

Abstract
The International Registry of Werner syndrome (www.wernersyndrome.org) has been providing molecular diagnosis of the Werner syndrome (WS) for the past decade. The present communication summarizes, from among 99 WS subjects, the spectrum of 50 distinct mutations discovered by our group and by others since the WRN gene (also called RECQL2 or REQ3) was first cloned in 1996; 25 of these have not previously been published. All WRN mutations reported thus far have resulted in the elimination of the nuclear localization signal at the C-terminus of the protein, precluding functional interactions in the nucleus; thus, all could be classified as null mutations. We now report two new mutations in the N-terminus that result in instability of the WRN protein. Clinical data confirm that the most penetrant phenotype is bilateral ocular cataracts. Other cardinal signs were seen in more than 95% of the cases. The median age of death, previously reported to be in the range of 46–48 years, is 54 years. Lymphoblastoid cell lines (LCLs) have been cryopreserved from the majority of our index cases, including material from nuclear pedigrees. These, as well as inducible and complemented hTERT (catalytic subunit of human telomerase) immortalized skin fibroblast cell lines are available to qualified investigators. Published 2006 Wiley-Liss, Inc.†
Keywords: Werner syndrome, WRN, RECQL2, RECQ3, Werner helicase, RecQ helicases, progeroid syndromes, aging, international registries, penetrance, aging
INTRODUCTION
Werner syndrome (WS; MIM# 277700) is a rare autosomal recessive disorder characterized by many features suggestive of accelerated aging [Epstein et al., 1966; Martin, 1978; Goto, 1997]. These include premature graying and loss of hair, bilateral ocular cataracts, type 2 diabetes mellitus, osteoporosis, various forms of arteriosclerosis including atherosclerosis, and hypogonadism. There are scleroderma-like skin changes and regional atrophy of subcutaneous fat. Deep ulcerations around the Achilles tendon and malleoli are common and virtually pathognomonic. WS subjects have an elevated risk of various cancers, particularly sarcomas [Goto et al., 1996]. As many as five different neoplasms have been found in a single individual (soft tissue sarcomas, thyroid carcinoma, osteosarcoma, acral lentigenous melanoma, and meningioma) [Goto et al., 1996].
WS subjects appear to develop normally until adolescence, when there is lack of a pubertal growth spurt. Subsequent signs and symptoms begin to emerge in the early 20 s. The median age of death has been reported to range from 46 years [Epstein et al., 1966] to 48 years [Goto, 1997], typically as a result of cancer or atherosclerotic cardiovascular disease. Because the WS phenotype overlaps with many but not all of the symptoms manifested during aging as it usually occurs (“normative” aging), it has been classified as a “segmental progeroid syndrome” [Martin, 1978; Martin et al., 1999].
Several cellular defects have been identified in cultured somatic cells derived from individuals with WS, including a much shorter replicative life span when compared to age-matched controls [Martin et al., 1970]. The genome of WS cells is highly unstable, with increased chromosomal rearrangements (“variegated transmosaicism”) [Salk et al., 1985; Melaragno et al., 1995], and large spontaneous DNA deletions [Fukuchi et al., 1989]. The WS cells are unable to optimally repair exogenous DNA with double strand breaks [Chen et al., 2003a]. Telomere length dynamics are abnormal in WS cells; telomeres shorten rapidly as cell population doublings increase [Schulz et al., 1996; Tahara et al., 1997]. WS cells also appear to be deficient in DNA replication and certain DNA repair pathways. DNA replication initiation sites are reduced and there is an extended S-phase of DNA replication [Poot et al., 1992]. WS cells are hypersensitive to a number of agents that damage DNA, including 4-nitroquinoline-1-oxide (4-NQO) [Ogburn et al., 1997; Poot et al., 2002], topoisomerase I inhibitors (camptothecin) [Okada et al., 1998; Poot et al., 1999], and DNA cross-linking agents [Poot et al., 2001]. In addition, WS cells are impaired in RNA polymerase II transcription [Balajee et al., 1999] and p53-mediated apoptosis [Spillare et al., 1999; Sommers et al., 2005]. The unstable genome of WS cells, and the associated limited replicative potential and cell loss may be of central significance to the pathogenesis of WS.
The gene underlying WS (WRN; MIM#] 604611) was identified in 1996 [Yu et al., 1996]. It was shown to be a member of the RecQ family of DNA helicases. Structural analysis suggests that the WRN protein contains a RecQ-type helicase domain within the central region of the polypeptide [Yu et al., 1996] and a nuclease domain in the N-terminal region [Moser et al., 1997; Mushegian et al., 1997]. Subsequent biochemical studies confirmed that the WRN protein displays both 3′->5′; helicase [Gray et al., 1997] and exonuclease [Huang et al., 1998] activities. Alternative DNA structures that can be accidentally generated during various DNA metabolic pathways are shown to be preferred substrates of the WRN protein [Huang et al., 2000; Mohaghegh et al., 2001].
A nuclear localization signal resides between residues 1369–1402 at the C-terminus [Matsumoto et al., 1997b; Suzuki et al., 2001]. An additional nucleolar localization signal is implicated between residue 949–1092 within the RecQ consensus domain [von Kobbe and Bohr, 2002]. The WRN protein is localized primarily in the nucleoli in human cells and relocates to form nucleoplasmic foci at sites of DNA damage [Sakamoto et al., 2001].
In addition, there are two consensus domains in the C-terminal region of the WRN protein: the RecQ helicase conserved region (RQC) and the “helicase, RNaseD, C-terminal conserved region” (HRDC) [Morozov et al., 1997]. Structural analysis predicts that WRN protein interacts with the substrate DNA and proteins via its RQC region [Bennett and Keck, 2004] and with DNA through the HRDC region [Liu et al., 1999]. Subsequent biochemical studies demonstrated that substrate-specific DNA binding activity occurs in three domains: N-terminal, RQC, and HRDC [von Kobbe et al., 2003]. The WRN protein was shown to accumulate at sites of double strand breaks via its HRDC domain [Lan et al., 2005].
Following the initial report of four mutations [Yu et al., 1996], additional mutations were reported in the WRN gene [Oshima et al., 1996; Goto et al., 1997; Meisslitzer et al., 1997; Yu et al., 1997; Matsumoto et al., 1997a]. The spectrum of 19 WRN mutations was reviewed in 1999 [Moser et al., 1999]. All mutations so far reported result in truncations of the WRN protein, with the loss of the nuclear localization signal. This precludes participation of the WRN protein in its nuclear functions. We now extend the Moser et al. [1999] report to a total of 50 WRN mutations, 25 of which have not been previously published.
MATERIALS AND METHODS
Samples
Samples were collected from individuals with Werner syndrome participating in our International Registry of Werner Syndrome (www.wernersyndrome.org), supplemented by materials banked at the Coriell Institute for Medical Research (Camden, NJ). The International Registry of Werner Syndrome has ongoing approval from the University of Washington Institutional Review Board.
Sample Preparations
Blood or skin samples were processed using the follow protocols: 1) cryopreservation of an aliquot of the primary cells as a resource for the cell bank; 2) isolation of DNA from primary cells; 3) establishment of lymphoblastoid cell line with Epstein-Barr virus or of fibroblast lines with hTERT transformation; 4) isolation of DNA, RNA, and protein from such lines; 5) Western analysis with positive and negative controls; 6) RT-PCR, with confirmation of successful amplification by agarose gel electrophoresis and sequencing of the entire coding region; and 7) verification of mutations by genomic DNA sequencing (Fig. 1).
FIGURE 1.

Diagram of standard protocols at the International Registry of Werner Syndrome. Typical processing steps of the blood samples are shown.
Generally, 20–30 ml of peripheral blood was received, from which 1–2 ml was used for DNA isolation and the rest was fractionated to obtain plasma and buffy coat. The plasmas were stored at −80°C for future studies and buffy coats were used to establish LCLs. Surplus white blood cells were frozen and stored in liquid nitrogen for future use. Once LCLs were established, a minimum of 10 frozen vials were stored in two different locations. DNA, RNA, and protein were isolated from all LCLs.
LCLs were cultured in RPMI-1640 (Invitrogen/GIBCO, Carlsbad, CA) containing 15% heat-inactivated fetal bovine serum (FBS; Atlanta Biologicals, Norcross, GA), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in an atmosphere of 5% CO2 and ambient oxygen concentrations. Fibroblast cells were similarly cultured except that the medium was Dulbecco's modified Eagle's medium (DMEM high glucose formulation, Invitrogen/GIBCO).
RT-PCR, PCR, and Sequencing
RT-PCR sequencing of the WRN gene and Western analysis of the WRN protein were performed in samples, together with positive and negative controls as previously described [Oshima et al., 1996]. RT-PCR rather than genomic PCR was used as our initial screen because it can detect splicing mutations; these were frequent. Moreover, the WRN gene has a total of 35 exons, making genomic screening rather cumbersome. When mutations were identified by RT-PCR, the PCR products of the mutated exons were sequenced in genomic DNA to confirm the mutation [Yu et al., 1997]. When exon skipping was identified by RT-PCR sequencing, the skipped exon and the two adjacent exons were sequenced with special attention to the exon/intron boundary mutations [Yu et al., 1997]. When mutations were not identified, RT-PCR sequencing was repeated to ensure the absence of mutations. In all cases, Western analyses were performed to confirm the sequencing results. Typical cases of WS are due to null mutations, in which case there is an absence of WRN protein. Any discrepant results would trigger a repeat set of assays.
When only genomic DNA was available, all the coding exons were sequenced, including 50–100 nucleotide of intron sequences flanking all exons [Yu et al., 1997]. When mutations were identified, the genomic PCR and sequencing analysis of the mutated exons were repeated to confirm the results. When mutations were not identified, all coding exons were resequenced before negative results were reported.
The reaction conditions and technical details of the RT-PCR and PCR sequencing has been described previously [Oshima et al., 1996; Yu et al., 1997]. All the data are independently examined and analyzed by two different individuals by comparisons with the wild type WRN sequence (GenBank accession number L76937.1, GI:1280207).
Generation and Detection of Recombinant WRN Proteins
Constructs containing WRN cDNA encoding either the wild type, E84A exonuclease mutant [Huang et al., 1998], K125N mutant, K135E mutant, or [K125N+K135E] double mutant N-terminal region (amino acids 1–333) of the WRN protein were expressed in baculoviruses according to the published protocol [Huang et al., 2000]. For the protein analysis, total protein was prepared from insect cells infected with baculoviruses carrying the indicated cDNAs, and purified by an affinity column [Huang et al., 2000]. The purified WRN N-terminal fragment proteins were resolved by a 10% PAGE followed by staining with Coomassie brilliant blue. For Western analysis, 30 μg total proteins were resolved by a 10% PAGE and then blotted to a nylon membrane and detected by a rabbit polyclonal anti-WRN antibody followed by chemiluminescent autography. For Northern blotting, total RNAs (5 μg) were loaded for each of the samples, blotted to a nylon membrane, and detected with a WRN-specific 32P-labelled cDNA probe followed by autoradiography.
RESULTS
International Registry of Werner Syndrome
The International Registry of Werner Syndrome at the Department of Pathology, University of Washington, Seattle, WA, was established in 1988. The original purpose of the Registry was to collect samples from WS patients for positional cloning of WRN. Between 1988 and the time of identification of the WRN gene in 1996, we collected a total of 81 index cases. During this early period, we focused on cases with a definite diagnosis of WS based on a set of clinical criteria [Nakura et al., 1994]. In order to identify unusual WRN mutations and to investigate other genes responsible for segmental progeroid syndromes whose features partially overlap with those of the WS, we expanded our Registry to include probable and possible diagnoses of WS [Nakura et al., 1994].
Clinicians of patients with WS symptoms have contacted us via several linkages: 1) review articles published by our group; 2) an internet search directing them to our website (www.wernersyndrome.org); 3) the GeneTests/GeneClinics online database (www.genetests.org); and 4) the Genetic Alliance patient advocacy listing (www.geneticalliance.org). Clinicians are asked to complete registry forms. Informed consent for the registry and repository was done by our genetic counselor or the clinician if local regulations allow (forms can be downloaded at www.wernersyndrome.org/registry/participate.html). As of November 2005, we have enrolled a total of 144 pedigrees, or a total of 274 individuals, including index cases and their family members.
In many cases, peripheral blood samples were available; rarely, skin biopsies were sent to our laboratory (see LCL or Fibroblast in Materials Available column in Supplementary Table S1 (available online at http://www.interscience.wiley.com/jpages/1059-7794/suppmat). We successfully established LCLs in more than 95% of the cases including blood samples that arrived at our laboratory several days after sampling. For samples that arrived 1 week or more after sampling (mostly from abroad), the success rate was approximately 30%. In some instances, the molecular genetic analysis was based upon previously established cell cultures or previously extracted DNA (DNA in Supplementary Table S1, Materials Available column).
We report clinical and molecular data compiled from a total of 99 WS subjects with confirmed mutations (50 were female, 44 male and 5 unknown). This is consistent with the expected gender ratios observed in autosomal recessive disorders. They represent 21 ethnic/geographic groups: German, French, Japanese, Italian, Swiss, Austrian, United States-Caucasian, Brazil, New Zealand, Sardinia, African, Indian, Ashkenazi Jewish, Korean, Taiwanese, Puerto Rican, Turk, Celtic, Dutch, Syrian, and Mexican.
WRN Mutations in WS Patients
Fifty individually distinct WRN mutations were observed, 25 of which have not been previously published. Of these 50 mutations, 41 occur in the coding sequence and nine are in introns. These are summarized in Supplementary Table S1 and their locations are mapped on the WRN gene in Figure 2. The mutations can be grouped into four categories: 1) nonsense mutations which change an amino acid codon to a stop codon and lead to the termination of protein translation; 2) insertions and/or deletions which lead to a reading frameshift and subsequent termination of protein translation; 3) substitutions at the splice junctions that cause the skipping of exons and a subsequent frameshift; and 4) missense mutations which lead to amino acid changes in the protein.
FIGURE 2.

Locations of WRN mutations. Exons are designated as boxes 1–35. Introns are indicated by thin lines. The numbers above the exons indicate the nucleotide positions in the WRN cDNA (GenBank accession no. L76937.) with the first ATG being 1. The localizations of the functional domains are indicated. Purple boxes, exonuclease domain; yellow boxes, helicase domain; green boxes, RQC, the RecQ helicase conserved region; pink boxes, HRDC, the Helicase, RnaseD, C-terminal conserved region; and gray box, NLS, the nuclear localization signal; CDS, coding sequence; FS/Ter, frameshift and premature translation termination; IVS, intervening sequence. I-VI indicate the motifs in exonuclease and helicase domains.
The mutations were located throughout the coding region of the WRN gene. There was one mutation at the 5′ region, six mutations in the 3′->5′ exonuclease domain, 10 mutations between the exonuclease and the helicase domains, 11 mutations within the helicase domain, 10 mutations in the RQC domain, five mutations between the RQC and HRDC domains, four mutations in the HRDC domain, and three mutations between HRDC and the nuclear localization signal region. The most frequent mutation among non-Japanese WS cases was a C to T transition substitution at nucleotide 1105, changing amino acid codon 369 from arginine (CGA) to stop codon (TGA) [Oshima et al., 1996; Yu et al., 1997; Matsumoto et al., 1997a]. This mutation occurred in 24 of the 99 WS subjects (24%) compiled in the study either as homozygotes or compound heterozygotes. The most common mutation among Japanese WS was a transversion substitution from G to C at the consensus splicing sequence at the junction of intron 25 and exon 26 that abolishes the splicing site and leads to deletion of exon 26 [Yu et al., 1997]. This c.3139–1G>C mutation was seen in 67% of our Japanese cases, and has not been seen in cases from other ethnic origins. The skipping of exon 26 was found in one French pedigree, due to the substitution from G to C at the splicing consensus sequence in intron 26 adjacent to exon 26 (c.3233+1G>C) [Moser et al., 2000].
Novel Mutations That Alter WRN Protein Stability
Two new missense mutations in the exonuclease domain, c.375A>T and c.403A>G, were identified as homozygous mutations in one Caucasian German subject (STUTT1010) ([c.375A>T + c.403A>G]+ [c.375A>T 1 c.403A>G]). A transversion substitution from A to T at nucleotide 375 changed amino acid codon 125 from lysine (AAA) to asparagine (AAT) (K125N). The other missense mutation in this subject was the result of a transversion (A to G at nucleotide 403, changing amino acid codon 135 from lysine (AAG) to glutamate (GAG) (K135E) ([K125N+K135E]+[K125N+K135E]).
Sequence and structural analysis predicted that the K125N and K135E mutations are unlikely to affect exonuclease activity, but are more likely to affect protein stability (Fig. 3). The three-dimensional structure of the Klenow fragment of E. coli DNA polymerase I complex with single-stranded DNA was used to interpret the potential impact of the two missense mutations [Mian, 1997]. The WRN mutations are predicted to occur towards the end of an α helix and the beginning of a β strand (Fig. 3A). The region of WRN protein containing these mutations (Fig. 3B, green side chains) would be distal from the substrate binding and catalytic sites and close to the surface, especially K125N; this face of the exonuclease domain would be available for interaction with another molecule. Inspection of the Klenow fragment structure indicates that the surface exposed residues K406 (K125 in WRN), E410 (E129 in WRN), and R436 (K156 in WRN) are aligned such that the positively and negatively side charged chains can form stabilizing charge-charge interactions (Fig. 3C). The NZ atom of Klenow residue K416 (K135 in WRN) can form stabilizing interactions with the peptide carbonyl oxygen atoms of the loop containing E410 and R436. Thus, the WRN mutations, K125N and K135E, are likely to perturb and/or eliminate this intricate network of stabilizing interactions thereby disrupting the local structure of the exonuclease domain without influencing exonuclease activity.
FIGURE 3.

Sequence and structural analysis of the N-terminal region of theWRN protein. A: A multiple sequence alignment of a 3′-5′ exonuclease domain present in the following selected proteins: EcDPOIII, Escherichia coli DNA polymerase III (RCSB code 1J54); EcExoI, E. coli exonuclease I (RCSB 1FXX); EcRNaseT, E. coli RNaseT (RefSeq NP_416169); EcKlenow_1QSL, Klenow fragment of E. coli DNA polymerase I (RCSB 1QSL); HsWRN: Homo sapiensWRN protein (RefSeq NP_000544). Numbers indicate the number of residues not shown explicitly; residues that are conserved in three of the five sequences are highlighted. The annotations below the alignment refer toWRN (HsWRN) and indicate the locations and names of mutations discussed in this study. The annotations above the alignment refer to DNA polymerase I (EcKlenow_1QSL) and refer to features observed in the crystal structure of the Klenow fragment complexed with single-stranded substrate and taken from the 1QSL entry in the PDBsum Resource (www.ebi.ac.uk/thorntonsrv/databases/pdbsum).The arrows and cylinders represent beta-strands and alpha-helices, respectively. Red and blue triangles indicate the locations and names of residues that interact with a metal ion in the active site and DNA substrate respectively. Green triangles mark the residues in the Klenow fragment (K406 and K416) predicted to be equivalent to the WRN mutations K125N and K135E. Yellow triangles mark residues and the boxed region indicates the region shown in the crystal structure. B: Images of the Klenow fragment shown in two different orientations using the same color scheme as in panel A.The thin gray tube is a smoothed representation of the alpha carbon backbone and the small gray spheres denote the N- and C-terminal residues.The N-termini are represented by the gray spheres at the bottoms of the molecules.The single-stranded DNA substrate is shown in cyan (all atom representation).The red and blue side amino acid chains drawn explicitly correspond to the metal and DNA binding residues.The green side chains (K406 and K416) are the positions equivalent to the WRN mutations K125N and K135E; the yellow side chains (E410 and R436) are discussed in the text. C: Images of part of the Klenow fragment shown in two different orientations and corresponding to the region boxed in the alignment. Only atoms in the peptide backbone of Klenow fragment residues 406 to 438 are shown (C: gray, N: blue, O: red).The side chains of four specific residues are drawn explicitly, K406 and K416 (green), and E410 and R436 (yellow).
In order to verify the structural analysis, we expressed the N-terminal regions of the mutant WRN proteins, including the exonuclease region, in eukaryotic cells to examine protein stability (Fig. 4). Following the baculovirus-mediated transfection of wild type, K125N mutant, K135E mutant, or [K125N+K135E] double mutant, mRNA and proteins were isolated from sf9 insect cells every 24 hr. As controls, wild type and E84A exonuclease mutant were employed. The latter is the mutant that had been shown to abolish exonuclease activity without affecting protein stability [Huang et al., 1998]. The mRNAs transcribed from the cDNAs containing the missense mutations (K125N, K135E, [K125N+K135E]) were at levels similar to those from the wild type and the E84A mutant (Fig. 4A). On the other hand, the protein levels produced from the single mutant cDNAs, either K125N or K135E, were much lower compared to the wild type and exonuclease mutant controls. [K125N+K135E] double mutant proteins were virtually undetectable with this method (Fig. 4B and C). This is not due to the overall reduction of protein production (Fig. 4B). We tested protein stability using full-length WRN cDNA. Because of its relatively large size, however, protein levels were overall low and the differences between the wild type and the mutants were not clearly seen (data not shown). We were unable to obtain blood or fibroblast samples from this patient to further examine these mutations. In conclusion, these results are consistent with the sequence and structural observations described above.
FIGURE 4.
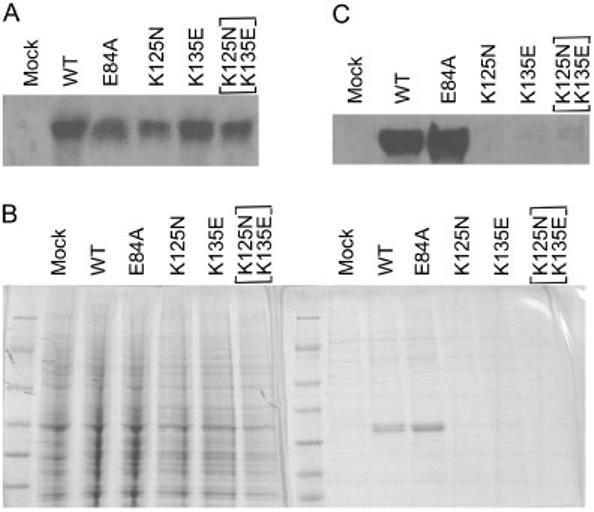
K125N and K135E mutations renderWRN proteins unstable. A: Northern blotting of total RNA isolated from insect cells expressing segments of the WRN protein with the indicated mutations. B: Coomassie blue staining of the proteins isolated from the culture. Left panel shows the patterns of 1 mg of total proteins. Right panel shows the patterns of 100 μg of the purified proteins. C:Western analysis of the purified proteins.
Clinical Features of Patients With WRN Mutations
Previous clinical reviews of WS cases have based the diagnosis on clinical signs and symptoms and did not include molecular data [Epstein et al., 1966; Tollefsbol and Cohen, 1984; Goto, 1997]. We therefore have evaluated the known signs and symptoms of WS in genetically diagnosed WS patients. The patients were referred to us by collaborating physicians who suspected a diagnosis of WS. Most of the clinical information was provided at the time of referral, along with biological samples. We have received periodic medical updates on some patients. The median age of referral was 46.4 years, the youngest being 15.9 years (Fig. 5). Table 1 summarizes the frequency of well-documented abnormalities of WS. Most notably, bilateral ocular cataracts were reported in all of the cases in which information was available. Skin alterations (scleroderma-like skin, tight skin, thin skin, hyperkeratosis, etc.), graying and/or loss of hair and short stature were reported in 98.6, 96.3, and 94.7% of cases, respectively; 90.9% of the cases presented all four cardinal signs of WS [Nakura et al., 1994]. Other signs that are known to develop at later stages of WS or ones that require additional laboratory tests to determine were reported in slightly lower percentages. Atherosclerosis and neoplasia, two major causes of death, were reported in 39.5 and 43.6% of the molecularly confirmed cases, respectively. Diabetes and osteoporosis were reported in 70.8 and 90.6% of the cases, respectively. The median age of death of WS patients in our registry of molecularly confirmed WS was 54.3 years (Fig. 5), 7 years older than the median age of death reported by Epstein et al. [1966]. The mean age of onset of cataracts was approximately 31 years, comparable to that reported by Epstein et al. [1966].
FIGURE 5.

Distribution of the ages of onset of ocular cataracts in molecularly confirmed WS (A), distribution of the ages of the patients at the time of referral to our registry (B), and the distribution of the ages of death (C). X-axes indicate the age in decades, and Y-axes show the number of cases.
TABLE 1.
Clinical Features of Werner Syndrome Patients With Molecular Documentation of the Diagnosis*
| Sign or symptom | Percent frequency |
|---|---|
| Bilateral cataractsa | 100 (87/87) |
| Skin alterationsa | 98.6 (72/73) |
| Thin limbs | 98.4 (60/61) |
| Premature graying/hair lossa | 96.3 (79/82) |
| Pinched facial features | 96.1 (49/51) |
| Short staturea | 94.7 (71/75) |
| Osteoporosis | 90.6 (48/53) |
| Voice change | 89.0 (65/73) |
| Hypogonadism | 79.5 (35/44) |
| Type 2 diabetesmellitus | 70.8 (46/65) |
| Soft tissue calcification | 66.7 (28/42) |
| Neoplasm(s) | 43.6 (24/55) |
| Atherosclerosis | 39.5(17/43) |
| All four cardinal signsa | 90.9 (50/55) |
Clinical information was obtained at the time of referrals. Actual numbers of the cases where information was available is given in the parentheses. Diagnosis of atherosclerosis is based on the primary physicians' reports.
Cardinal signs (bilateral ocular cataracts, characteristic dermatological pathology, short stature, premature graying and/or thinning of scalp hair).
DISCUSSION
The WRN gene consists of 35 exons that encode a multi-functional protein of 1,432 amino acids (Fig. 2; Supplementary Table S1). The WRN mutations so far identified are located all across the entire coding region, including six mutations within the exonuclease domain, eleven mutations within the helicase domain, 10 mutations within the RQC domain, and four mutations within the HRDC domain. All the mutations so far identified in WS patients, except for the two missense mutations in the exonuclease domain, are nonsense mutations or insertion/deletion/substitution mutations, which result in truncation of protein translation before the nuclear localization signal. These truncated proteins cannot be transported into the nucleus to perform their functions [Matsumoto et al., 1997b; Suzuki et al., 2001]. Mutated mRNAs are known to be degraded via an alternative pathway [Jacobson and Peltz, 1996]. Mutant WRN mRNA has been shown to have a shorter half-life than that of the wild type [Yamabe et al., 1997]. These appear to be satisfactory explanations as to how the mutations in WRN abolish enzymatic activities and why all the mutations are functionally null.
The two missense mutations in the exonuclease domain, K125N and K135E, are located close to exonuclease motif II [Mian, 1997] (Fig. 3A, green triangles). Both of these missense mutations appear to render the WRN protein unstable (Fig. 4), an experimental finding that can be rationalized by inspection of the three-dimensional structure of the Klenow fragment of E. coli DNA polymerase I (Fig.3B and C). Therefore, these missense mutations may also act as null mutations. Our preliminary studies showed that the K125N and K135E mutations do not significantly alter the activities of WRN protein, as predicted by the structural analysis. It is possible, however, that these mutations may affect other properties of the WRN protein, such as its interactions with other proteins or protein complexes. A recent publication demonstrated that site directed mutation of lysine at codon 1016 in the RQC domain significantly reduces helicase activity and the binding activity for various DNA substrates [Lee et al., 2005].
Two highly prevalent mutations were noted. One is c.1105C>T, which changes codon 369 CGA to stop codon TGA. This mutation accounted for approximately 25% of our cases, depending on whether pedigrees, cases or affected chromosomes were counted. The ethnicities of this mutation include French, Austrian, Japanese, German, and US subjects. The other is c.3139–1G>C, which leads to the deletion of exon 26 (r.3139_3233del95) and truncation of the protein immediately after the RQC domain. This mutation occurred in 22 Japanese WS subjects all as homozygotes, and accounts for 67% of Japanese WS cases among our Registry cases and 62% in another study [Satoh et al., 1999]. This mutation has been seen exclusively in Japanese WS patients and a founder effect has been postulated [Matsumoto et al., 1997a]. This can be also partly attributed to an increased awareness of the syndrome among Japanese physicians as well as to higher consanguinity among the Japanese population as compared to the U.S. population. Other mutations only occurred in one or a few WS subjects and appear to be due to sporadic mutations.
Out of 99 cases with clinical presentation of WS and/or absent WRN protein by Western blot, indicating two mutant alleles, six have only one mutation identified (indicated as 1mut in Supplementary Table 1, Mutation Status column). In four cases, this was due to insufficient DNA or low quality of the DNA. Based upon this, we estimated that approximately 1% (2/198) of mutations might have been missed in our screening.
The WRN protein is thought to be a “caretaker of the genome” [Hickson, 2003]. The studies on interacting proteins suggest that WRN protein is able to participate in multiple DNA metabolic pathways when recruited to the site of gene action by binding proteins. At least 16 proteins interacting with WRN protein have been identified to date, many of which bind at the overlapping RQC and HRDC regions. Most noticeably, the WRN protein is involved in DNA double strand break repair by nonhomologous end-joining in concert with the DNA-PK complex (DNA-PKcs, Ku70 and 80) [Cooper et al., 2000; Li and Comai, 2001; Yannone et al., 2001]. Physical and functional interactions with FEN-1 [Brosh et al., 2001; Sharma et al., 2005], DNA polymerase β [Harrigan et al., 2003; Ahn et al., 2004], PARP-1 [Li et al., 2004; von Kobbe et al., 2004], and APE1 [Ahn et al., 2004] suggest a role of WRN in base excision repair. WRN protein may modulate DNA replication via interaction with Topo I [Laine et al., 2003] and DNA polymerases δ [Szekely et al., 2000]. Physical association of TRF-2 and WRN protein [Johnson et al., 2001], in conjunction with biochemical data demonstrating the displacement of D-loops by WRN protein [Orren et al., 2002], suggests that WRN may participate in the maintenance of telomeres. WRN protein has been shown to be involved in the synthesis of lagging strand of telomeres [Crabbe et al., 2004]. The tumor suppressor, p53, binds to and inhibits WRN helicase activity, while signaling downstream transcriptional activity that regulates the initiation of apoptosis following DNA damage [Sommers et al., 2005]. Taken together, WRN exonuclease and helicase activities may be used for various DNA transactions, particularly during various modalities of DNA repair. The specifics of the DNA transaction may depend on the protein complexes which direct WRN protein to the site of DNA damage and coordinate its enzymatic activities.
We have reported four pedigrees among our Werner Registry families with wild-type WRN but with heterozygous mutations at the LMNA locus [Chen et al., 2003b]. It is possible that other “atypical WS” cases with wild-type WRN and LMNA are due to mutations in these various WRN interacting proteins.
WS cases followed up by our registry presented as either probands or nuclear families. Figure 6 shows the WS pedigrees in which samples from more than one individual have been examined and biological samples were obtained. These materials can be made available to investigators upon request.
FIGURE 6.
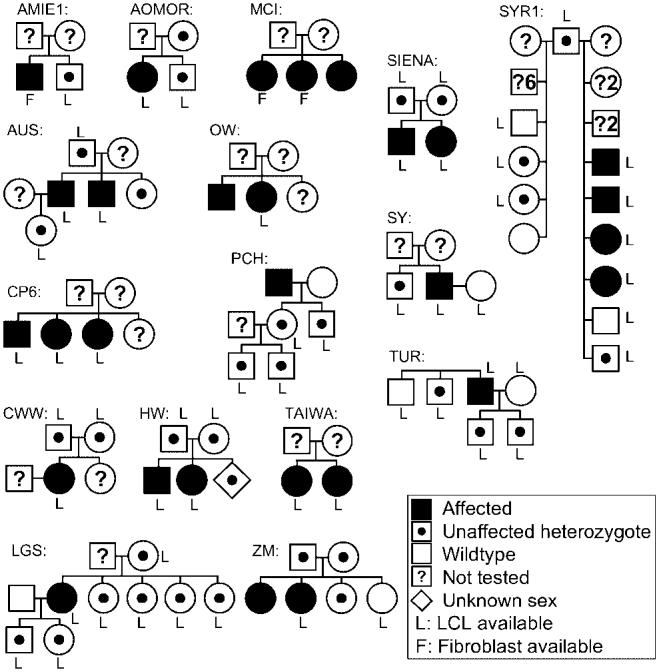
Pedigrees of the WS patients with documented WRN mutations. Family names correspond to those given in Supplementary Table S1.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mark Steele, MD, for his efforts in identifying the PCH family, Johannes Ring, MD, for his contributions with the MUNCH family, and Dr Hassani Benyouness, an endocrinologist in Oujda, Morocco, for facilitating enrollment of MOROCCO1010. This work was jointly supported by the National Cancer Institute and the National Institute on Aging (CA78088 to G.S.M.), and the California Breast Cancer Research Program (to I.S.M.).
Footnotes
The Supplementary Material referred to in this article can be accessed at http://www.interscience.wiley.com/jpages/1059-7794/suppmat.
Grant sponsor: National Cancer Institute; California Breast Cancer Research Program; Grant sponsor: National Institute of Aging; Grant number: CA78088.
This article is a US government work, and as such, is in the public domain in the United States of America.
REFERENCES
- Ahn B, Harrigan JA, Indig FE, Wilson DM, 3rd, Bohr VA. Regulation of WRN helicase activity in human base excision repair. J Biol Chem. 2004;279:53465–53474. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM, Nehlin JO, Brosh R, Orren DK, Bohr VA. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999;10:2655–2668. doi: 10.1091/mbc.10.8.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, Keck JL. Structure and function of RecQ DNA helicases. Crit Rev Biochem Mol Biol. 2004;39:79–97. doi: 10.1080/10409230490460756. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–5801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang S, Lee L, Davalos A, Schiestl RH, Campisi J, Oshima J. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003a;2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J. LMNA mutations in atypical Werner's syndrome. Lancet. 2003b;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–912. [PMC free article] [PubMed] [Google Scholar]
- Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome: a review of its symptomatology, natural history, pathologic features, genetics and relationships to the natural aging process. Medicine. 1966;45:172–221. doi: 10.1097/00005792-196605000-00001. [DOI] [PubMed] [Google Scholar]
- Fukuchi K, Martin GM, Monnat RJ., Jr Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc Natl Acad Sci USA. 1989;86:5893–5897. [Google Scholar]
- Goto M, Miller RW, Ishikawa Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria) Cancer Epidemiol Biomarkers Prev. 1996;5:239–246. [PubMed] [Google Scholar]
- Goto M. Hierarchical deterioration of body systems in Werner's syndrome: implications for normal ageing. Mech Ageing Dev. 1997;98:239–254. doi: 10.1016/s0047-6374(97)00111-5. [DOI] [PubMed] [Google Scholar]
- Goto M, Imamura O, Kuromitsu J, Matsumoto T, Yamabe Y, Tokutake Y, Suzuki N, Mason B, Drayna D, Sugawara M, Sugimoto M, Furuichi Y. Analysis of helicase gene mutations in Japanese Werner's syndrome patients. Hum Genet. 1997;99:191–193. doi: 10.1007/s004390050336. [DOI] [PubMed] [Google Scholar]
- Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. The Werner syndrome protein is a DNA helicase. Nat Genet. 1997;17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, Wilson SH, Bohr VA. The Werner syndrome protein stimulates DNA polymerase beta strand displacement synthesis via its helicase activity. J Biol Chem. 2003;278:22686–22695. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- Huang S, Li B, Gray MD, Oshima J, Mian IS, Campisi J. The premature ageing syndrome protein, WRN, is a 3′–45′ exonuclease. Nat Genet. 1998;20:114–116. doi: 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Characterization of the human and mouse WRN 3′–45′ exonuclease. Nucleic Acids Res. 2000;28:2396–2405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson A, Peltz SW. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem. 1996;65:693–739. doi: 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L. The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J. 2001;20:905–913. doi: 10.1093/emboj/20.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine JP, Opresko PL, Indig FE, Harrigan JA, von Kobbe C, Bohr VA. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 2003;63:7136–7146. [PubMed] [Google Scholar]
- Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, Yasui A. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci. 2005;118:4153–4162. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- Lee JW, Kusumoto R, Doherty KM, Lin GX, Zeng W, Cheng WH, von Kobbe C, Brosh RM, Jr, Hu JS, Bohr VA. Modulation of Werner syndrome protein function by a single mutation in the conserved RecQ domain. J Biol Chem. 2005;280:39627–39636. doi: 10.1074/jbc.M506112200. [DOI] [PubMed] [Google Scholar]
- Li B, Comai L. Requirements for the nucleolytic processing of DNA ends by the Werner syndrome protein-ku70/80 complex. J Biol Chem. 2001;276:9896–9902. doi: 10.1074/jbc.M008575200. [DOI] [PubMed] [Google Scholar]
- Li B, Navarro S, Kasahara N, Comai L. Identification and biochemical characterization of a Werner's syndrome protein complex with Ku70/80 and poly(ADP-ribose) polymerase-1. J Biol Chem. 2004;279:13659–13667. doi: 10.1074/jbc.M311606200. [DOI] [PubMed] [Google Scholar]
- Liu Z, Macias MJ, Bottomley MJ, Stier G, Linge JP, Nilges M, Bork P, Sattler M. The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteins. Structure. 1999;7:1557–1566. doi: 10.1016/s0969-2126(00)88346-x. [DOI] [PubMed] [Google Scholar]
- Martin GM. Genetic syndromes in man with potential relevance to the pathobiology of aging. Birth Defects Orig Artic Ser. 1978;14:5–39. [PubMed] [Google Scholar]
- Martin GM, Oshima J, Gray MD, Poot M. What geriatricians should know about the Werner syndrome. J Am Geriatr Soc. 1999;47:1136–1144. doi: 10.1111/j.1532-5415.1999.tb05240.x. [DOI] [PubMed] [Google Scholar]
- Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Effects of donor's age, tissue, and genotype. Lab Invest. 1970;23:86–92. [PubMed] [Google Scholar]
- Matsumoto T, Imamura O, Yamabe Y, Kuromitsu J, Tokutake Y, Shimamoto A, Suzuki N, Satoh M, Kitao S, Ichikawa K, Kataoka H, Sugawara K, Thomas W, Mason B, Tsuchihashi Z, Drayna D, Sugawara M, Sugimoto M, Furuichi Y, Goto M. Mutation and haplotype analyses of the Werner's syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum Genet. 1997a;100:123–130. doi: 10.1007/s004390050477. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Shimamoto A, Goto M, Furuichi Y. Impaired nuclear localization of defective DNA helicases in Werner's syndrome. Nat Genet. 1997b;16:335–336. doi: 10.1038/ng0897-335. [DOI] [PubMed] [Google Scholar]
- Meisslitzer C, Ruppitsch W, Weirich-Schwaiger H, Weirich HG, Jabkowsky J, Klein G, Schweiger M, Hirsch-Kauffmann M. Werner syndrome: characterization of mutations in the WRN gene in an affected family. Eur J Hum Genet. 1997;5:364–370. [PubMed] [Google Scholar]
- Melaragno MI, Pagni D, Smith MA. Cytogenetic aspects of Werner's syndrome lymphocyte cultures. Mech Ageing Dev. 1995;78:117–122. doi: 10.1016/0047-6374(94)01530-y. [DOI] [PubMed] [Google Scholar]
- Mian IS. Comparative sequence analysis of ribonucleases HII, III, II PH and D. Nucleic Acids Res. 1997;25:3187–3195. doi: 10.1093/nar/25.16.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov V, Mushegian AR, Koonin EV, Bork P. A putative nucleic acid-binding domain in Bloom's and Werner's syndrome helicases. Trends Biochem Sci. 1997;22:417–418. doi: 10.1016/s0968-0004(97)01128-6. [DOI] [PubMed] [Google Scholar]
- Moser MJ, Holley WR, Chatterjee A, Mian IS. The proofreading domain of Escherichia coli DNA polymerase I and other DNA and/or RNA exonuclease domains. Nucleic Acids Res. 1997;25:5110–5118. doi: 10.1093/nar/25.24.5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser MJ, Oshima J, Monnat RJ., Jr WRN mutations in Werner syndrome. Hum Mutat. 1999;13:271–279. doi: 10.1002/(SICI)1098-1004(1999)13:4<271::AID-HUMU2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Moser MJ, Bigbee WL, Grant SG, Emond MJ, Langlois RG, Jensen RH, Oshima J, Monnat RJ., Jr Genetic instability and hematologic disease risk in Werner syndrome patients and heterozygotes. Cancer Res. 2000;60:2492–2496. [PubMed] [Google Scholar]
- Mushegian AR, Bassett DE, Jr, Boguski MS, Bork P, Koonin EV. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci USA. 1997;94:5831–5836. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakura J, Wijsman EM, Miki T, Kamino K, Yu CE, Oshima J, Fukuchi K, Weber JL, Piussan C, Melaragno MI, Epstein CJ, Scappaticci S, Fraccaro M, Matsumura T, Murano S, Yoshida S, Fujiwara Y, Saida T, Ogihara T, Martin GM, Schellenberg GD. Homozygosity mapping of the Werner syndrome locus (WRN) Genomics. 1994;23:600–608. doi: 10.1006/geno.1994.1548. [DOI] [PubMed] [Google Scholar]
- Ogburn CE, Oshima J, Poot M, Chen R, Hunt KE, Gollahon KA, Rabinovitch PS, Martin GM. An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum Genet. 1997;101:121–125. doi: 10.1007/s004390050599. [DOI] [PubMed] [Google Scholar]
- Okada M, Goto M, Furuichi Y, Sugimoto M. Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner's syndrome patients. Biol Pharm Bull. 1998;21:235–239. doi: 10.1248/bpb.21.235. [DOI] [PubMed] [Google Scholar]
- Orren DK, Theodore S, Machwe A. The Werner syndrome helicase/exonuclease (WRN) disrupts and degrades D-loops in vitro. Biochemistry. 2002;41:13483–13488. doi: 10.1021/bi0266986. [DOI] [PubMed] [Google Scholar]
- Oshima J, Yu CE, Piussan C, Klein G, Jabkowski J, Balci S, Miki T, Nakura J, Ogihara T, Ells J, Smith M, Melaragno MI, Fraccaro M, Scappaticci S, Matthews J, Ouais S, Jarzebowicz A, Schellenberg GD, Martin GM. Homozygous and compound heterozygous mutations at the Werner syndrome locus. Hum Mol Genet. 1996;5:1909–1913. doi: 10.1093/hmg/5.12.1909. [DOI] [PubMed] [Google Scholar]
- Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res. 1992;202:267–273. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- Poot M, Gollahon KA, Rabinovitch PS. Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum Genet. 1999;104:10–14. doi: 10.1007/s004390050903. [DOI] [PubMed] [Google Scholar]
- Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, Rabinovitch PS. Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J. 2001;5:5. doi: 10.1096/fj.00-0611fje. [DOI] [PubMed] [Google Scholar]
- Poot M, Gollahon KA, Emond MJ, Silber, Rabinovitch PS. Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J. 2002;16:757–758. doi: 10.1096/fj.01-0906fje. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Nishikawa K, Heo SJ, Goto M, Furuichi Y, Shimamoto A. Werner helicase relocates into nuclear foci in response to DNA damaging agents and co-localizes with RPA and Rad51. Genes Cells. 2001;6:421–430. doi: 10.1046/j.1365-2443.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- Salk D, Au K, Hoehn H, Martin GM. Cytogenetic aspects of Werner syndrome. Adv Exp Med Biol. 1985;190:541–546. doi: 10.1007/978-1-4684-7853-2_27. [DOI] [PubMed] [Google Scholar]
- Satoh M, Imai M, Sugimoto M, Goto M, Furuichi Y. Prevalence of Werner's syndrome heterozygotes in Japan. Lancet. 1999;353:1766. doi: 10.1016/S0140-6736(98)05869-3. [DOI] [PubMed] [Google Scholar]
- Schulz VP, Zakian VA, Ogburn CE, McKay J, Jarzebowicz AA, Edland SD, Martin GM. Accelerated loss of telomeric repeats may not explain accelerated replicative decline of Werner syndrome cells. Hum Genet. 1996;97:750–754. doi: 10.1007/BF02346184. [DOI] [PubMed] [Google Scholar]
- Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hubscher U, Brosh RM., Jr The interaction site of Flap Endonuclease-1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–6781. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommers JA, Sharma S, Doherty KM, Karmakar P, Yang Q, Kenny MK, Harris CC, Brosh RM., Jr p53 modulates RPA-dependent and RPA-independent WRN helicase activity. Cancer Res. 2005;65:1223–1233. doi: 10.1158/0008-5472.CAN-03-0231. [DOI] [PubMed] [Google Scholar]
- Spillare EA, Robles AI, Wang XW, Shen JC, Yu CE, Schellenberg GD, Harris CC. p53-mediated apoptosis is attenuated in Werner syndrome cells. Genes Dev. 1999;13:1355–1360. doi: 10.1101/gad.13.11.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Shiratori M, Furuichi Y, Matsumoto T. Diverged nuclear localization of Werner helicase in human and mouse cells. Oncogene. 2001;20:2551–2558. doi: 10.1038/sj.onc.1204344. [DOI] [PubMed] [Google Scholar]
- Szekely AM, Chen YH, Zhang C, Oshima J, Weissman SM. Werner protein recruits DNA polymerase delta to the nucleolus. Proc Natl Acad Sci USA. 2000;97:11365–11370. doi: 10.1073/pnas.97.21.11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara H, Tokutake Y, Maeda S, Kataoka H, Watanabe T, Satoh M, Matsumoto T, Sugawara M, Ide T, Goto M, Furuichi Y, Sugimoto M. Abnormal telomere dynamics of B-lymphoblastoid cell strains from Werner's syndrome patients transformed by Epstein-Barr virus. Oncogene. 1997;15:1911–1920. doi: 10.1038/sj.onc.1201377. [DOI] [PubMed] [Google Scholar]
- Tollefsbol TO, Cohen HJ. Werner's syndrome: an under-diagnosed disorder resembling premature aging. Age. 1984;7:75–88. [Google Scholar]
- von Kobbe C, Bohr VA. A nucleolar targeting sequence in the Werner syndrome protein resides within residues 949–1092. J Cell Sci. 2002;115:3901–3907. doi: 10.1242/jcs.00076. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, Thoma NH, Czyzewski BK, Pavletich NP, Bohr VA. Werner syndrome protein contains three structure-specific DNA binding domains. J Biol Chem. 2003;278:52997–53006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- von Kobbe C, Harrigan JA, Schreiber V, Stiegler P, Piotrowski J, Dawut L, Bohr VA. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32:4003–4014. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamabe Y, Sugimoto M, Satoh M, Suzuki N, Sugawara M, Goto M, Furuichi Y. Down-regulation of the defective transcripts of the Werner's syndrome gene in the cells of patients. Biochem Biophys Res Commun. 1997;236:151–154. doi: 10.1006/bbrc.1997.6919. [DOI] [PubMed] [Google Scholar]
- Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA- dependent protein kinase. J Biol Chem. 2001;276:38242–38248. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S, Nakura J, Miki T, Ouais S, Martin GM, Mulligan J, Schellenberg GD. Positional cloning of the Werner's syndrome gene. Science. 1996;272:258–262. doi: 10.1126/science.272.5259.258. [DOI] [PubMed] [Google Scholar]
- Yu CE, Oshima J, Wijsman EM, Nakura J, Miki T, Piussan C, Matthews S, Fu YH, Mulligan J, Martin GM, Schellenberg GD. Mutations in the consensus helicase domains of the Werner syndrome gene. Werner's Syndrome Collaborative Group. Am J Hum Genet. 1997;60:330–341. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.