basic fibroblast growth factor overexpression in endometrial HEC-1-B cells: an export-dependent mechanism of action. Cancer Res 1995, 55:4729-4738 [PubMed] [Google Scholar]
- 40.Konerding MA, Fait E, Dimitropoulou C, Malkusch W, Ferri C, Giavazzi R, Coltrini D, Presta M: Impact of fibroblast growth factor-2 on tumor microvascular architecture. A tridimensional morphometric study. Am J Pathol 1998, 152:1607-1616 [PMC free article] [PubMed] [Google Scholar]
- 41.Giavazzi R, Giuliani R, Coltrini D, Bani MR, Ferri C, Sennino B, Tosatti MP, Stoppacciaro A, Presta M: Modulation of tumor angiogenesis by conditional expression of fibroblast growth factor-2 affects early but not established tumors. Cancer Res 2001, 61:309-317 [PubMed] [Google Scholar]
- 42.Belletti B, Ferraro P, Arra C, Baldassarre G, Bruni P, Staibano S, De Rosa G, Salvatore G, Fusco A, Persico MG, Viglietto G: Modulation of in vivo growth of thyroid tumor-derived cell lines by sense and antisense vascular endothelial growth factor gene. Oncogene 1999, 18:4860-4869 [DOI] [PubMed] [Google Scholar]
- 43.Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ: Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284:1994-1998 [DOI] [PubMed] [Google Scholar]
- 44.Smith KA, Hill SA, Begg AC, Denekamp J: Validation of the fluorescent dye Hoechst 33342 as a vascular space marker in tumours. Br J Cancer 1988, 57:247-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM: Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 2002, 160:985-1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS: Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res 1999, 59:1592-1598 [PubMed] [Google Scholar]
- 47.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E: Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394:485-490 [DOI] [PubMed] [Google Scholar]
- 48.Semenza GL: Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol 2000, 59:47-53 [DOI] [PubMed] [Google Scholar]
- 49.Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW: Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999, 59:5830-5835 [PubMed] [Google Scholar]
- 50.Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL: The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 2000, 157:411-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, Bohlen P: Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer Metastasis Rev 1998, 17:155-161 [DOI] [PubMed] [Google Scholar]
- 52.Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS: Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 2000, 105:R15-R24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK: Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 1996, 93:14765-14770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM: Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 2000, 156:1363-1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Daldrup H, Shames DM, Wendland M, Okuhata Y, Link TM, Rosenau W, Lu Y, Brasch RC: Correlation of dynamic contrast-enhanced MR imaging with histologic tumor grade: comparison of macromolecular and small-molecular contrast media. Am J Roentgenol 1998, 171:941-949 [DOI] [PubMed] [Google Scholar]
- 56.Jain RK: Integrative pathophysiology of solid tumors: role in detection and treatment. Cancer J Sci Am 1998, 4(Suppl 1):S48-S57 [PubMed] [Google Scholar]
- 57.Beasley NJ, Leek R, Alam M, Turley H, Cox GJ, Gatter K, Millard P, Fuggle S, Harris AL: Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res 2002, 62:2493-2497 [PubMed] [Google Scholar]
- 58.Hlatky L, Hahnfeldt P, Folkman J: Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us. J Natl Cancer Inst 2002, 94:883-893 [DOI] [PubMed] [Google Scholar]
- 59.Goto F, Goto K, Weindel K, Folkman J: Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels. Lab Invest 1993, 69:508-517 [PubMed] [Google Scholar]
- 60.Asahara T, Bauters C, Zheng LP, Takeshita S, Bunting S, Ferrara N, Symes JF, Isner JM: Synergistic effect of vascular endothelial growth factor and basic fibroblast growth factor on angiogenesis in vivo. Circulation 1995, 92:II365-II371. [DOI] [PubMed] [Google Scholar]
- 61.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, Huber J, Nakatani T, Tsujinoue H, Yanase K, Imazu H, Fukui H: Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002, 35:834-842 [DOI] [PubMed] [Google Scholar]
- 62.Konerding MA, Miodonski AJ, Lametschwandtner A: Microvascular corrosion casting in the study of tumor vascularity: a review. Scanning Microsc 1995, 9:1233-1243 [PubMed] [Google Scholar]
- 63.Vaupel P, Gabbert H: Evidence for and against a tumor type-specific vascularity. Strahlenther Onkol 1986, 162:633-638 [PubMed] [Google Scholar]
- 64.Drake CJ, Little CD: Exogenous vascular endothelial growth factor induces malformed and hyperfused vessels during embryonic neovascularization. Proc Natl Acad Sci USA 1995, 92:7657-7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG: Heterogeneity of angiogenesis and blood vessel maturation in human tumors: implications for antiangiogenic tumor therapies. Cancer Res 2000, 60:1388-1393 [PubMed] [Google Scholar]
- 66.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES: Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol 2001, 188:359-368 [DOI] [PubMed] [Google Scholar]
- 67.Hellstrom M, Kaln M, Lindahl P, Abramsson A, Betsholtz C: Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126:3047-3055 [DOI] [PubMed] [Google Scholar]
- 68.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J: Vascular-specific growth factors and blood vessel formation. Nature 2000, 407:242-248 [DOI] [PubMed] [Google Scholar]
- 69.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL: Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res 1996, 56:4625-4629 [PubMed] [Google Scholar]
- 70.Leek RD, Hunt NC, Landers RJ, Lewis CE, Royds JA, Harris AL: Macrophage infiltration is associated with VEGF and EGFR expression in breast cancer. J Pathol 2000, 190:430-436 [DOI] [PubMed] [Google Scholar]
- 71.Duyndam MC, Hilhorst MC, Schluper HM, Verheul HM, van Diest PJ, Kraal G, Pinedo HM, Boven E: Vascular endothelial growth factor-165 overexpression stimulates angiogenesis and induces cyst formation and macrophage infiltration in human ovarian cancer xenografts. Am J Pathol 2002, 160:537-548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D: Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 1996, 87:3336-3343 [PubMed] [Google Scholar]
- 73.Marumo T, Schini-Kerth VB, Busse R: Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 1999, 48:1131-1137 [DOI] [PubMed] [Google Scholar]
- 74.Mantovani A: Tumor-associated macrophages in neoplastic progression: a paradigm for the in vivo function of chemokines. Lab Invest 1994, 71:5-16 [PubMed] [Google Scholar]
- 75.Yano S, Shinohara H, Herbst RS, Kuniyasu H, Bucana CD, Ellis LM, Davis DW, McConkey DJ, Fidler IJ: Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res 2000, 60:4959-4967 [PubMed] [Google Scholar]
- 76.Cheng SY, Huang HJ, Nagane M, Ji XD, Wang D, Shih CC, Arap W, Huang CM, Cavenee WK: Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA 1996, 93:8502-8507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW: Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 1996, 380:439-442 [DOI] [PubMed] [Google Scholar]
- 78.Brizel DM, Dodge RK, Clough RW, Dewhirst MW: Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiother Oncol 1999, 53:113-117 [DOI] [PubMed] [Google Scholar]
- 79.Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, Chapman W, Levin W, Manchul L, Keane TJ, Hill RP: Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol 1998, 48:149-156 [DOI] [PubMed] [Google Scholar]
- 80.Teicher BA, Sotomayor EA, Huang ZD: Antiangiogenic agents potentiate cytotoxic cancer therapies against primary and metastatic disease. Cancer Res 1992, 52:6702-6704 [PubMed] [Google Scholar]
- 81.Jain RK: Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 2001, 7:987-989 [DOI] [PubMed] [Google Scholar]
- 82.Griscelli F, Li H, Cheong C, Opolon P, Bennaceur-Griscelli A, Vassal G, Soria J, Soria C, Lu H, Perricaudet M, Yeh P: Combined effects of radiotherapy and angiostatin gene therapy in glioma tumor model. Proc Natl Acad Sci USA 2000, 97:6698-6703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teicher BA: A systems approach to cancer therapy. Antioncogenics + standard cytotoxics–>mechanism(s) of interaction. Cancer Metastasis Rev 1996, 15:247-272 [DOI] [PubMed] [Google Scholar]
- 84.Miller KD, Sweeney CJ, Sledge GW, Jr: Redefining the target: chemotherapeutics as antiangiogenics. J Clin Oncol 2001, 19:1195-1206 [DOI] [PubMed] [Google Scholar]