

Abstract
Enzymatic activities of the proteins encoded in nuclear genome are regulated by transcriptional, translational and post-transcriptional level. Enzymatic activities of proteins encoded in mitochondrial DNA (mtDNA) have been considered to be regulated by the same steps although detailed mechanisms might differ. However, dynamic change of the number of mtDNA, from some hundred to more than ten thousand, should be considered as another novel mechanism to regulate mtDNA-encoded proteins. Recently, we showed the connection of mtDNA depletion and deletion to cancer progression (Higuchi et al., 2006). This review focuses and describes the possible connections of the mitochondrial DNA depletion and deletion to cancer.
Introduction
Mitochondria are essential organelles that generate cellular energy (ATP) through oxidative phosphorylation and this process is accomplished by a series of protein complexes, mitochondrial respiratory chains (MRC), encoded by nuclear DNA and mtDNA. Although ATP generation through oxidative phosphorylation is most efficient, it is not only way to produce cellular energy. Glycolysis can also generate ATP and provides compensatory mechanisms when oxidative phosphorylation becomes inefficient because of defects in respiratory chain. Human mtDNA is remarkably small (16,569 bp) compared with nuclear DNA (approximately 109 bp). Mitochondrial DNA encodes only 13 polypeptides in the MRC and the majority of mitochondrial respiratory proteins (at least 74 proteins) are encoded by nuclear DNA that are translated in the cytoplasm and then imported into mitochondria.
More than 50 years ago, Warburg pioneered the research on mitochondrial respiratory alterations in the context of cancer (Warburg, 1956). In his series of publications, he hypothesized that a key event in carcinogenesis involved the development of an ‘injury’ to the respiratory machinary, resulting in compensatory increases in glycolytic ATP production. Several reports showed that mutations of mtDNA have been identified in various types of cancer including breast cancer, colon carcinoma, prostate cancer, pancreatic cancer, and others (Carew and Huang, 2002). Decrease for mtDNA in renal cancer (Selvanayagam and Rajaraman, 1996), hepatocellular carcinoma (Lee et al., 2004; Yin et al., 2004) and gastric cancer (Wu et al., 2005) has also been reported. Additionally, Simonnet et al. showed that a decrease in mitochondrial respiratory function was observed in accordance with increased invasiveness of cancer (Simonnet et al., 2002). It is very likely that mtDNA mutation, deletion or depletion might induce the changes which Warburg hypothesized. Additionally, since the inheritance of mitochondrial haplotype U is associated with approximately 2 –fold increased risk of prostate cancer and 2.5-fold increased risk of renal cancer in white North America individuals (Booker et al., 2006), mtDNA definitely affects initiation of cancer. Growth advantage of the cancer cells with specific mutant mtDNA were also demonstrated in vivo mouse model system using cybrid (transmitochondrial hybrid) cells (Petros et al., 2005; Shidara et al., 2005), indicating that specific mutations of mtDNA give advantage of survival to cancer cells. However, Coller et al. (Coller et al., 2001) showed that cancer progenitor cells already achieve homoplasmy through stochastic redistribution of the mitochondrial mutation and claimed that replicative advantage of point mutant mtDNA and selective expansion of cancer cells with specific mutant mtDNA are not always necessary to explain homoplasmy of mutant mtDNA in cancer.
Our recent finding indicate that the depletion and deletion of mtDNA is important for cancer progression (Higuchi et al., 2006). Here, we review the roles of mtDNA depletion and deletion on cancer and the possible causes for mtDNA depletion and deletion.
Depletion of mtDNA and cancer progression
Mitochondrial DNA deficient cells (ρ0) were established by long-term exposure of low concentration of ethidium bromide (King and Attardi, 1989). Roles of mtDNA and mitochondrial respiratory function on biological function including apoptosis using ρ0 cells has been published (Chandel and Schumacker, 1999). We showed that TNF and serum starvation could not induce apoptosis in respiration-deficient cells, whereas they induced apoptosis in parental cells and cells reconstituted with normal mtDNA (Higuchi et al., 1997). These results indicate that depletion of mtDNA leads cells resistant to a certain apoptosis pathway. Avadhani and colleagues (Amuthan et al., 2001) demonstrated that mtDNA depleted murine skeletal myoblasts C2C12 cells showed invasive phenotypes and overexpression of the tumor-specific markers cathepsin L and transforming growth factor β indicating that the loss of mtDNA could contribute to tumor progression and metastasis. Furthermore, they also demonstrated that mtDNA depletion activated NFκB/Rel factors through the inactivation of IκBβ through calcineurin-mediated dephosphorylation and caused the phenotype changes (Biswas et al., 2003). It is thus likely that mtDNA depletion could affect progression and metastasis of cancer cells by preventing apoptosis and generating cancer-related proteins.
The concept of a mutator phenotype in cancer was formulated to account for the disparity between the rarity of mutations in normal cells and the large number of mutations present in a variety of human malignancies (Loeb, 2001). Singh and colleagues (Rasmussen et al., 2003) reported that mitochondrial dysfunction leads to a nuclear mutator phenotype (i) through ROS-dependent pathway induced by complex III inhibitor antimycin A and (ii) through ROS-independent pathway induced by the depletion of mtDNA. Thus, depletion or deletion of mtDNA in cancer cells can be responsible for the generation of mutator phenotype leading to further progression and/or initiation of cancer through ROS-independent pathway. Thus, two hypothesized pathway exist, i.e., reversible pathway by Avadhani, and irreversible by Singh for cancer progression.
MtDNA determines androgen dependence in prostate cancer cell line
Androgen-dependent cells can become androgen independent following androgen ablation in vivo and in vitro leading to a more progressed prostate cancer (Kokontis et al., 1998; Thalmann et al., 2000). Such changes in prostate cancers induced by androgen ablation must be caused by a change in nuclear DNA (possibly mutation or epigenetic effects) or mtDNA (mutation, deletion or depletion of mtDNA). Prostate cancer is associated with aging and mtDNA depletion and deletion mutations accumulate with age in many tissues of the body, suggesting a potential link. Heteroplasmic large deletion mutant mtDNA is very common in prostate cancer (Jessie et al., 2001). We investigated the roles of mtDNA by using human androgen-dependent prostate cancer cell line LNCaP and androgen-independent C4-2, sub clone derived by inoculation of LNCaP into castrated mice (Wu et al., 1994), mtDNA deficient cells derived from LNCaP, and cybrids which has nuclear DNA from LNCaP with healthy mtDNA. We demonstrated that decrease in mtDNA and mitochondrial respiratory function induced by androgen ablation made androgen-dependent prostate cancer to androgen independent by the following observations (Higuchi et al., 2006). 1) The amount of normal mtDNA was greatly reduced after androgen ablation (Figure 1). In addition, the amount of large deletion mutant mtDNA was greatly increased. 2) These changes lead to the inhibition of mitochondrial respiratory function. 3) Depletion of mtDNA (LNρ0–8 Figure 1) from androgen-dependent human prostate cancer cell line LNCaP resulted in a loss of androgen dependence in vitro and in vivo. 4) Reconstitution of normal mtDNA to mtDNA-depleted clone reversed androgen-dependence. The role of mtDNA in androgen dependence was illustrated in Figure 2.
Figure 1.
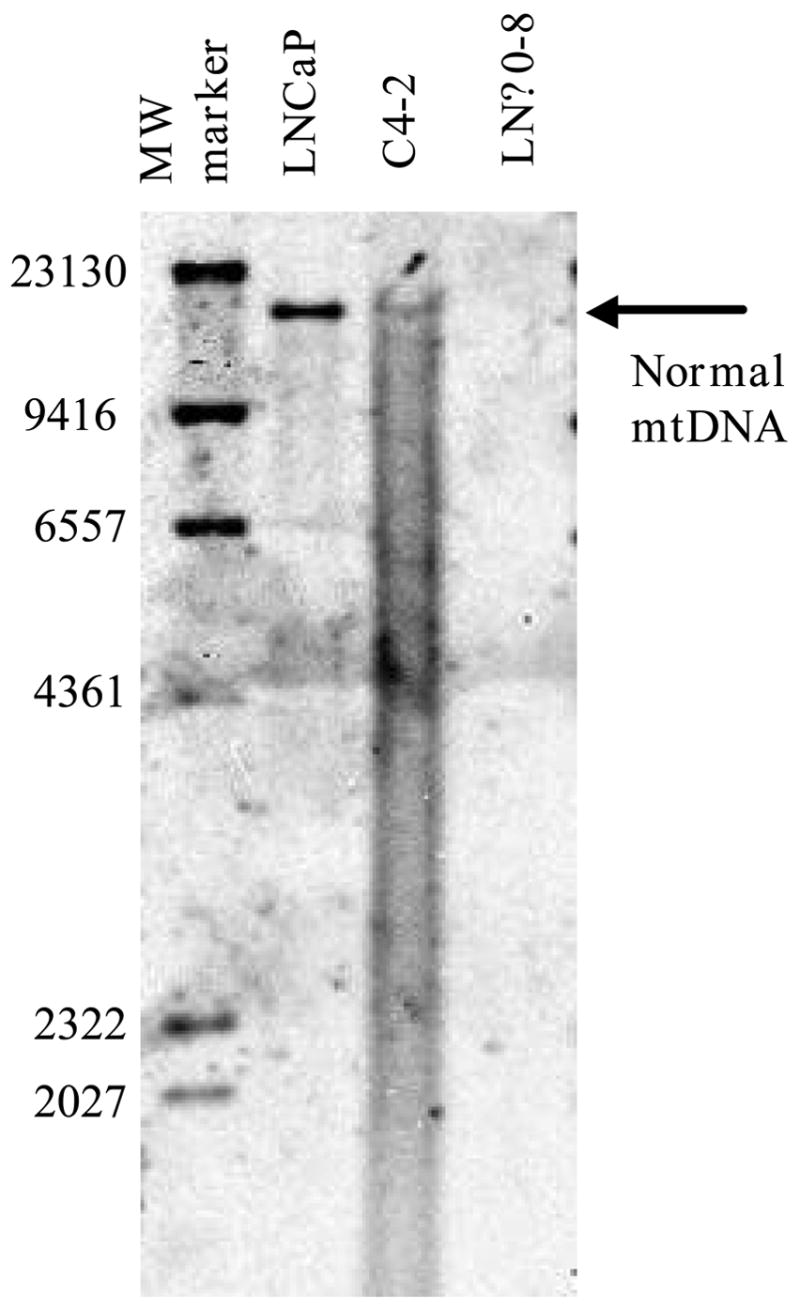
mtDNA amount of LNCaP, C4-2 and LNρ0-8 detected by Southern Blotting.
Figure 2.
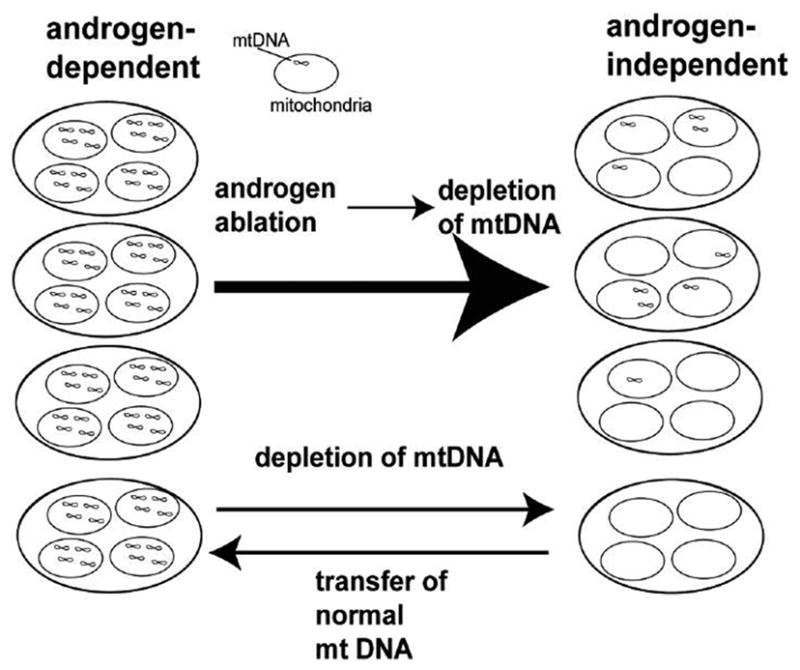
Roles of mitochondrial DNA in androgen-dependence
Causes for mtDNA depletion and deletion
The only non-coding segment of mtDNA is the displacement loop (D-loop), a region of 1121 bp that contains the origin of replication of the H-strand (OH) and the promoters for L and H-strand transcription. Mutation of D-loop region in cancer cells is very common (Carew and Huang, 2002). D-loop region is responsible for the control of replication and transcription of mtDNA. Thus, the D-loop alterations may interfere with sequence in the promoter regions and modify the binding affinities of the inducers and/or modulators of mtDNA transcription, and thus changes the rate of transcription and replication of mtDNA (Clayton, 1991). Wei and his colleagues showed that 39.3% of the hepatocellular cancer had mutation in D-loop region of mtDNA and that the copy number of mtDNA was significantly reduced in 70.8% of the cancer with mutation in D-loop region (Lee et al., 2004). Therefore, it is likely that mtDNA mutation in D-loop region might be one of the causes for the depletion of mtDNA. Turner et. al. showed that a well-characterized pathological mutation at nucleotide position 3243 of human mtDNA induces the depletion of mtDNA (Turner et al., 2005). They introduced mutant A3243G mtDNA to human mtDNA-deficient teratocarcinoma cells and found the mitotic segregation toward increasing level of mutant mtDNA followed by the loss of mtDNA. This report indicates that a point mutation other than the D-loop region can be the cause for mtDNA depletion.
It is suggested that point mutation to mtDNA can be the cause for multiple large-deletion mutant mtDNA and depletion of mtDNA (Nishigaki et al., 2004). In 1989, Zeviani and colleagues described autosomal dominant progressive external ophthalmoplegia (adPEO) with multiple mtDNA deletion, the first of several diseases attributed to defects of nuclear DNA leading to the disorder of mtDNA (Zeviani et al., 1989). Mutations in the mitochondrial proteins adenine nucleotide translocator 1 (ANT1) (Kaukonen et al.), Twinkle (Spelbrink et al.) and polymerase γ (Van Goethem et al.) have been found to cause autosomal dominant progressive external ophthalmoplegia with multiple deletion of mtDNA. Mitochondrial Neurogastrointestinal Encephalomyopathy (MINGIE) is an autosomal recessive disorder due to loss-of-function mutations in the gene encoding thymidine phosphorylase, associated with multiple deletions, depletion and site-specific point mutations of mtDNA (Hirano et al., 1994; Nishigaki et al., 2003; Papadimitriou et al., 1998). ANT1 forms a homodimeric inner mitochondrial membrane channel that translocates ADP into ATP out of the mitochondrial matrix. Therefore, this protein regulates concentration of adenine nucleotides in the cytoplasm and mitochondria. Twinkle is a mitochondrial protein with homology to phage T7 primase/helicase, and the mutation to Twinkle enhances dNTPase activity (Washington et al., 1996). The identification of mutations of genes encoding ANT1 and Twinkle in patients with adPEO and thymidine phosphorylase in patients with MINGIE indicate that imbalance of mitochondrial nucleotide pools may cause multiple deletion of mtDNA and depletion of mtDNA. Individuals lacking deoxycitidine kinase were described and they showed severe hepatocerebral syndromes due to mtDNA depletion in the affected tissues (Mandel et al., 2001). Mutation of thymidine kinase 2 causes described in patients with severe mtDNA depletion myopathy (Saada et al., 2001). In contrast, mutations of polymerase γ and Twinkle leading to the change of mtDNA may be caused by the defects in the mtDNA repair and replication machinery.
Another possible pathway to induce mtDNA mutation is the ROS-associated pathway. The mitochondrial genome is extremely susceptible to damages from constant exposure to ROS produced endogenously from MRC. Mitochondrial DNA has been shown to accumulate high levels of 8-hydroxy-2’-deoxyguanosine (8-oxo-G), the product of hydroxylation of guanine at carbon 8, which is a mutagenic lesion. The base excision repair pathway repairs most of these small-base modifications. The 8-oxoguanine-DNA glycosylase 1 (OGG1) protein is the major DNA glycosylase for the repair of 8-oxo-G lesions in the DNA. Inactivation of OGG1 leads to the accumulation of point mutations and deletion mutations in mtDNA (Singh et al., 2001). Polymerase γ is another key enzyme in the repair of 8-oxo-G lesions in the DNA induced by ROS, and transgenic mice expressing a proofreading-deficient polymerase γ exhibit accumulation of point and deletion mutations in mtDNA (Zhang et al., 2000). To protect against the effects of ROS, mitochondria metabolize superoxide and hydrogen peroxide with MnSOD and Se-containing glutathione peroxidase, respectively. ROS have been thought to be involved in the increase in the proportion of both point mutant and deletion mutant mtDNA (Ozawa, 1997). Given these observations, it is likely that the inhibition of the repair system for ROS-mediated damage to mtDNA, detoxification of ROS, or increase in ROS generation might be possible causes for point mutations of mtDNA, accumulation of large-deletion mutant mtDNA, and the depletion of mtDNA. Additionally, the report indicates that oxidative stress generated by myocardial infraction (Ide et al., 2001) and TNF (Suematsu et al., 2003) rapidly and directly induced depletion of mtDNA. It is likely that mtDNA depletion can be induced independent of mtDNA mutation.
The p53 tumor suppressor protein plays a central role in response to DNA damage, cell cycle regulation, and apoptosis. More than 50% of human cancers carry mutations in p53 (Vogelstein et al., 2000). In addition to its role as a transcription factor, p53 protein can translocate to the mitochondria in response to certain stimuli, and induces apoptosis (Arima et al., 2005). Achanta G. et.al. (Achanta et al., 2005) reported that p53 has a novel role in maintaining mtDNA stability through its ability to translocate to mitochondria and interacts with polymerase γ in response to mtDNA damage induced by exogenous and endogenous insults including ROS. The p53 protein physically interacts with mtDNA and polymerase γ, and enhances the DNA replication function of polymerase γ. Loss of p53 results in a significant increase in mtDNA damage and possibly leads to the depletion and the deletion of mtDNA.
Therefore, mutation of the nuclear DNA or the change in microenvironment, which induce the change in nucleotide pool, mtDNA repair mechanisms and ROS generation, might be responsible for the mtDNA mutation and depletion or deletion of mtDNA leading to cancer progression. It is very likely that the same mechanisms might be working in the delayed-onset and progressive course of the age-related diseases that is hypothesized by Wallace (Wallace, 2005).
Acknowledgments
This work has been supported by NIH grant CA100846.
Nonstandard abbreviation used
- mtDNA
mitochondrial DNA
- MRC
mitochondrial respiratory chain
- ROS
reactive oxygen species
- adPEO
autosomal dominant progressive external ophthalmoplegia
- MINGIE
mitochondrial neurogastrointestinal Encephalomyopathy
- 8-oxo-G
8-hydroxy-2’-deoxyguanosine
- ANT1
adenine nucleotide translocator 1
- OGG1
8-Oxoguanine-DNA glycosylase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, Huang P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO Journal. 2005;24:3482–3492. doi: 10.1038/sj.emboj.7600819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. Embo J. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. Journal of Biological Chemistry. 2005;280:19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG. Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. Journal of Cell Biology. 2003;161:507–519. doi: 10.1083/jcb.200211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker LM, Habermacher GM, Jessie BC, Sun QC, Baumann AK, Amin M, Lim SD, Fernandez-Golarz C, Lyles RH, Brown MD, Marshall FF, Petros JA. North American white mitochondrial haplogroups in prostate and renal cancer. Journal of Urology. 2006;175:468–472. doi: 10.1016/S0022-5347(05)00163-1. [DOI] [PubMed] [Google Scholar]
- Carew JS, Huang P. Mitochondrial defects in cancer. Molecular Cancer. 2002;1:1–12. doi: 10.1186/1476-4598-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Schumacker PT. Cells depleted of mitochondrial DNA (rho0) yield insight into physiological mechanisms. FEBS Letters. 1999;454:173–176. doi: 10.1016/s0014-5793(99)00783-8. [DOI] [PubMed] [Google Scholar]
- Clayton DA. Replication and transcription of vertebrate mitochondrial DNA. Annu Rev Cell Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- Coller HA, Khrapko K, Bodyak ND, Nekhaeva E, Herrero-Jimenez P, Thilly WG. High frequency of homoplasmic mitochondrial DNA mutations in human tumors can be explained without selection. Nat Genet. 2001;28:147–150. doi: 10.1038/88859. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Aggarwal BB, Yeh ET. Activation of CPP32-like protease in tumor necrosis factor-induced apoptosis is dependent on mitochondrial function. Journal of Clinical Investigation. 1997;99:1751–1758. doi: 10.1172/JCI119339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Kudo T, Suzuki S, Evans TT, Sasaki R, Wada Y, Shirakawa T, Sawyer JR, Gotoh A. Mitochondrial DNA determines androgen dependence in prostate cancer cell lines. Oncogene. 2006;25:1437–1445. doi: 10.1038/sj.onc.1209190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, Hays AP, Lovelace RE, Butler I, Bertorini TE. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–727. doi: 10.1212/wnl.44.4.721. [DOI] [PubMed] [Google Scholar]
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circulation Research. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- Jessie BC, Sun CQ, Irons HR, Marshall FF, Wallace DC, Petros JA. Accumulation of mitochondrial DNA deletions in the malignant prostate of patients of different ages. Exp Gerontol. 2001;37:169–174. doi: 10.1016/s0531-5565(01)00153-x. [DOI] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, Keranen S, Peltonen L, Suomalainen A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kokontis JM, Hay N, Liao S. Progression of LNCaP prostate tumor cells during androgen deprivation: hormone-independent growth, repression of proliferation by androgen, and role for p27Kip1 in androgen-induced cell cycle arrest. Mol Endocrinol. 1998;12:941–953. doi: 10.1210/mend.12.7.0136. [DOI] [PubMed] [Google Scholar]
- Lee HC, Li SH, Lin JC, Wu CC, Yeh DC, Wei YH. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutation Research. 2004;547:71–78. doi: 10.1016/j.mrfmmm.2003.12.011. [DOI] [PubMed] [Google Scholar]
- Loeb LA. A mutator phenotype in cancer. Cancer Research. 2001;61:3230–3239. [PubMed] [Google Scholar]
- Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, Shalata A, Anbinder Y, Berkowitz D, Hartman C, Barak M, Eriksson S, Cohen N. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nature Genetics. 2001;29:337–341. doi: 10.1038/ng746. [DOI] [PubMed] [Google Scholar]
- Nishigaki Y, Marti R, Copeland WC, Hirano M. Site-specific somatic mitochondrial DNA point mutations in patients with thymidine phosphorylase deficiency. Journal of Clinical Investigation. 2003;111:1913–1921. doi: 10.1172/JCI17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishigaki Y, Marti R, Hirano M. ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Human Molecular Genetics. 2004;13:91–101. doi: 10.1093/hmg/ddh010. [DOI] [PubMed] [Google Scholar]
- Ozawa T. Genetic and functional changes in mitochondria associated with aging. Physiol Rev. 1997;77:425–464. doi: 10.1152/physrev.1997.77.2.425. [DOI] [PubMed] [Google Scholar]
- Papadimitriou A, Comi GP, Hadjigeorgiou GM, Bordoni A, Sciacco M, Napoli L, Prelle A, Moggio M, Fagiolari G, Bresolin N, Salani S, Anastasopoulos I, Giassakis G, Divari R, Scarlato G. Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology. 1998;51:1086–1092. doi: 10.1212/wnl.51.4.1086. [DOI] [PubMed] [Google Scholar]
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, Lim S, Issa MM, Flanders WD, Hosseini SH, Marshall FF, Wallace DC. mtDNA mutations increase tumorigenicity in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen AK, Chatterjee A, Rasmussen LJ, Singh KK. Mitochondria-mediated nuclear mutator phenotype in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:3909–3917. doi: 10.1093/nar/gkg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nature Genetics. 2001;29:342–344. doi: 10.1038/ng751. [DOI] [PubMed] [Google Scholar]
- Selvanayagam P, Rajaraman S. Detection of mitochondrial genome depletion by a novel cDNA in renal cell carcinoma. Laboratory Investigation. 1996;74:592–599. [PubMed] [Google Scholar]
- Shidara Y, Yamagata K, Kanamori T, Nakano K, Kwong JQ, Manfredi G, Oda H, Ohta S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Research. 2005;65:1655–1663. doi: 10.1158/0008-5472.CAN-04-2012. [DOI] [PubMed] [Google Scholar]
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- Singh KK, Sigala B, Sikder HA, Schwimmer C. Inactivation of Saccharomyces cerevisiae OGG1 DNA repair gene leads to an increased frequency of mitochondrial mutants. Nucleic Acids Res. 2001;29:1381–1388. doi: 10.1093/nar/29.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, Wanrooij S, Garrido N, Comi G, Morandi L, Santoro L, Toscano A, Fabrizi GM, Somer H, Croxen R, Beeson D, Poulton J, Suomalainen A, Jacobs HT, Zeviani M, Larsson C. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nature Genetics. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M, Pathak S, Chung LW. LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate. 2000;44:91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. Jul 101;144(102) [DOI] [PubMed] [Google Scholar]
- Turner CJ, Granycome C, Hurst R, Pohler E, Juhola MK, Juhola MI, Jacobs HT, Sutherland L, Holt IJ. Systematic segregation to mutant mitochondrial DNA and accompanying loss of mitochondrial DNA in human NT2 teratocarcinoma Cybrids. Genetics. 2005;170:1879–1885. doi: 10.1534/genetics.105.043653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nature Genetics. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origine of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Washington MT, Rosenberg AH, Griffin K, Studier FW, Patel SS. Biochemical analysis of mutant T7 primase/helicase proteins defective in DNA binding, nucleotide hydrolysis, and the coupling of hydrolysis with DNA unwinding. J Biol Chem. 1996;271:26825–26834. doi: 10.1074/jbc.271.43.26825. [DOI] [PubMed] [Google Scholar]
- Wu CW, Yin PH, Hung WY, Li AF, Li SH, Chi CW, Wei YH, Lee HC. Mitochondrial DNA mutations and mitochondrial DNA depletion in gastric cancer. Genes, Chromosomes & Cancer. 2005;44:19–28. doi: 10.1002/gcc.20213. [DOI] [PubMed] [Google Scholar]
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. Int J Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- Yin PH, Lee HC, Chau GY, Wu YT, Li SH, Lui WY, Wei YH, Liu TY, Chi CW. Alteration of the copy number and deletion of mitochondrial DNA in human hepatocellular carcinoma. British Journal of Cancer. 2004;90:2390–2396. doi: 10.1038/sj.bjc.6601838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Construction of transgenic mice with tissue-specific acceleration of mitochondrial DNA mutagenesis. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]