

Abstract
Latent membrane protein 2A (LMP2A) of Epstein-Barr virus (EBV) plays a key role in regulating viral latency and EBV pathogenesis by functionally mimicking signals induced by the B-cell receptor (BCR) altering normal B cell development. LMP2A specifically associates with Nedd4-family ubiquitin-protein ligases which downmodulate LMP2A activity by ubiquitinating LMP2A and LMP2A-associated protein tyrosine kinases (PTKs). Since specific ubiquitin tags provide an endocytic sorting signal for plasma membrane proteins which traffic to membrane vesicles, we examined LMP2A localization and trafficking. We found that LMP2A is secreted through exosomes, small endocytic membrane vesicles, as previously demonstrated for LMP1. Interestingly, the treatment of cells with methyl-beta-cyclodextrin (MCD), which depletes cholesterol from plasma membrane, dramatically increased LMP2A abundance and LMP2A exosome secretion. Cholesterol depletion also blocked LMP2A endocytosis resulting in the accumulation of LMP2A on plasma membrane. LMP2A phosphorylation and ubiquitination were blocked by cholesterol depletion. LMP2A in the exsosomal fraction was ubiquitinated but not phosphorylated. These results indicate that cholesterol-dependent LMP2A trafficking determines the fate of LMP2A degradation.
Keywords: Epstein-Barr virus (EBV), Latent membrane protein 2A (LMP2A), ubiquitin, Nedd4-family ubiquitin-protein ligase, choresterol, methyl-beta-cyclodextrin, endocytosis, exosome
Introduction
Latent membrane protein 2A (LMP2A) is an Epstein-Barr virus (EBV) encoded protein that has been implicated in regulating viral latency and pathogenesis in EBV infected cells. The LMP2A amino-terminal domain interacts and activates protein tyrosine kinases (PTKs) such as Lyn and Syk (Fruehling and Longnecker, 1997; Fruehling et al., 1998) in a constitutive manner mimicking a BCR providing development and survival signals for B cells in the absence of corresponding antigens (Caldwell et al., 1998). Two PY motifs (PPXY) in the LMP2A amino-terminus specifically associate with Nedd4-family ubiquitin-protein ligases resulting in the downmodulation of LMP2A activity by ubiquitinating both LMP2A and LMP2A-associated PTKs (Ikeda et al., 2000; Ikeda et al., 2001; Winberg et al., 2000). LMP2A ubiquitination negatively regulates LMP2A signal transduction in B cell development (Ikeda et al., 2003; Ikeda et al., 2004). LMP2A ubiquitin-dependent processes are likely important for LMP2A function in EBV latent infection such as the modulation of LMP2A-induced signals altering normal B cell development (Casola et al., 2004; Ikeda et al., 2004).
Recently, new roles in normal cellular processes and virus replication for ubiquitination have been determined besides the well-established role of ubiquitination as a marker for proteins to be degraded by the proteosome. Ubiquitination plays an essential role in the budding process of virus particles in retroviruses and rhabdoviruses by recruitment of ubiquitin ligases to PY motifs found in viral encoded proteins (Morita and Sundquist, 2004). The PY motif in viral structural proteins has been shown to function as the late assembly domain (L domain). For example, mutations in the PY-motif-type L domains of the Rous sarcoma virus (RSV) Gag protein and the vesicular stomatitis virus (VSV) M protein result in the absence of viral budding at late stages of virus replication (Jayakar et al., 2000; Xiang et al., 1996). Nedd4-family ubiquitin-protein ligases bind to the L domain and ubiquitinate these viral proteins (Vana et al., 2004). The importance of ubiquitination in the trafficking of plasma membrane proteins also has recently been demonstrated. Multi-ubiquitin tagged proteins are recognized by proteasome to be degraded, while mono-ubiquitin tags provide an endocytic sorting signal for plasma membrane proteins which traffic to intracellular membrane vesicles (Hicke, 2001). In this study, we examined LMP2A localization to investigate factors important for LMP2A trafficking.
Materials and Methods
Cell culture
All cell lines were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin (complete RPMI1640 medium). LCL1 is a B95-8 EBV-transformed LCL expressing wild-type LMP2A (Longnecker and Kieff, 1990). ES1 is a LCL harboring LMP2A-deleted EBV genome (Longnecker et al., 1993). BJAB is an EBV negative B lymphoma cell line obtained from ATCC (Rockville, MD). BJAB cell lines expressing LMP2A were generated by infection with retrovirus stocks using a previously described LMP2A retroviral expression construct (Longnecker and Kieff, 1990). Various inhibitors were purchased and used at the concentrations shown in Table 1.
Table 1.
Various inhibitors used in study.
| Inhibitor | Cellular target | Concentration | Vendor |
|---|---|---|---|
| Lactacystin | Proteasome | 2, 10 μM | Calbiochem |
| Chloroquine | Lysosomal protease | 10, 50 μM | Sigma |
| Methyl-beta-cyclodextrin (MCD) | Cholesterol | 0.75, 4 mM | Sigma |
| Sodium vanadate | Protein tyrosine phosphatase | 20, 100 μM | Fisher |
| Genistein | Pan-PTK | 10, 50 μM | Sigma |
| PP2 | Src-PTK | 2, 10 μM | Calbiochem |
Antibodies
The anti-LMP2A rat monoclonal antibody (14B7) (Fruehling et al., 1996) and the anti-LMP2A rabbit polyclonal antibody (Longnecker and Kieff, 1990) have been previously described. The anti-HLA-DR mouse monoclonal antibody was purchased from DAKO. The anti-LAMP1 mouse monoclonal antibody was from BD. The anti-GAPDH mouse monoclonal antibody was from Abcam. The anti-phosphotyrosine mouse monoclonal antibody (PY20) and the anti-ubiquitin mouse monoclonal antibody were from Santa Cruz. The anti-LMP1 mouse monoclonal antibody (S12) was described previously (Mann et al., 1985). All horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Amersham. All FITC- and Cy3-conjugated secondary antibodies were purchased from Jackson ImmunoResearch Laboratory.
Exosome preparation
Growing cells were inoculated into fresh complete medium with a density of 5×105 cells/ml. Cells were grown for 0–6 days without changing the media, and then pelleted at 1,500 rpm (cell fraction). The resulting supernatant, collected on different days, was cleared by centrifugation at 3,000 rpm and exosomes were isolated by centrifugation at 30,000 rpm (exosome fraction). Pelleted cell and exosome fractions were solubilized and analyzed.
Immunoprecipitation
A total of 1×107 cells were harvested and lysed in 1 ml of Triton X-100 lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 10% glycerol, 10 mM NaF, 1 mM Na3VO4, 2 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride, 10 μg/ml pepstatin, 10 μg/ml leupeptin). Cleared lysates were incubated with the appropriate antibody for 1 hour at 4°C. Immune complexes were captured with 20 μl of Protein A or G-Sepharose (Pharmacia) for 1 hour at 4°C. Following, three washes with lysis buffer, immunoprecipitated proteins were re-suspended in 2X SDS-PAGE sample buffer.
Immunoblotting
Protein samples were resolved by SDS-PAGE, transferred to Immobilon, and blocked with 4% skim milk or 4% bovine serum albumin (BSA) at room temperature for 1 hour. Membranes were then incubated in 4% skim milk or 4% BSA in TBST (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.05% Tween 20) with primary antibody for 1 hour, and then with appropriate secondary antibody for 1 hour. Following incubation, the membranes were washed with TBST, and the blot was visualized using ECL (Amersham).
Immunofluorescence
Suspended cells were washed with 1X PBS, dried on slides rapidly at 37°C, and fixed in methanol at −20°C for 10 min. The fixed cells were incubated with primary antibody in 1X PBS with 20% goat serum (GIBCO) for 24 hours at room temperature, and then incubated with appropriate secondary antibody for 1 hour. The slides mounted with Gelvatol (16.7% polyvinyl alcohol and 33.3% glycerol in PBS) were visualized by Leica DMIRE2 inverted microscope and analyzed with Improvision Openlab 4.0.
Results
LMP2A is secreted in exosomes
To investigate LMP2A endocytic trafficking, LMP2A localization to exosomes was examined. Exosomes are small membrane vesicles of endocytic origin that are secreted in the extracellular medium in most cells. These vesicles are formed by invagination followed by budding into the lumen of endocytic compartments, leading to the formation of multivesicular bodies (MVBs). Upon fusion of MVB with the plasma membrane, internal exosome vesicles are released into the medium. Exosomes are thought to be involved in the protein sorting and/or the regulation of the immune response (de Gassart et al., 2004). To determine if LMP2A is present in exosomes, extracellular medium was purified by ultracentririfugation after clearing cells by low-speed centrifugation. Protein levels of LMP2A, an exosomal control protein HLA-DR, and a lysosomal control protein LAMP1 were investigated by immunoblot in wild-type EBV infected LCLs (WT) and LMP2A-mutated EBV infected LCLs (ΔLMP2A). LMP2A exosomal secretion was observed in a WT LCL after 1 day and was continuously accumulated for the six days of the analysis (Fig. 1A). Similar secretion in the extracellular medium was observed for HLA-DR but not for LAMP1 (Fig. 1A) indicating that LMP2A observed in extracellular medium was formed via normal exosomal secretion. As expected, LMP2A was not detected in the ΔLMP2A LCL. Next, LMP2A exosomal secretion was examined in BJAB cells, an immortalized B lymphoma cell line not infected with EBV. In BJAB cell lines expressing LMP2A from a retroviral vector promoter, LMP2A was secreted in exosomes similarly to the EBV infected WT LCL (Fig. 1B), indicating that the LMP2A exosomal secretion was not dependent on other EBV gene products.
Figure 1. LMP2A exosomal secretion.
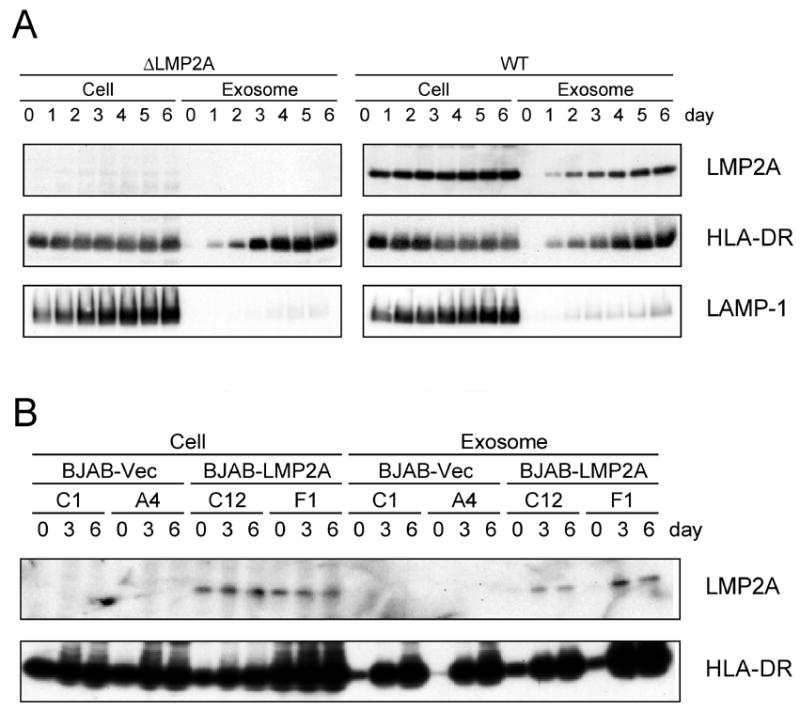
(A) LMP2A-mutated (ΔLMP2A) or LMP2A-expressing (WT) LCL and (B) LMP2A-expressing BJAB cell lines, C12 and F1, and control cell lines, C1 and A4, were grown for six days without changing the media. Cell culture media was removed from 0–6 days and cells were pelleted at 1,500 rpm (cellular fraction). The resulting supernatant was cleared by centrifugation at 3,000 rpm and exosomes were isolated by centrifugation at 30,000 rpm (exosomal fraction). Pelleted cell and exosomal fractions were solubilized and immmunoblotted with LMP2A, HLA-DR, and LAMP1. The reduction in HLA-DR expression in the late time points is likely due to MCD toxicity.
Cholesterol depletion increases LMP2A cellular expression and exosomal secretion
To further define the requirements for LMP2A exosomal secretion, an EBV WT infected LCL was treated with various chemicals (Table 1) and LMP2A expression and exosomal secretion was monitored following treatment. Previous studies have shown that LMP2A localizes to lipid rafts (Dykstra et al., 2001). MCD specifically depletes cholesterol from the plasma membrane disrupting lipid rafts. Interestingly, of all the inhibitors tested, 4 mM methyl-beta-cyclodextrin (MCD) dramatically increased the abundance and secretion of LMP2A (Fig. 2), whereas each of the other inhibitors had little effect on LMP2A abundance or secretion in exosomes (Fig. 2). LMP2A abundance in the cellular fraction increased almost five fold with MCD treatment as determined by densitometry when compared with the control treated cells (Fig. 2). There was more than ten-fold increase in LMP2A exosomal secretion in the presence of MCD in comparison with the control treated cells (Fig. 2). This increase indicates that cholesterol is important for LMP2A cellular abundance and exosomal secretion further suggesting the LMP2A localization to lipid rafts plays an important LMP2A function. A modest increase of LMP2A in cellular fractions was observed with 10 μM of the proteasome inhibitor lactacystine (LC) and 50 μM of the lysosomal protease inhibitor chloroquine (CQ) (Fig. 2). However, the increases by LC and CQ were 1.5-fold and two-fold when compared with the control, respectively (Fig. 2). These results are compatible for the role of the proteasome and the lysosome in LMP2A degradation. In contrast, the protein tyrosine phosphatase inhibitor sodium vanadate (Van), protein tyrosine kinase (PTK) inhibitor genistein (Gen), and Src-PTK-specific inhibitor PP2 did not alter the amount of LMP2A cellular fraction or the exosomal fraction indicating that LMP2A phosphorylation is not involved in the cellular abundance of LMP2A and LMP2A exosomal secretion.
Figure 2. The effect of various chemicals on the abundance of LMP2A in the cellular and exosomal fractions.
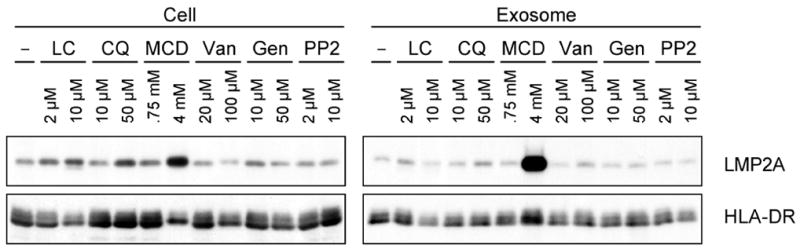
WT LCL was treated with indicated chemicals for 3 days. Cell and exosome fractions were immunoblotted with LMP2A and HLA-DR. Lactacystin (LC), Chloroquine (CQ), Methyl-beta-cyclodextrin (MCD), Sodium vanadate (Van), Genistein (Gen), and Src inhibitor PP2.
To further examine the effect of MCD on LMP2A cellular abundance and exosomal secretion, a time course of LMP2A in the two fractions was done after MCD treatment (Fig. 3A). The amount of LMP2A increased six hours after MCD addition and continued to increase for the 24 hour duration of the time course. However, no change in the abundance was observed for the exosomal control proteins LMP1 and HLA-DR. LMP1 was previously shown to be secreted in exosomes (Flanagan et al., 2003). Similar to the increase of LMP2A seen in the cellular fraction, exosomal secretion of LMP2A was also increased becoming readily apparent at six hours. A similar pattern of expression in the exosomal fraction was also observed for LMP1 and HLA-DR. However, the relative amount of LMP1 and HLA-DR in the exosomal fractions compared to the cellular fractions was significantly lower than that of LMP2A. The levels of these two proteins were similar to or even lower than their cellular expression at 0 hour. In contrast, the amount of LMP2A secretion at 24 hours was approximately five-fold to the amount present in the cellular fraction at 0 hour. These results indicate that MCD specifically increases the relative abundance and exosomal secretion of LMP2A when compared to LMP1 or HLA-DR. Next, we examined whether the effect of MCD on LMP2A levels relied on cholesterol depletion. Cholesterol was added to the medium to restore depleted cholesterol from the MCD treated cells. As shown in Fig. 3B, the increases of LMP2A observed in the cellular fraction and exosomal fraction were completely suppressed by the addition of 30 μg/ml cholesterol. Thus, cholesterol plays a pivotal role in LMP2A cellular abundance and exosomal secretion.
Figure 3. MCD specifically increases LMP2A in the cellular fraction and the exosomal fraction.
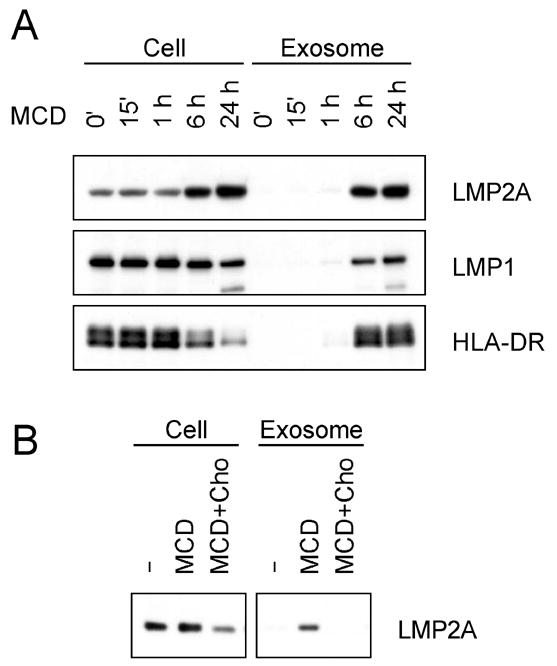
(A) WT LCL was treated with 4 mM MCD for indicated times. Cellular and exosomal fractions were immunoblotted for LMP2A, LMP1 and HLA-DR. (B) WT LCL was treated with 4 mM MCD or 4 mM MCD plus 30 μg/ml soluble choresterol (Cho) for 24 hours. Cellular and exosomal fractions were immunoblotted with LMP2A.
MCD increases the half-life and accumulation in the plasma membrane of LMP2A
To further investigate the alteration in the abundance and exosomal secretion of LMP2A by MCD, the half-life of LMP2A was determined when de novo protein synthesis was blocked by cycloheximide (CHX). A WT infected LCL was treated with CHX and cultured with or without MCD. The abundance of cellular LMP2A was densitometrically determined from the LMP2A immunoblot shown in Fig. 4A and graphed in Fig. 4B. From this analysis, it is readily apparent that LMP2A is more stable when cells are treated with MCD. The half-life of LMP2A was approximately 16 hours when cells were treated with MCD, while it was approximately nine hours without MCD treatment.
Figure 4. Increased abundance of LMP2A by MCD is due to the protein stability and not de novo protein synthesis.
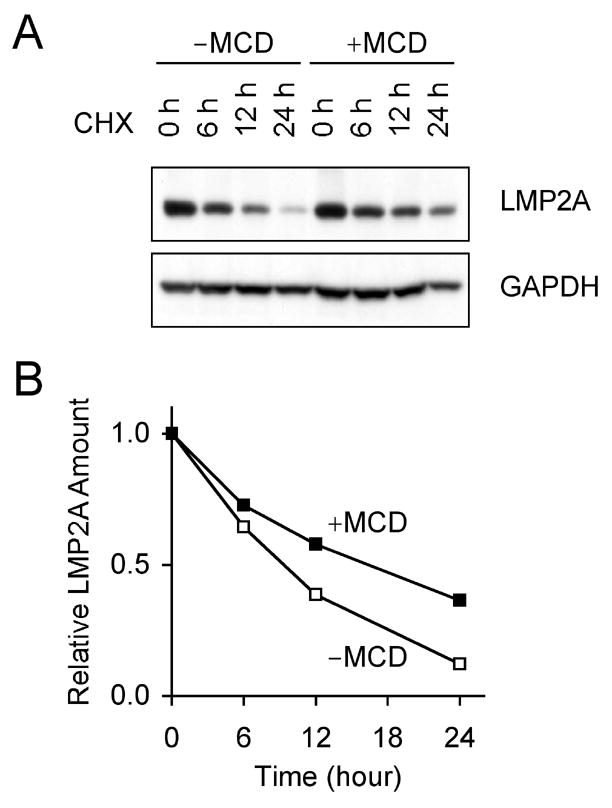
(A) WT LCL was treated with 4 mM MCD in the presence of 100 μg/ml cycloheximide (CHX). Cell lystates were isolated at indicated times and immunoblotted with LMP2A and GAPDH. (B) The amount of LMP2A was densitometrically determined and plotted as a relative ratio. One of three comparable experiments is shown.
The cellular localization of LMP2A was also determined by immunofluorescence microscopy using HLA-DR as a marker for the plasma membrane and LAMP1 as a marker for lysosomes and late endosomes. Without the MCD treatment, LMP2A was observed in intracellular regions as well as in the plasma membrane (Fig. 5) consistent with previous studies (Longnecker and Kieff, 1990). When cells were treated with MCD, the abundance of LMP2A in the plasma membrane was greatly enhanced and was much more diffuse in staining when compared with the localization of LMP2A in the untreated cells (Compare merge images of LMP2A staining and HLA-DR staining with and without MCD treatment.). HLA-DR staining also became more diffuse in pattern in the plasma membrane following MCD treatment (Fig. 5). There was little alteration of LAMP1 following MCD treatment. A similar fraction of LM2A localized with LAMP1 with and without MCD treatment. Taken together, we conclude that cholesterol depletion increases LMP2A stability from our analysis of the half-life of LMP2A and enhances expression of LMP2A on the plasma membrane.
Figure 5. MCD translocates LMP2A to the plasma membrane.
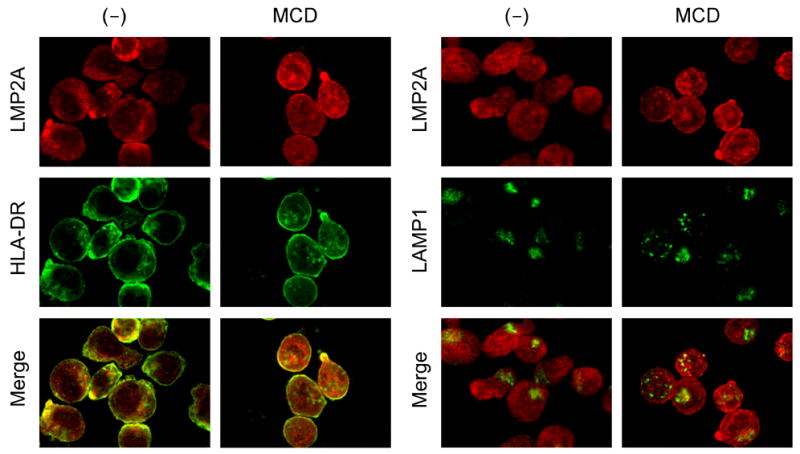
WT LCL was treated with 4 mM MCD for six hours and fixed with methanol. Localization of LMP2A and HLA-DR was visualized by reacting with anti-LMP2A or anti-HLA-DR, and followed by staining with FITC-conjugated goat anti-rat-immunoglobulin antibody or Cy3-conjugated goat anti-mouse-immunoglobulin antibody, respectively.
MCD inhibits LMP2A cellular phosphorylation and ubiquitination
Phosphorylation and ubiquitination of LMP2A in MCD treated cells was investigated to clarify the role of these two modifications in LMP2A localization. LMP2A immunoprecipitates of cell lysates from the WT infected LCL treated with or without MCD were blotted for tyrosine phosphorylation and ubiquitin conjugation. In the cell fraction, LMP2A from non-treated cells was highly phosphorylated and ubiquitinated as previously reported (Fig. 6). However, MCD treatment resulted in a decrease of LMP2A phosphorylation and ubiquitination in the cellular fraction. This indicates that cholesterol is required for the phosphorylation and ubiquitination of LMP2A contained in the cellular fraction. Next, exosomal fractions were tested. Exosomal LMP2A from MCD-treated cells was not phosphorylated but interestingly was ubiquitinated (Fig. 6). Although LMP2A secretion is relatively modest when cells are not treated with MCD (Fig. 6), exosomal LMP2A was not phosphorylated but ubiquitinated similar to MCD treatment on longer exposure of the immunoblot (data not shown). Thus, MCD treatment increases the overall amount of LMP2A that is secreted in exosomes without altering the modification of LMP2A in the exosomal fraction. Moreover, this result suggests that phosphorylation is not important for the LMP2A trafficking into exosomes while ubiquitination is important.
Figure 6. Phosphorylation and ubiquitination of MCD-treated LMP2A.
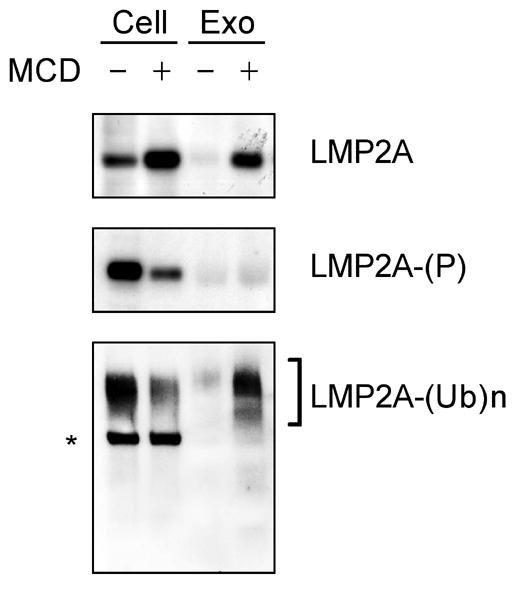
WT LCL was treated with 4 mM MCD for 48 hours. Cellular and exosomal fractions were immunoprecipitated with rabbit anti-LMP2A, and immunoblotted with LMP2A, phosphotyrosine and ubiquitin. One of three comparable experiments is shown. * indicates cell-derived immunoglobulin heavy chain.
Discussion
Cell surface proteins are endocytosed to early endosomes where they can be recycled back to the plasma membrane or targeted to late endosomes and lysosomes for degradation. Cholesterol in the plasma membrane is enriched in specific microdomains termed lipid rafts. Early endosomes contain cholesterol and have an overall lipid composition very similar to that found in the plasma membrane, whereas cholesterol is highly enriched in the recycling endosomes (Kobayashi et al., 2001). Thus, recycling endosomes are enriched with raft components. The late endosomes exhibit morphologically visible membrane domains, which appear as multivesicular and/or multilamellar structures which accumulate within the endosomal lumen. Unlike early endosomes, late endosomes normally do not contain significant amounts of cholesterol but contain large amounts of neutral lipids (Kobayashi et al., 2001). With respect to membrane lipid composition, late endosomes on the degradation pathway can be clearly distinguished from early endosomes on the recycling pathway. These observations indicate the importance of lipid raft domains for membrane dynamics.
Lipid rafts are involved in the internalization of cell surface molecules. Studies using drugs that sequester cholesterol reveal the essential role of cholesterol in lipid raft-dependent endocytosis (Kobayashi et al., 2001). We demonstrated that cholesterol depletion caused LMP2A accumulation in the plasma membrane resulting an increase of LMP2A in the cellular fraction. This is explained by the inhibition of LMP2A endocytosis in a lipid raft-dependent pathway. Thus, cholesterol depletion prevents LMP2A transport to endosomes and subsequent degradation in lysosomes indicating that cholesterol-dependent trafficking is a critical step for LMP2A degradation. Since cholesterol is found primarily in early endosomes but not in late endosomes, the effect of cholesterol depletion on LMP2A degradation may be restricted to early endosomes in the internalization as well as recycling pathway. It is unlikely that the increase of LMP2A in cellular fractions is due to alteration in LMP2A gene expression because the LMP2A increase was also observed in LMP2A-expressing BJAB cells where a retroviral promoter regulates LMP2A expression in contrast to the LMP2A endogenous promoter in EBV infected cells. Interestingly, the necessity of cholesterol-dependent trafficking for degradation is more readily apparent for LMP2A than other endocytic proteins such as HLA-DR and LMP1. This suggests that the regulation of LMP2A protein trafficking is unique and different from that of other proteins.
Previous LMP2A localization studies have demonstrated that LMP2A expression is observed abundantly in intracellular organelles but very little in plasma membrane (Dawson et al., 2001; Longnecker and Kieff, 1990; Lynch et al., 2002). These intracellular organelles include perinuclear regions and trans-Golgi network as well as early endosomes and lysosomes (Dawson et al., 2001; Lynch et al., 2002). However, results in this study clearly show that LMP2A localizes in the plasma membrane when endocytosis is blocked by cholesterol depletion. Thus, this finding suggests that the localization of LMP2A in intracellular vesicles is the consequence of cholesterol-dependent LMP2A endocytosis from lipid rafts in the plasma membrane. A model for LMP2A trafficking is proposed based on the results in this study (Fig. 7). In this model, LMP2A localization in the lipid rafts is essential for LMP2A endocytosis and trafficking to the early endosomes. LMP2A is then transported to MVB where it can be recycled to the plasma membrane or shuttled to lysosomes for degradation. LMP2A that is invaginated into the lumen of endosomes is released to extracellular medium as exosomes. Thus, LMP2A localization in the plasma membrane is a critical step in the fate of LMP2A in cellular trafficking and degradation.
Figure 7. Model for LMP2A trafficking.
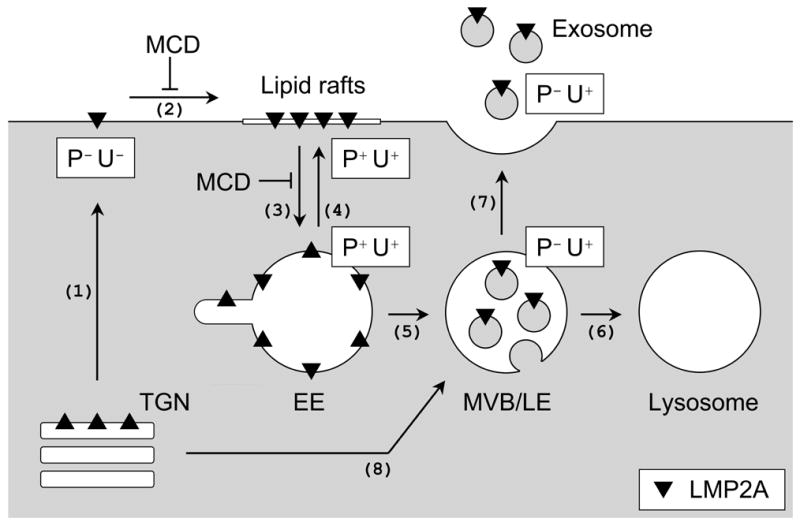
(1) LMP2A is synthesized in the endoplasmic reticulumn and transported to the trans-Golgi network (TGN) prior to being transported to the plasma membrane. LMP2A is not phosphorylated and ubiquitinated (P−U−). (2) Once in the plasma membrane, LMP2A localizes in the lipid rafts forming multi-protein complexes with the Lyn and Syk PTKs and the Nedd4-family ubiquitin ligases resulting in the phosphorylation and ubiquitination of LMP2A (P+U+). (3) The LMP2A recruitment into the lipid rafts and subsequent phosphorylation and ubiquitination results in internalization and trafficking of LMP2A to early endosomes (EE). The MCD-mediated increase in LMP2A in cellular fractions is likely explained by LMP2A accumulation in the plasma membrane due to a block in LMP2A internalization. (4) Some LMP2A is recycled back to the plasma membrane through recycling endosomes. (5) LMP2A is further transported to multivesicular bodies (MVB)/late endosomes (LE) where LMP2A-containing membrane is invaginated into the lumen of MVB/LE. During the transportation from EE to MVB/LE, LMP2A dephosphorylation occurrs. However, whether LMP2A dephosphorylation induces this transportation to MVB/LE or LMP2A is dephosphorylated as it is transported, is still unclear. (6) LMP2A destined to lysosomes is degraded. (7) Some LMP2A is released to extracellular domains by fusing to the plasma membrane. In the exosome fractions, LMP2A is highly ubiquitinated but not phosphorylated (P−U+). When LMP2A endocytosis is blocked at the plasma membrane by MCD, increases in LMP2A exosomal secretion may be mediated by a poorly described mechanism in common for other proteins as described in the discussion and/or by the transport of LMP2A directly from TGN to MVB/LE (8).
LMP2A is tyrosine phosphorylated and ubiquitinated. As shown in this study, cholesterol depletion decreased LMP2A turnover and LMP2A phosphorylation in the cellular fraction indicating that cholesterol is required for LMP2A phosphorylation and ubiquitination. In the exosomal fraction, cholesterol depletion increased the amount of LMP2A in the exosomal fraction and LMP2A in this fraction was highly ubiquitinated. Interestingly, LMP2A in the exosome fraction was not phosphorylated. Since exosomes originate from the internal vesicles in MVB, it is likely that LMP2A is dephosphorylated in MVB prior to being released as exosomes. Immunofluorescence microscopic analysis demonstrated that LMP2A is observed in early endosome-like intracellular compartments. Thus, it seems that LMP2A is dephosphorylated during transport from the early endosomes to MVB. However, the necessity of LMP2A dephosphorylation for the trafficking from early endosomes to MVB is still unclear.
As shown in this study, LMP2A transition into lipid rafts is necessary for LMP2A phosphorylation and ubiquitination. This translocalization may be necessary prior to LMP2A association with Nedd4-family ubiquitin ligases or Src-family PTKs. Once LMP2A is recruited into lipid rafts, it becomes susceptible for phosphorylation and ubiquitination. The domains essential for LMP2A lipid raft localization are not known. Previous studies have suggested that other factors besides fatty acid modification are important (Katzman and Longnecker, 2004). It has been suggested that LMP2A localization in lipid rafts is related to its self-aggregation. The importance of the LMP2A carboxyl-terminal domain for self-aggregation has been suggested by several studies. An LMP2A mutant lacking the last ten transmembrane and carboxyl-terminal domains localizes diffusely on plasma membrane (Longnecker et al., 1991). In addition, the carboxyl terminus has been suggested to mediate self-association of LMP2A in 293 cells (Matskova et al., 2001), although a deletion mutant of LMP2A that lacks the LMP2A carboxyl-terminus and last seven transmembrane domains is not totally deficient in the block of B cell activation (Miller et al., 1994). The deletion mutant lacking the carboxyl-terminal tail and last ten transmembrane domains is still tyrosine phosphorylated (Longnecker et al., 1991). Thus, both the transmembrane domains and carboxyl-terminal domain are likely critical for LMP2A to self-cluster on the plasma membrane. These domains may be essential for LMP2A endocytosis as well. Previous studies with LMP1 identified a FWLY domain from amino acids 38 to 41 within the first LMP1 transmembrane domain to be important for raft localization (Yasui et al., 2004). Interestingly, this domain is important for LMP1 intermolecular association and NF-κB activation (Soni et al., 2006; Yasui et al., 2004). Similar domains may also be in LMP2A.
The increase in LMP2A exosomal secretion is similar to what has been observed in other studies in which a poorly defined mechanism results in increased exosomal secretion (Claas et al., 2001). Exosomes originate from small vesicles present in lumen of late endosomes. In late endosomes, the lipid composition of internal membrane is distinct from that of other membranes. A unique phospholipid, lysobisphosphatidic acid (LBPA) is restricted to the late endosome membranes as well as exosomal membranes (Kobayashi et al., 2001). Recent studies have provided insights into the modulation of intracellular cholesterol transportation. In Niemann-Pick disease type C (NPC) disease, an autosomal recessive disorder, large amounts of cholesterol and LBPA accumulate in the late endosomes (Kobayashi et al., 1998). The egress of late endosome cholesterol to the plasma membrane or the endoplasmic reticulum is defective in this disease leading to aberrant regulation of cellular cholesterol homeostasis and the redistribution of membrane proteins that control intracellular protein sorting and trafficking. In this disorder, cholesterol depletion in the plasma membrane may be compensated by mobilizing cholesterol from late endosomes to plasma membrane. Similarly, LMP2A in early and late endosomes, which would normally be targeted for degradation, may be transported to extracellular medium when cholesterol is depleted by agents such as MCD.
In these studies, we demonstrated that LMP2A is secreted in exosomes in a cholesterol dependent manner. Interestingly, LMP1, another EBV latent membrane protein, is also secreted in exosomes (Flanagan et al., 2003). LMP1 is an essential protein for EBV-driven cell growth and immortalization by mimicking an activated CD40 receptor. As well as exosomal secretion, LMP1 and LMP2A have other similar characteristics. They share structural similarities having cytosolic amino- and carboxyl-terminal domains and twelve or six transmembrane domains, respectively. They are colocalized in lipid raft microdomains by forming aggregates (Dykstra et al., 2001; Higuchi et al., 2001; Longnecker and Kieff, 1990). Moreover, both are ubiquitinated at the amino terminus which has been reported for three other proteins (Aviel et al., 2000; Bloom et al., 2003; Breitschopf et al., 1998; Ikeda et al., 2002; Reinstein et al., 2000). Thus, the functions of LMP1 and LMP2A may be regulated in a similar fashion. These similarities may provide a clue to elucidate common mechanisms important for the regulation of EBV latent infection and EBV-associated disease with the human population.
Acknowledgments
We would like to thank the members of the Longnecker laboratory for their help with these studies. R. L. is supported by Public Health Service grants CA62234 and CA73507 from the National Cancer Institute. R.L. is a Stohlman Scholar of the Leukemia and Lymphoma Society of America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aviel S, Winberg G, Massucci M, Ciechanover A. Degradation of the epstein-barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J Biol Chem. 2000;275(31):23491–9. doi: 10.1074/jbc.M002052200. [DOI] [PubMed] [Google Scholar]
- Bloom J, Amador V, Bartolini F, DeMartino G, Pagano M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell. 2003;115(1):71–82. doi: 10.1016/s0092-8674(03)00755-4. [DOI] [PubMed] [Google Scholar]
- Breitschopf K, Bengal E, Ziv T, Admon A, Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO Journal. 1998;17(20):5964–73. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9(3):405–11. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC, Rajewsky K. B cell receptor signal strength determines B cell fate. Nat Immunol. 2004;5(3):317–27. doi: 10.1038/ni1036. Epub 2004 Feb 1. [DOI] [PubMed] [Google Scholar]
- Claas C, Stipp CS, Hemler ME. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. Journal of Biological Chemistry. 2001;276(11):7974–7984. doi: 10.1074/jbc.M008650200. [DOI] [PubMed] [Google Scholar]
- Dawson CW, George JH, Blake SM, Longnecker R, Young LS. The Epstein-Barr virus encoded latent membrane protein 2A augments signaling from latent membrane protein 1. Virology. 2001;289(2):192–207. doi: 10.1006/viro.2001.1142. [DOI] [PubMed] [Google Scholar]
- de Gassart A, Geminard C, Hoekstra D, Vidal M. Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic. 2004;5(11):896–903. doi: 10.1111/j.1600-0854.2004.00223.x. [DOI] [PubMed] [Google Scholar]
- Dykstra ML, Longnecker R, Pierce SK. Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR. Immunity. 2001;14(1):57–67. doi: 10.1016/s1074-7613(01)00089-9. [DOI] [PubMed] [Google Scholar]
- Flanagan J, Middeldorp J, Sculley T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. Journal of General Virology. 2003;84:1871–1879. doi: 10.1099/vir.0.18944-0. [DOI] [PubMed] [Google Scholar]
- Fruehling S, Lee SK, Herrold R, Frech B, Laux G, Kremmer E, Grasser FA, Longnecker R. Identification of latent membrane protein 2A (LMP2A) domains essential for the LMP2A dominant-negative effect on B-lymphocyte surface immunoglobulin signal transduction. J Virol. 1996;70(9):6216–26. doi: 10.1128/jvi.70.9.6216-6226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruehling S, Longnecker R. The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction. Virology. 1997;235(2):241–51. doi: 10.1006/viro.1997.8690. [DOI] [PubMed] [Google Scholar]
- Fruehling S, Swart R, Dolwick KM, Kremmer E, Longnecker R. Tyrosine 112 of latent membrane protein 2A is essential for protein tyrosine kinase loading and regulation of Epstein-Barr virus latency. J Virol. 1998;72(10):7796–806. doi: 10.1128/jvi.72.10.7796-7806.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L. A new ticket for entry into budding vesicles-ubiquitin. Cell. 2001;106(5):527–30. doi: 10.1016/s0092-8674(01)00485-8. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Izumi KM, Kieff E. Epstein-Barr virus latent-infection membrane proteins are palmitoylated and raft-associated: protein 1 binds to the cytoskeleton through TNF receptor cytoplasmic factors. Proc Natl Acad Sci U S A. 2001;98(8):4675–80. doi: 10.1073/pnas.081075298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Caldwell RG, Longnecker R, Ikeda M. Itchy, a Nedd4 ubiquitin ligase, downregulates latent membrane protein 2A activity in B-cell signaling. J Virol. 2003;77(9):5529–34. doi: 10.1128/JVI.77.9.5529-5534.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda A, Merchant M, Lev L, Longnecker R, Ikeda M. Latent membrane protein 2A, a viral B cell receptor homologue, induces CD5+ B-1 cell development. J Immunol. 2004;172(9):5329–37. doi: 10.4049/jimmunol.172.9.5329. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ikeda A, Longan LC, Longnecker R. The Epstein-Barr virus latent membrane protein 2A PY motif recruits WW domain-containing ubiquitin-protein ligases. Virology. 2000;268(1):178–91. doi: 10.1006/viro.1999.0166. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ikeda A, Longnecker R. PY motifs of Epstein-Barr virus LMP2A regulate protein stability and phosphorylation of LMP2A-associated proteins. J Virol. 2001;75(12):5711–8. doi: 10.1128/JVI.75.12.5711-5718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Ikeda A, Longnecker R. Lysine-independent ubiquitination of Epstein-Barr virus LMP2A. Virology. 2002;300(1):153–9. doi: 10.1006/viro.2002.1562. [DOI] [PubMed] [Google Scholar]
- Jayakar HR, Murti KG, Whitt MA. Mutations in the PPPY motif of vesicular stomatitis virus matrix protein reduce virus budding by inhibiting a late step in virion release. Journal of Virology. 2000;74(21):9818–9827. doi: 10.1128/jvi.74.21.9818-9827.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman RB, Longnecker R. LMP2A does not require palmitoylation to localize to buoyant complexes or for function. Journal of Virology. 2004;78(20):10878–10887. doi: 10.1128/JVI.78.20.10878-10887.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature. 1998;392(6672):193–197. doi: 10.1038/32440. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yamaji-Hasegawa A, Kiyokawa E. Lipid domains in the endocytic pathway. Seminars in Cell & Developmental Biology. 2001;12(2):173–182. doi: 10.1006/scdb.2000.0234. [DOI] [PubMed] [Google Scholar]
- Longnecker R, Druker B, Roberts TM, Kieff E. An Epstein-Barr virus protein associated with cell growth transformation interacts with a tyrosine kinase. J Virol. 1991;65(7):3681–92. doi: 10.1128/jvi.65.7.3681-3692.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longnecker R, Kieff E. A second Epstein-Barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J Virol. 1990;64(5):2319–26. doi: 10.1128/jvi.64.5.2319-2326.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longnecker R, Miller CL, Tomkinson B, Miao XQ, Kieff E. Deletion of DNA encoding the first five transmembrane domains of Epstein-Barr virus latent membrane proteins 2A and 2B. J Virol. 1993;67(8):5068–74. doi: 10.1128/jvi.67.8.5068-5074.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch DT, Zimmerman JS, Rowe DT. Epstein-Barr virus latent membrane protein 2B (LMP2B) co-localizes with LMP2A in perinuclear regions in transiently transfected cells. J Gen Virol. 2002;83(Pt 5):1025–35. doi: 10.1099/0022-1317-83-5-1025. [DOI] [PubMed] [Google Scholar]
- Mann KP, Staunton D, Thorley-Lawson DA. Epstein-Barr virus-encoded protein found in plasma membranes of transformed cells. Journal of Virology. 1985;55(3):710–20. doi: 10.1128/jvi.55.3.710-720.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matskova L, Ernberg I, Pawson T, Winberg G. C-terminal domain of the Epstein-Barr virus LMP2A membrane protein contains a clustering signal. J Virol. 2001;75(22):10941–9. doi: 10.1128/JVI.75.22.10941-10949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Lee JH, Kieff E, Burkhardt AL, Bolen JB, Longnecker R. Epstein-Barr virus protein LMP2A regulates reactivation from latency by negatively regulating tyrosine kinases involved in sIg-mediated signal transduction. Infect Agents Dis. 1994;3(2–3):128–36. [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annual Review of Cell and Developmental Biology. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Reinstein E, Scheffner M, Oren M, Ciechanover A, Schwartz A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19(51):5944–50. doi: 10.1038/sj.onc.1203989. [DOI] [PubMed] [Google Scholar]
- Soni V, Yasui T, Cahir-McFarland E, Kieff E. LMP1 Transmembrane Domain 1–2 FWLY mediates Intermolecular Interactions with 3–6 to activate NF-{kappa}B. J Virol. 2006;80(21):10787–93. doi: 10.1128/JVI.01214-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vana ML, Tang Y, Chen AP, Medina G, Carter C, Leis J. Role of Nedd4 and ubiquitination of Rous sarcoma virus gag in budding of virus-like particles from cells. Journal of Virology. 2004;78(24):13943–13953. doi: 10.1128/JVI.78.24.13943-13953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg G, Matskova L, Chen F, Plant P, Rotin D, Gish G, Ingham R, Ernberg I, Pawson T. Latent membrane protein 2A of Epstein-Barr virus binds WW domain E3 protein-ubiquitin ligases that ubiquitinate B-cell tyrosine kinases. Mol Cell Biol. 2000;20(22):8526–35. doi: 10.1128/mcb.20.22.8526-8535.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Cameron CE, Wills JW, Leis J. Fine mapping and characterization of the Rous sarcoma virus Pr76(gag) late assembly domain. Journal of Virology. 1996;70(8):5695–5700. doi: 10.1128/jvi.70.8.5695-5700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui T, Luftig M, Soni V, Kieff E. Latent infection membrane protein transmembrane FWLY is critical for intermolecular interaction, raft localization, and signaling. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(1):278–83. doi: 10.1073/pnas.2237224100. [DOI] [PMC free article] [PubMed] [Google Scholar]