

Abstract
Deoxyhypusine hydroxylase (DOHH) catalyzes the final step in the post-translational synthesis of hypusine [Nε-(4-amino-2-hydroxybutyl)lysine] in eIF5A. DOHH is a HEAT-repeat protein with eight tandem helical hairpins in a symmetrical dyad. It contains two potential iron coordination sites (one on each dyad) comprised of two strictly conserved His-Glu motifs. The purified human recombinant DOHH was a mixture of active holoenzyme containing 2 mol of iron per mol of DOHH and inactive metal-free apoenzyme. The two species could be distinguished by their different mobilities upon native gel electrophoresis. The DOHH apoenzyme exhibited markedly reduced levels of iron and activity. DOHH activity could be restored only by addition of Fe(II) to the apoenzyme, but not by other metals including Cd, Co, Cr, Cu, Mg, Mn, Ni and Zn. The role of the strictly conserved His-Glu residues was evaluated by site-directed mutagenesis. Substitution of any single amino acid in the four His-Glu motifs with alanine abolished the enzyme activity. Of these eight alanine substitutions, six, including His56A, His89A, Glu90A, His207A, His240A and Glu241A, caused a severe reduction in the iron content. Our results provide strong evidence that Fe(II) is the active-site bound metal critical for DOHH catalysis and that the strictly conserved His-Glu motifs are essential for iron binding and catalysis. Furthermore, the iron to DOHH stoichiometry and dependence of iron binding on each of the four conserved His-Glu motifs suggest a binuclear iron mediated reaction mechanism, distinct from that of other Fe(II)-dependent protein hydroxylases, such as prolyl 4-hydroxylase or lysyl hydroxylases.
Eukaryotic translation initiation factor 5A (eIF5A) is the only protein in nature that contains an unusual amino acid, hypusine [Nε-4-amino-(2-hydroxybutyl)lysine] (see a recent review (1)). Hypusine is derived from one specific lysine residue of the eIF5A precursor protein by a posttranslational modification reaction that involves two enzymes. In the first step, deoxyhypusine synthase catalyzes the NAD-dependent cleavage and transfer of the aminobutyl moiety of the polyamine spermidine to the ε-amino group of the lysine residue to form an intermediate, deoxyhypusine [Nε-4-amnobutyl-lysine] residue (2,3). This intermediate is subsequently hydroxylated by deoxyhypusine hydroxylase (DOHH) (4) (5) to form the hypusine residue in mature eIF5A. eIF5A and its modification are essential for eukaryotic cell proliferation (1,6–11)
eIF5A is a highly conserved protein that occurs in all eukaryotes. Its homolog is found in archaea (12) and a distant homolog, elongation factor P (EF-P), in bacteria (13,14). The essential nature of eIF5A and deoxyhypusine synthase in eukaryotic cell proliferation was established from gene disruption studies in Saccharomyces cerevisiae (15–18). A DOHH homolog gene is found in all eukaryotic species. Although it is not an essential gene in S. cerevisiae (as the DOHH null strain is viable (4,19)), inactivation of the DOHH gene is recessively lethal in multicellular eukaryotes, e.g. Caenorhabditis elegans (20) and Drosophila melanogaster (21), suggesting a requirement for the fully modified eIF5A in higher eukaryotes.
DOHH from mammalian cells and tissues shares similarities with other protein hydroxylases, such as prolyl hydroxylase and lysyl hydroxylases. Like these enzymes, DOHH is inhibited by a panel of metal chelators including mimosine, 2,2’-dipirydyl, deferiprone, deferoxamine and ciclopirox olamine (5,22–24) and it recognizes a similar amino acid sequence surrounding the modification site, a G-X-Y-G motif (where X or Y being the deoxyhypusine, lysine, or proline residue that undergoes modification) in the substrate proteins. Because of these similarities, DOHH had been assumed to be a member of the super family of Fe(II) and 2-oxoacid-dependent dioxygenases. Enzymes of this family share a common beta jelly roll structure, termed the double stranded beta helix (DSBH) (25–27). DSBH enzymes display universal dependence on Fe(II) and contain a consensus Fe(II) coordination sequence. They catalyze a wide variety of reactions that include hydroxylation of amino acid residues, such as proline, lysine, aspartic acid and asparagines, in the substrate proteins, repair of alkylated DNA/RNA, biosynthesis of antibiotics and plant products and metabolism of small molecules (25,26). Most representative hydroxylases utilize molecular oxygen as the source of the hydroxyl group and couple the oxidative decomposition of a co-substrate, α-ketoglutarate, to hydroxylation of the substrate, leading to formation of the hydroxylated product, CO2 and succinate (25,27,28). In the case of DOHH, however, neither the addition of the three cofactors of DSBH enzymes, Fe(II), ascorbic acid and α-ketoglutarate, enhanced the activity of the partially purified rat testis enzyme, nor could the release of 14CO2 from α-keto[14C]glutarate be detected with the same enzyme (5). Although the inhibition of DOHH activity in cells by known iron chelators was shown to be overcome by addition of either Fe(II) or Fe(III) in HeLa cell extracts (22), there has been no definitive evidence on the identity of the metal involved in DOHH catalysis. Thus, the relationship between the DOHH and DSBH enzymes has remained obscure.
Recently, our group has identified and cloned the yeast DOHH gene (YJR070C) (by screening a S. cerevisiae GST-ORF library (29)) and also the human DOHH gene (4). DOHH homolog exists in all eukaryotes as a single gene in each species and the amino acid sequence is highly conserved. Examination of the sequence alignment of DOHH revealed that this enzyme belongs to a family of HEAT-repeat proteins, named for human huntingtin (H), elongation factor 3 (E), a subunit of protein phosphatase 2A (A) and the target of rapamycin (TOR), that are commonly involved in protein-protein interactions (30). In these proteins, the HEAT-motif, composed of an α-helical hairpin (a pair of α-helices) of roughly 50 amino acids, is tandemly repeated to form superhelical structures. Computer modeling of DOHH predicted a structure consisting of eight HEAT-repeats in a symmetrical dyad of four HEAT motifs connected by a variable region, a structure that is totally different from the beta jelly roll structures of Fe(II)-and 2-oxoacid-dependent dioxygenases. The model predicts that the N- and C- terminal halves of DOHH form a symmetric shell-like structure with four conserved His-Glu pairs potentially contributing to metal-chelation (4). However, this homology-based model represents only an approximate structure for the protein and its possible metal interactions.
In an effort to validate the proposed model and to gain insight into the reaction mechanism of DOHH, we performed structural analyses of the wild type and mutant enzymes by CD spectroscopy and investigated the roles of iron and the conserved His-Glu residues in DOHH catalysis. Our results demonstrate that the four strictly conserved His-Glu pairs indeed constitute the iron coordinating active site for DOHH. Furthermore, we present definitive evidence that Fe(II), but not Fe(III) or other divalent transition metals, is the critical metal for DOHH catalysis.
Experimental procedures
Materials
L-Glutathione (GSH) was purchased from Sigma, Glutathione Sepharose 4B from Amersham Biosciences, Thrombin Cleavage Capture Kit from Novagen, and Complete protease inhibitor cocktail from Roche. [1,8-3H]Spermidine.3HCl (~20 Ci/mmol) was purchased from Dupont/NEN. Precast Tris-glycine and NuPAGE (Bis-Tris) gels, MOPS running buffer and Simply Blue staining solution, were purchased from Invitrogen. The Quick Change Site-Directed Mutagenesis Kit was purchased from Stratagene. The bacterial expression vector (pGEX-4T-3/hDOHH) expressing human DOHH was previously described (4). The radiolabeled deoxyhypusine-containing protein, human eIF5A([3H]Dhp) was prepared in an in vitro deoxyhypusine synthase reaction using human eIF5A precursor, eIF5A(Lys) and [1,8-3H]spermidine as described previously (31). The oligonucleotide primers were synthesized by Integrated DNA Technologies, Inc.
Methods
Site-Directed Mutagenesis
Human recombinant mutant DOHH enzymes with a single amino acid replaced with alanine (H56A, E57A, H89A, E90A, H207A, E208A, H240A and E241A) and four double mutant enzymes with His-Glu pair replaced with an Ala-Ala pair (H56A/E57A, H89A/E90A, H207A/E208A, and H240A/E241A) were generated using the Quick Change Site-Directed Mutagenesis Kit. The bacterial vector encoding human wild type DOHH as a GST-fusion protein, pGEX-4T-3/hDOHH, was used as a template for PCR and the primer sets were designed for substitution of His or Glu with alanine. The entire ORF of the mutated DOHH was sequenced for confirmation of the intended mutation. The mutated plasmids were introduced into BL21(DE3) competent cells for overexpression of the mutant enzymes.
Overexpression and purification of the human recombinant wild type and mutant DOHH
The selected clones were grown in 120 ml of LB medium containing 100 μg/ml of ampicillin. Protein expression was induced at a density of 0.6 (OD 600nm) by addition of 1 mM isopropyl β-D-thiogalactoside (IPTG) for 2 h. Cell pellets were resuspended in 2.4 ml of buffer A (50 mM Tris.HCl, pH 7.5, 1 mM dithiothreitol (DTT)) containing a protease inhibitor cocktail (EDTA free) and lysed by sonication using an Ultrasonic Processor. After centrifugation of the lysate at 15,000 x g for 30 min, the clarified supernatant was rotated with 0.6 ml of GSH-sepharose for 3 h at 4°C. The resins were washed with buffer B (50 mM Tris.HCl, pH 7.5, 1 mM DTT, 0.1 M NaCl) three times and divided into two tubes. One half was used for preparation of GST-fusion enzymes by elution with buffer C (50 mM Tris.HCl, 1 mM DTT, 30 mM GSH, final pH. 8.0). The other half of the resin was treated with thrombin using Thrombin Cleavage Capture Kit to release free DOHH enzymes. The enzyme was equilibrated in buffer A for activity assays, in HPLC metal free water for metal analysis, or 50 mM sodium phosphate buffer, pH 7.5 for analysis of CD spectra.
Large-scale preparation of wild type DOHH (WT1) and apoenzymes, WTa1 and WTa2, was carried out as follows. E. coli BL21(DE3) cells transformed with pGEX-4T-3/hDOHH were grown in 6 liter of Superbroth at 37 °C. When the cell density reached an OD of 0.6 at 600 nm, protein expression was induced with 0.1 mM isopropyl β-D-thiogalactoside (IPTG) for 4 h at 37°C. Cells were lysed by sonication and the clarified supernatant was incubated with GSH-sepharose (30 ml) by rotation at 4 °C for 2 h. The resin was packed into a column and washed with 500 ml of PBS, 0.5 M NaCl, 10 mM DTT. The resin was resuspended in 50 ml of buffer D (20 mM Tris.HCl, pH 7.5, 0.5 M NaCl, 1 mM DTT) containing 1000 U of thrombin and incubated 2 h at 4°C to release DOHH. Thrombin was removed by passing the DOHH solution over benzamidine resin and any residual thrombin was inactivated by treatment with 1 mM phenylmethylsulfonyl fluoride (PMSF), resulting in WT1. For the WTa1 preparation, 4 mM EDTA was included in the sonication buffer. DOHH released from the GSH resin was applied to a Superdex 200 gel filtration column. Human recombinant DOHH migrated as one major peak with a leading edge shoulder. The early fractions of this peak were pooled, concentrated and designated as WTa1, later shown to contain very low levels of iron (0.02 mol per mol). A second apoenzyme sample, WTa2, was prepared by overnight dialysis of WT1 against 0.1 M sodium citrate buffer, pH 4.5. The enzyme precipitated after dialysis in the acidic citrate buffer and the precipitated protein, after redissolving in buffer A, was found to contain only 0.02 mol iron per mol of protein.
Analysis of secondary structures of DOHH by circular dichroism
Circular dichroic spectra were measured in a Jasco J-715 spectropolarimeter, using quartz cuvettes, thermostated at 25°C. Far ultraviolet spectra were measured from 260–190 nm, with four scans at 50 nm/min, time constant = 1s, bandwidth = 1nm and slit width = 500 μm using 200 μl of solution in a 1mm pathlength cuvette. Protein concentrations were in the range 100–200 μg/ml. Visible spectra were measured from 600–250 nm with a 1 cm pathlength cuvette. The protein concentration was 2.7 mg/ml. The instrument was continually flushed with nitrogen and was calibrated every two weeks with a solution of ammonium (−)-10-camphorsulphonate. Protein concentrations were measured spectrophotometrically, assuming that a 1mg/ml solution has an absorbance of 0.657 at 280 nm. Data were converted into mean residue ellipticities, using a mean residue weight of 109. Estimates of secondary structure were made using the CONTIN program (32)
Deoxyhypusine hydroxylase (DOHH) assay
A typical DOHH reaction mixture contained 25 mM Tris.HCl pH 7.5, 6 mM DTT, 25 μg BSA, 1–2 pmol of the radiolabeled protein substrate (2–4x104 dpm), human eIF5A([3H]Dhp), and enzyme (cell lysate or 0.02–3 μg of purified enzyme) in 20 μl. After incubation at 37°C for 1h, 500 μg of carrier BSA was added to each sample, followed by precipitation with 10 % trichloroacetic acid (TCA). The precipitates were hydrolyzed in 6N HCl at 110°C for 18 h. The content of [3H]hypusine and [3H]deoxyhypusine was determined after ion exchange chromatographic separation as described earlier (24,33).
Analysis of the metal content of DOHH
The buffers for the wild type DOHH and the mutant enzymes were prepared using metal free HPLC water. In order to remove extraneous metals, protein solutions were exchanged with either HPLC water or with 50 mM Tris. HCl buffer (pH 7.5) pretreated with CHELEX 100 resin. The HPLC water or the Tris buffer was also analyzed for any metal contamination. The protein samples were analyzed for metal content (Cd, Co, Cr, Cu, Fe, Mg, Mn, Ni, and Zn), using inductively coupled plasma-high resolution mass spectrometry, Thermo Finnigan Element1 or Element2 (conducted by Dr. Ted Huston, W. M. Keck Elemental Geochemistry Laboratory, Department of Geological Sciences, University of Michigan).
Separation of DOHH apoenzyme and holoenzyme by native gel electrophoresis and their identification by amino acid sequencing
DOHH holo- and apoenzymes were separated by analytical native gel electrophoresis on precast 8–16 % Tris-glycine polyacrylamide gels (Invitrogen) at 4°C using a Tris-glycine buffer, pH 8.3, at 125 V. Samples were applied after 30 min of pre-electrophoresis. For amino acid sequencing, WT1 was separated as above except that a neutral Tris-glycine running buffer (pH 7.2) was used. The proteins were blotted onto PVDF membranes, and briefly stained using Ponceau S dye solution (0.1 %). Protein bands were excised for amino acid sequencing by Edman degradation (CBER, FDA, Bethesda, MD). For identification of active enzyme bands, the enzyme (4 μg) was separated by electrophoresis as above and the lane containing DOHH was sliced in 2.0 mm thickness. Half of each slice was stained with Simply Blue stain (Invitrogen) and the other half was minced and protein extracted with 50 μl of 50 mM Tris.HCl pH 7.5, 1 mM DTT. After incubation at 4°C for 18 h, 10 μl of extract was used for activity assays. In order to determine the metal content, the holo- and apoenzymes were isolated in liquid fractions by preparative gel electrophoresis of WT1 (1 mg) using Mini Prep Cell (Bio-Rad). 0.2 ml fractions were collected and proteins in each fraction were analyzed by SDS PAGE, native gel electrophoresis and for DOHH activity. Appropriate fractions of each protein peak were pooled and used for metal analysis.
Results
The helical structure of DOHH wild type and mutant enzymes
We have previously proposed that the DOHH structure would consist of eight HEAT-repeats in a symmetrical dyad of four HEAT motifs connected by a variable region (1,4). In an effort to validate the super helical structure of DOHH, its secondary structure was determined by CD spectroscopy (Fig. 1). The purified human recombinant wild type enzyme (WT1) displayed a CD spectra typical of an alpha helix rich protein (Fig. 1) and was found to contain 77± 1% alpha helix, 0±2% beta sheet, 5±1% turn and 18±1% remainder. This experimentally determined alpha-helical content is in close agreement with the calculated value obtained from the predicted HEAT-repeat model (76–78 %) of DOHH.
Fig. 1. Circular dichroic spectrum of DOHH.
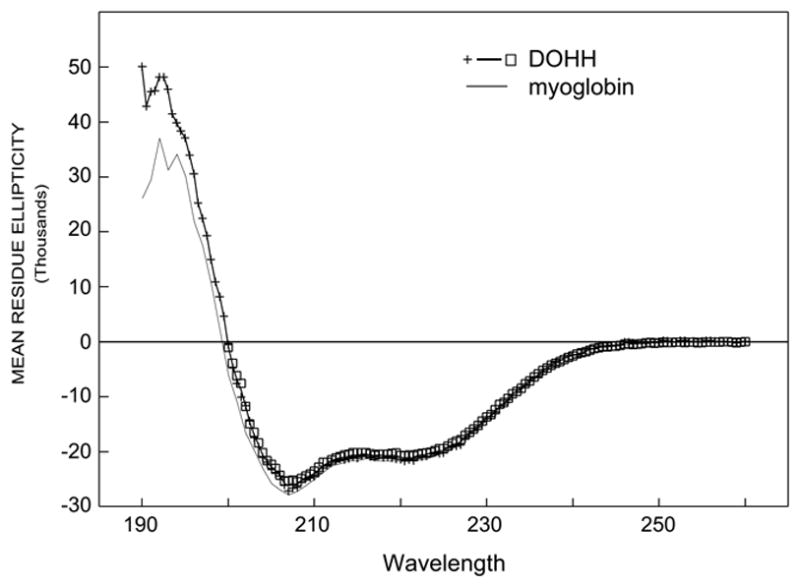
The analysis of the CD spectrum of the wild type enzyme (WT1) was performed as described in Methods. WT1 has a CD spectrum almost identical to that of myoglobin which contains 80 % alpha helix. Similar spectra were obtained for the mutant enzymes and wild type apoenzyme.
DOHH contains four characteristic, strictly conserved His-Glu motifs that were proposed as the metal binding sites. To investigate the role of these residues in structure, metal binding and catalysis, we generated mutant enzymes with substitution of the His-Glu pairs with Ala-Ala pairs and also mutants with the eight His or Glu residues individually replaced with alanine. Alanine substitution was chosen, because it effectively negates sidechain effects and was predicted not to alter the secondary structure of mainchains. As expected, all of the mutant enzymes with alanine substitution of the conserved His-Glu residues gave CD spectra similar to that of the wild type DOHH (data not shown). Furthermore, the CD spectrum of the wild type apoenzyme, WTa1, was also similar to that of the iron-containing enzyme WT1 (data not shown). These findings suggest that the α-helical secondary structure of DOHH does not depend on the conserved His-Glu motifs or metal binding.
Activities and metal content of the wild type DOHH and mutant enzymes; identification of Fe(II) as the catalytic metal for DOHH
Purified human recombinant DOHH catalyzed the conversion of deoxyhypusine-containing eIF5A, eIF5A(Dhp), to the hypusine containing protein, in a time- and concentration dependent manner (Fig. 2, A and B). The pH dependence activity measurement indicates that the purified recombinant enzyme has optimal activity at pH 8–8.5 (Fig. 2C).
Fig. 2. Time (A), concentration (B) and pH (C) dependence of DOHH reaction.
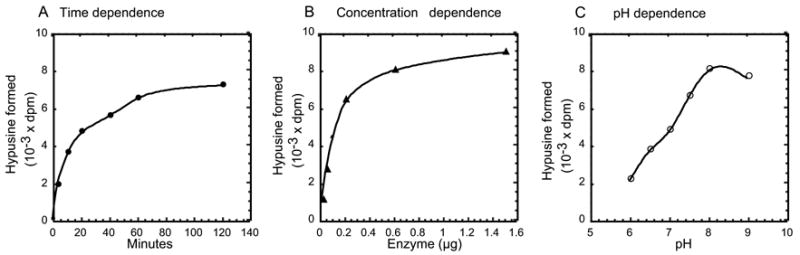
DOHH reactions were carried out as described under Methods using 2.5x104 dpm of labeled substrate protein. The hypusine formed is that from the half of the reaction mixture. The nonlinearity of the reaction in A and B is due to depletion of the substrate protein. In A, 0.2 μg of purified human recombinant enzyme was used and pH of the reaction was 7.5. In B, The reaction time was 60 min at pH 7.5. In C, 0.2 μg of enzyme was used and incubation was 1 h.
When the metal content of the wild type DOHH (purified by one-step affinity chromatography on GSH-Sepharose followed by cleavage of the GST-tag) was measured by inductively coupled plasma-high resolution mass spectrometry, iron was found to be the major metal. In the early preparations of wild type enzyme, its content varied from 0.7 to 1.3 mol per mol (not shown). The variation in the iron content in different preparations of wild type enzyme appears to be due to a gradual dissociation of iron during purification and due to the differences in the extent of buffer exchange or dialysis prior to the metal analysis. Although a low level of Zn was detected in some DOHH preparations, of the nine metals analyzed (Cd, Co, Cr, Cu, Fe, Mg, Mn, Ni, and Zn), no metal other than iron was consistently found to be associated with DOHH at a significant level (>0.07 mol per mol).
The importance of iron in DOHH activity is evident from the reduction or loss of activity upon removal of enzyme-bound iron. Compared to the activity of WT1 (the wild type enzyme purified by one-step affinity chromatography on GSH-Sepharose), WTa1, prepared using a lysis buffer containing 4 mM EDTA followed by gel filtration, and WTa2, prepared from WT1 by removal of iron by extensive dialysis in acidic sodium citrate buffer (pH 4.5), were found to contain a very low level of iron (0.02 mol iron per mol DOHH) (Fig. 3A). Both preparations (WTa1 and WTa2) showed a marked reduction in DOHH activity (Fig. 3B). Addition of ferrous ion (2 μM) increased the activity of a normal wild type DOHH preparation (WT1) and restored the activities of WTa1 and WTa2 enzymes to a large extent, indicating that the lack of iron was the cause for the reduced activities of the two apoenzyme preparations.
Fig 3. Iron content (A) and DOHH activities (B) of the wild type and mutant enzymes.
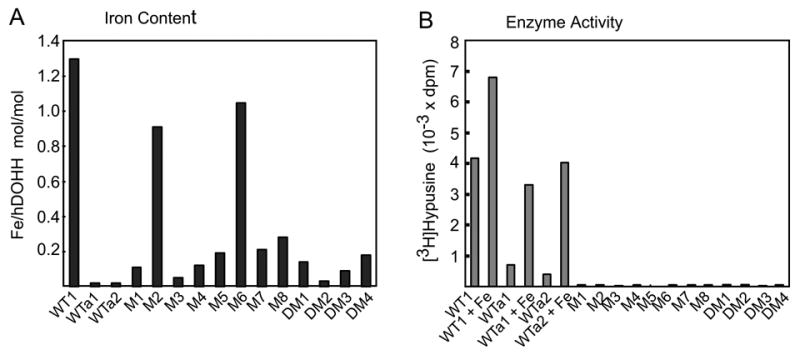
(A) The iron content was determined for WT1, WTa1 and WTa2, and twelve mutant enzymes using approximately 100 μg of purified proteins. (B), The DOHH reaction was carried out as described under Methods. Wild type enzymes (WT1, WTa1 and WTa2) were used at 0.1 μg with or without 2 μM ferrous ammonium sulfate. The twelve mutant enzymes (M1; H56A, M2; E57A, M3; H89A, M4; E90A, M5; H207A, M6; E208A, M7; H240A, M8; E241A, DM1; H56A/E57A, DM2; H89A/E90A, DM3; H207A/E208A and DM4; H240A/E241A) were assayed at 0.3 and 3.0 μg levels (bars shown for 3.0 μg only)
In contrast to the wild type enzyme (WT1), all the mutant enzymes with alanine substitutions of the conserved His-Glu residues at the predicted metal chelation sites, (including eight single mutants, M1; H56A, M2; E57A, M3; H89A, M4; E90A, M5; H207A, M6; E208A, M7; H240A, M8; E241A, and four double mutants, DM1; H56A/E57A, DM2; H89A/E90A, DM3; H207A/E208A, DM4; H240A/E241A) were totally devoid of DOHH activity (Fig. 3B). For these mutant enzymes, no activity was detected in the BL21(DE3) lysate expressing the GST-fusion proteins, the purified GST-fusion proteins or the free mutant proteins after removal of the GST-tag, even at the level of 0.3–3.0 μg, suggesting a critical role for the conserved His-Glu residues in catalysis.
Of the eight mutant enzymes with substitution of a single conserved His or Glu residue with Ala, six (H56A, H89A, E90A, H207A, H240A and E241A) had markedly reduced iron content (0.05–0.28 mol per mol) (Fig 3A), indicating that the six conserved His or Glu residues are involved in the coordination of iron at the active site of DOHH. The iron content was especially low (0.05– 0.15 mol iron per mol of enzyme, less than 12 % of that of the wild type enzyme) in three mutants, H56A, H89A and E90A. The almost complete loss of metal binding by a single mutation on only one dyad arm would suggest that the two metal binding sites (the two sites formed by H56, H89 and E90 for the N-terminal dyad, and H207, H240 and E241 for the C-terminal dyad) work cooperatively. For these six mutant enzymes, the deficiency in iron binding appears to be the cause of loss of activity. In contrast, the other two mutant enzymes, E57A and E208A, which were totally inactive, contained iron at a level (~1 mol per mol) comparable to that of the wild type enzyme. Thus, E57 and E208, while not required for iron binding, are still essential for DOHH catalysis. Addition of Fe(II) (1–1000 μM) to these eight mutant enzymes (His or Glu to Ala) had little or no stimulatory effects on DOHH activity (data not shown).
The separation of iron-containing holoenzyme and iron free apoenzyme of the wild type and mutant DOHH by native gel electrophoresis and gel filtration
The three preparations of the wild type enzyme (WT1, WTa1 and WTa2) and mutant enzymes appeared similarly pure and displayed one predominant band of 32 kDa upon SDS-PAGE (Fig. 4A, Coomassie Blue staining). However, upon electrophoresis under non-denaturing conditions, heterogeneity was observed. The wild type enzyme, WT1, resolved into two major species (Fig. 4B, lane 1): the first, a well focused, fast moving band (Band 1, solid arrow head) and the second, a more diffuse, slow moving band (Band 2, open arrowhead). The wild type enzyme preparations with low iron content, WTa1 and WTa2, migrated at the position of Band 2, indicating that this second diffuse band represents the iron-free apoenzyme. Upon incubation of WTa1 with 0.2 mM ferrous ammonium sulfate for 2 h, a portion of Band 2 protein (apoenzyme) was converted to Band 1, indicating a partial reconstitution of apoenzyme with iron to form the holoenzyme (Fig. 4B, compare lanes 3 and 3’). When the lane containing the WT1 enzyme was sliced and proteins extracted from the gel slices for the DOHH assay, activity was found at the position of Band 1 (holoenzyme monomer), but no activity was detectable at the position of Band 2 (apoenzyme monomer) (data not shown). In order to confirm that the two bands of the WT1 proteins are indeed DOHH holo- and apoenzymes, the proteins were transferred to a PVDF membrane after native gel electrophoresis, and subjected to amino acid sequencing. After nine cycles of Edman degradation, both bands yielded the identical amino acid sequence, GSMVTEQEV in which MVTEQEV is the first 7 amino acids of human DOHH. (The leading sequence, Gly-Ser is derived from the pGEX-4T-3 vector after thrombin cleavage of the R-G bond to remove the GST tag.) Furthermore, when the WTa2 enzyme was incubated with various transition metals for reconstitution, only Fe(II) and Fe(III) were found to convert a portion of the Band 2 form to the Band 1 form (Fig 4C, lanes 7 and 8), suggesting that iron is the only metal that effectively binds to the active site of DOHH.
Fig.4.
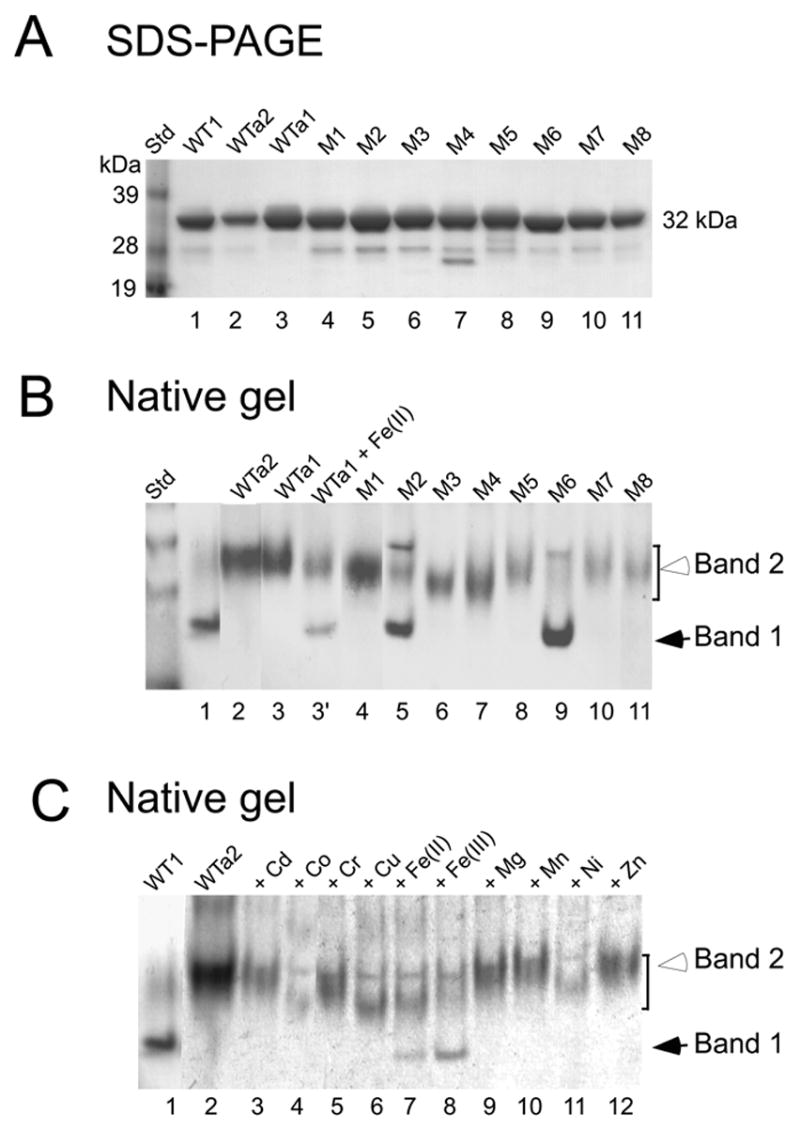
Analysis of wild type (WT1, WTa1 and WTa2) and the mutant enzymes by SDS-PAGE (A), by native gel electrophoresis (B) and the reconstitution of wild type apoenzyme WTa2 with various metals (C). (C), The apoenzyme WTa2 (1.5 μg) was incubated in 3.5 μl, 50 mM Tris. HCl pH, 7.5 with 0.2 mM of the indicated metal at room temperature for 2.5 h prior to native gel electrophoresis. The eight mutant enzymes are M1; H56A, M2; E57A, M3; H89A, M4; E90A, M5; H207A, M6; E208A, M7; H240A, M8; E241A.
All the mutant enzymes with reduced iron content (H56A, H89A, E90A, H207A, H240A and E241A) migrated mainly as the slow-moving diffuse band (Band 2), consistent with the wild type apoenzyme bands of WTa1 and WTa2. The two mutant enzymes that contained iron at the level of the wild type enzyme (~ 1 mol per mol), E57A and E208A, showed both, the holoenzyme (Band 1) and apoenzyme (Band 2), like the wild type DOHH.
DOHH is an acidic protein with a calculated pI of 4.74. Since the binding of Fe(II) or Fe(III) will reduce the negative charge of the enzyme, the iron-containing holoenzyme is expected to migrate slower than the apoenzyme, if there is no conformational difference. That the electrophoretic mobility of the iron-containing holoenzyme (Band 1) is faster than that of the iron-free apoenzyme (Band 2) suggests that the binding of iron to the DOHH active site causes a significant conformational change, giving rise to a more compact form. An indication of a larger hydrodynamic size of the apoenzyme than the holoenzyme was also given from their differential migration on gel filtration (Fig.5). DOHH wild type enzyme (purified by affinity chromatography) eluted as one major peak (at a position expected for 32 kDa monomer) with a leading edge shoulder (Fig.5A). When the protein in each fraction was analyzed by SDS-PAGE (Fig 5B) and native gel electrophoresis (Fig. 5C), the early shoulder fractions (Fr, 22-24) were found to contain the apoenzyme (Band 2 form) and the center peak fractions (Fr 25-28) largely the holoenzyme (Band 1 form) with a small amount of apoenzyme. DOHH activity in the fractions corresponded to the intensity of the holoenzyme band; there was no detectable activity in the apoenzyme fractions (Fig. 5D).
Fig.5.
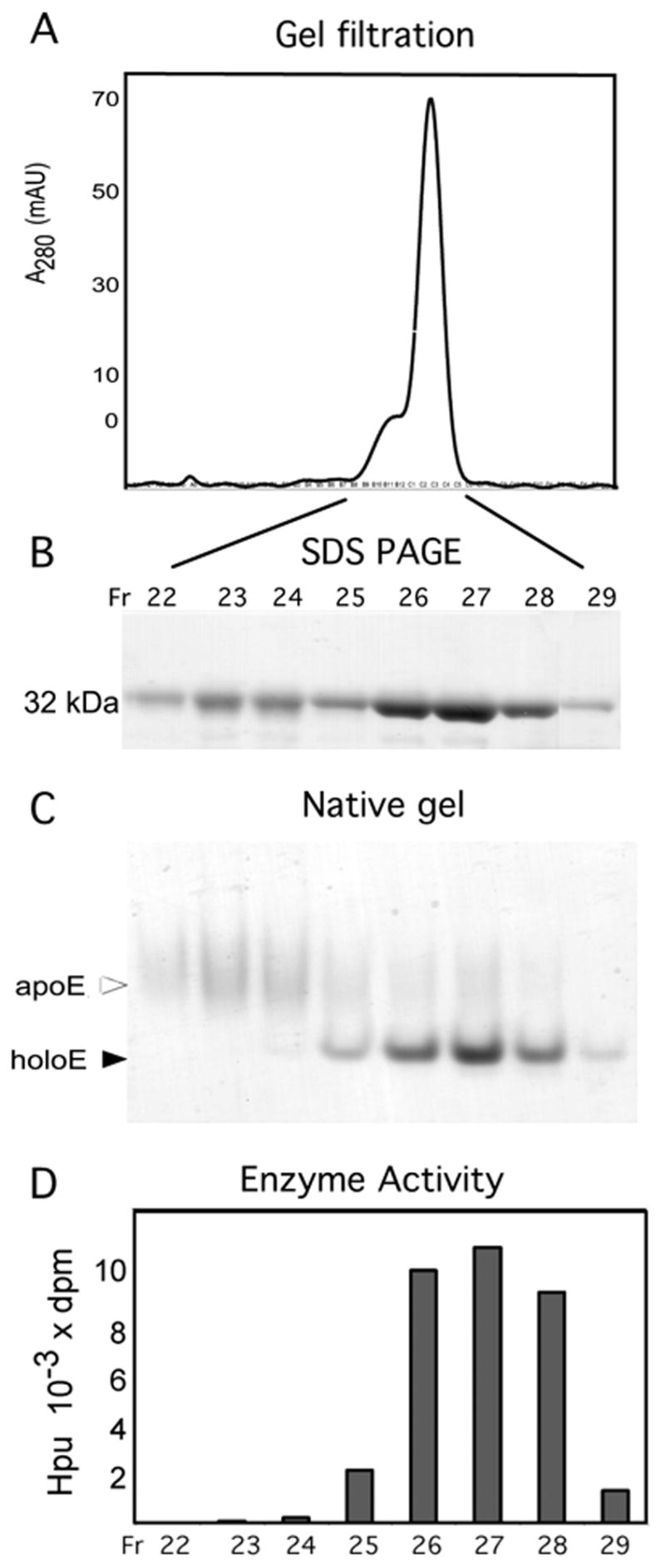
Gel filtration of wild type DOHH on Superdex 200 column. (A) absorbance at 280 nm, (B) SDS-PAGE, (C) native gel electrophoresis, and (D) activity. The wild type enzyme was purified by affinity chromatography on GSH Sepharose and GST affinity tag was removed as described in the Methods. 15 mg of the resulting enzyme was applied to Superdex 200 gel filtration column (2.6cm x 60 cm, 320 ml bed volume) equilibrated in buffer D (20 mM Tris.HCl pH 7.5, 1 mM DTT, 0.5 M NaCl and 5 % glycerol). 5 ml fractions were collected. 10 μl of fractions 22–24 and 5 μl of fractions 25–29 were used for SDS-PAGE (B) and native gel electrophoresis (C). 0.3 μl of each fraction was used for DOHH activity assay (D).
In order to obtain a homogenous DOHH preparation and to determine the iron stoichiometry of the holoenzyme, we performed preparative gel electrophoresis under non-denaturing conditions using the Mini Prep Cell (Bio-Rad). The DOHH wild type enzyme (WT1) resolved into two peaks, peak 1 (fractions 17–21) and peak 2 (fractions 30–36), each of which migrated as a single band of 32kDa upon SDS-PAGE (Fig. 6A). Upon native gel electrophoresis, fractions in the first peak contained mainly Band 1 and only trace amounts of Band 2. The second peak contained largely Band 2 (Fig. 6B), with a small amount of Band 1 in later fractions (fractions 33–35). The iron content of peak 1 was determined (in two preparations) to be 1.8 ± 0.09. This finding indicates that the majority of the peak 1 protein is indeed the holoenzyme, optimally containing two mol of iron per mol of enzyme monomer. Little iron was found in early fractions of peak 2 (fractions 30–32), confirming that the diffuse Band 2 (open arrowhead) is the apoenzyme. DOHH activity was found mainly in the first peak (DOHH holoenzyme monomer) (Fig 6C), in accordance with the data from assays of gel slices and gel filtration fractions (described above). DOHH activity was undetectable in fractions 30–32 from peak 2, a confirmation that the apoenzyme is inactive. A low level of activity in fractions 33 and 34, is likely due to a small contamination with the holoenzyme (Fig. 6B, seen as faint bands in fractions 33 and 34) which presumably derived from a third band (gray arrowhead, Fig. 6B, first lane) that has DOHH activity and DOHH sequence (data not shown) and is most likely a dimer of the DOHH holoenzyme. This third band was not consistently detected in different native gels, but its level seemed to increase at high DOHH concentration and in the absence of DTT. Overall, these results provide strong evidence that human recombinant DOHH is an iron-dependent enzyme optimally containing two moles of iron per mol of protein.
Fig.6.
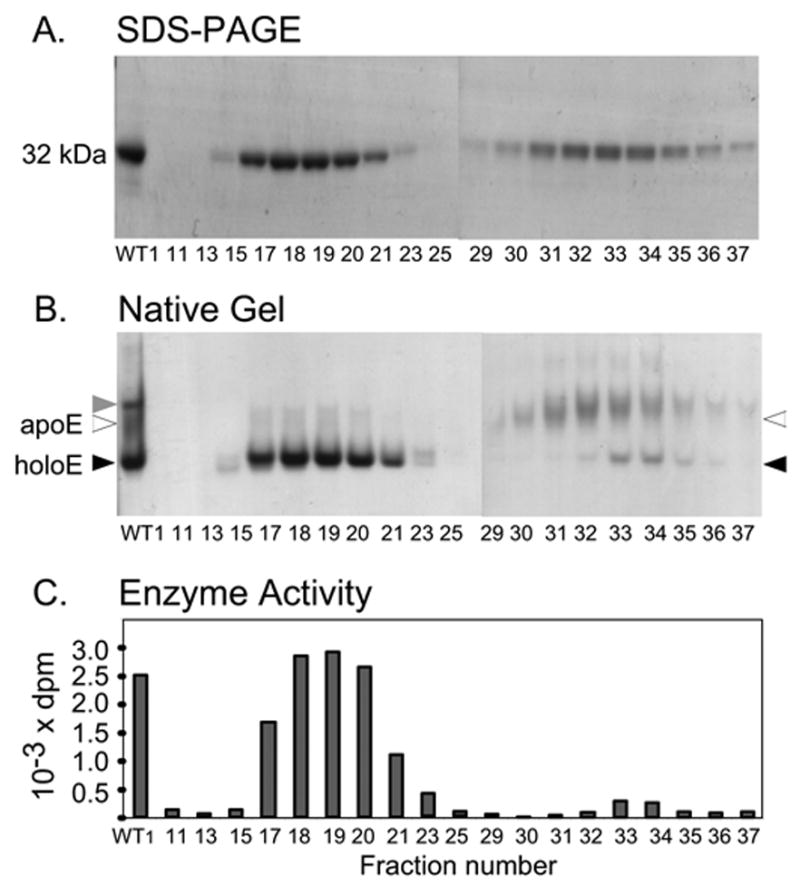
Separation of wild type holo-and apoenzymes by preparative gel electrophoresis. SDS-PAGE (A), native gel electrophoresis (B), and DOHH activities (C) of fractions containing proteins.
WT1 preparation (1 mg) in native gel sample buffer (0.4 ml) was applied to a disc gel (10 % Tris-Glycine polyacrylamide gel, 7 mm in diameter x 6 cm long running gel, 1.5 cm of stacking gel) and electrophoresis was conducted at 300 V using Mini Prep Cell (Bio-Rad) with cooling. 0.2 ml fractions (3 min) were collected and aliquots of each fraction were analyzed by SDS PAGE (10 μl), native gel electrophoresis (10 μl) and for DOHH activity (0.15 μl). The pooled fractions of each protein peak (pools of fractions 17–20, 31–32 and 33–34) were used for metal analysis.
The effects of various metal ions on DOHH activity
The specificity of metal-induced changes in the electrophoretic mobility of DOHH apoenzymes (Fig. 4C) suggests that the human enzyme binds only Fe(II) or Fe(III) with high affinity at the active site. Therefore, we examined the effects of various metal salts (CdCO3, CoCl2.6H2O , CrK(SO4)2, CuSO4.5H2O, Fe(NH4)2(SO4)2, FeCl3 , MgCl2, MnCl2.4H2O, NiCl2.6H2O and ZnCl2, at 1–1000 μM) on the enzymatic activity of the apoenzyme WTa2 (Fig. 7A) and the iron-containing enzyme WT1 (Fig. 7B). Fe(II) was the only metal that exerted a dramatic enhancement (>10 fold) of the apoenzyme activity at 1–10 μM. At higher concentrations of Fe(II) (>100 μM), the stimulation was diminished. Unlike Fe(II), Fe(III) exhibited little stimulation at 1–10 μM. A relatively small increase in the activity by Fe(III) at 100 μM may be due to a conversion of a small portion of Fe(III) to Fe(II) in the reaction mixture containing 6 mM DTT. These results indicate the oxidation state-specific catalytic function of Fe(II) in DOHH catalysis. When the same panel of metal ions was added to the iron-containing DOHH (WT1), only Fe(II) stimulated the activity at 1–3 μM concentration; it caused inhibition at high concentrations (>100 μM). Other metal ions, including Co, Cu, Mn, Zn, and Cr, also displayed a significant inhibition at 1 mM whereas Cd, Mg, and Ni did not.
Fig. 7. The effects of various metals on DOHH iron-containing enzyme WT1 (A) and the apoenzyme (WTa2) (B).
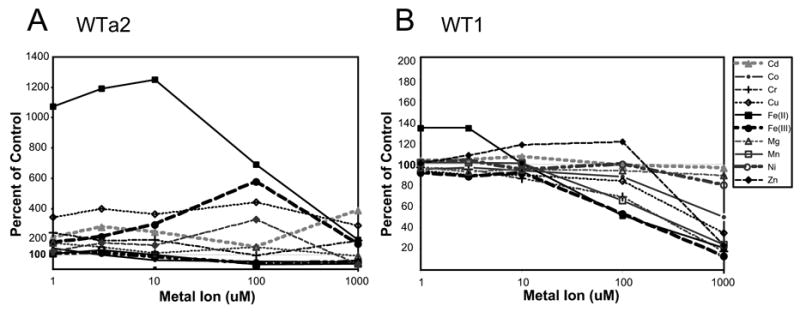
The activity assays were carried out as described in the Methods using 0.15 μg each of WT1 (A) or WTa2. The metal ions were added at 1, 3, 10, 100 and 1000 μM to the reaction mixture. The Y axis represents the percentage of control (no addition).
Discussion
Deoxyhypusine hydroxylase belongs to a family of proteins that contain tandem repeats of α-helical hairpins known as HEAT motifs. Sequence analysis of DOHH showed that it is comprised of two symmetrical domains (N- and C-terminal), each containing four HEAT-repeats, connected by a variable loop (4). Furthermore, it has four strictly conserved His-Glu pairs, two on each dyad, that were proposed as metal coordination sites. A homology model of DOHH, based on the known crystal structure of a HEAT-repeat protein, the E. coli protein YibA (Protein Data Bank # 1OYZ), presents a solenoid structure with a concave inner surface (4). However, this model did not predict a precise distance between the presumed metal-binding His-Glu pairs on the two dyad arms, nor the stoichiometry of bound metal to protein. In an effort to experimentally validate the proposed DOHH model and/or to refine it, we investigated the role of iron and the strictly conserved His-Glu residues in DOHH structure and catalysis. We have identified Fe(II) as the active site metal of DOHH and His56, His89, Glu90, His207, His240 and Glu241 as the iron coordination sites. Furthermore, we provide strong evidence for the critical role of each residue of the His-Glu motifs in DOHH catalysis. Thus, our results support a super helical DOHH model in which the two metal binding sites are closely interactive for binuclear iron binding and catalysis (Scheme 1A).
Scheme 1. Hypothetical structures of DOHH holo- and apoenzymes.
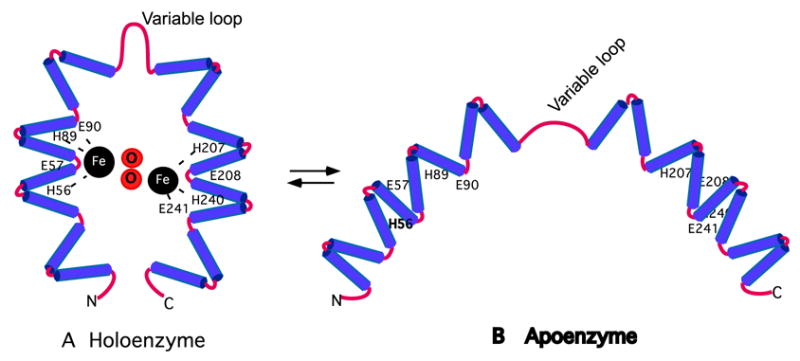
(A). Schematic view of the holoenzyme with two iron atoms at the active site. The eight conserved His and Glu residues critical for enzyme activity are indicated. Oxygen atoms are inserted between the iron atoms as projected for the reaction with dioxygen. Though consistent with the characteristics of the enzymes reported here, it is a tentative model. Refined from Fig. 5 of Ref (4)
(B). Schematic view of the apoenzyme with open dyad arms consistent with a larger hydrodynamic size. Apoenzyme would be a mixture with varying distances between the dyad arms.
The binding of iron does not affect the secondary structure of DOHH and its α-helical content, since there is no clear distinction between the CD spectra of iron-containing DOHH and the iron-deficient forms. In spite of similarities in the α-helical content between the two forms of DOHH, the iron binding does have a profound effect on the tertiary structure of DOHH, as a difference in the hydrodynamic size was clearly noted for the apo- and holoenzymes. Electrophorectic mobility on native gel depends on both protein charge and hydrodynamic size. Based on the charge effects alone, an iron-containing holoenzyme is expected to migrate more slowly than the iron-free apoenzyme. On the contrary, the DOHH holoenzyme migrates faster than the apoenzyme indicating that the iron-containing DOHH is in a more compact conformation than the apoenzyme (Fig. 4B, Fig 6B). Consistent with this notion, the apoenzyme was found to elute in earlier fractions than the holoenzyme upon gel filtraton (larger hydrodynamic size) (Fig. 5). In relating this observation the DOHH model, the two arms of the apoenzyme would be further separated than in the holoenzyme (Scheme 1B). In addition, unlike the holoenzyme which migrates as a well focused band on native gels, the apoenzyme migrates as a diffuse band suggesting a heterogeneous mixture of different conformations. This mixture may represent apoenzyme molecules with varying distances between the two dyad arms, probably due to a greater flexibility of the dyad arms around the variable hinge loop in the absence of bound metal. It seems likely that the binding of iron bridges the two dyad arms of the DOHH monomer, resulting in a homogeneous population with a compact, concave structure, as depicted in Scheme 1A.
The separation of holoenzyme from apoenzyme enabled us to determine the stoichiometry of iron binding to DOHH and to identify the active form of DOHH. Since the iron content of early preparations of the wild type DOHH (mixtures of apo- and holoenzymes) ranged from 0.7 to 1.3, it was not clear whether a DOHH monomer could bind one atom of iron only, or two atoms only, or both one and two atoms. Attempts to reconstitute DOHH to a fully charged holoenzyme were not successful, since incubation of DOHH (WT1) with iron (0.1–1.0 mM) only slightly increased the holoenzyme fraction (not shown) and iron at this high concentration inhibits DOHH activity. By preparative gel electrophoresis, the holoenzyme, containing close to two moles of iron per mole of protein, was isolated and was identified as the active form of DOHH. No DOHH peak with one mol per mol of iron was identified. Although the DOHH monomer contains two iron coordination sites (one on each dyad arm), it is likely that binuclear iron binding occurs coordinately to form one active center. If the two iron coordination sites (the two sites formed by H56, H89 and E90 for the N-terminal dyad, and H207, H240 and E241 for the C-terminal dyad) were to operate as two separate active centers, one would expect a 50% reduction in iron binding or in DOHH activity by a single alanine substitution at either site. However, a single alanine substitution at any of the His or Glu residues in the four His-Glu motifs abolished DOHH activity. Furthermore, substitution of alanine for any one of the six residues H56, H89, E90, H207, H240 or E241 caused a 70–90% reduction in the iron content. A tempting model to explain our overall data is a DOHH active center with a diiron coordinated by the six His and Glu residues from both dyad arms. (Scheme 1A).
Binuclear iron binding has been characterized in several non-heme enzymes, and iron-binding proteins such as hemerythrin and ferritin (34,35). A well studied group of diiron enzymes, including the ribonucleotide reductase R2 subunit, methane monooxygenase and Δ9-desaturases, have an active site diiron, located in a four helix bundle, typically liganded with two His and four Glu residues. Two of the glutamate/aspartate residues form a bidentate carboxylate bridge to the two iron atoms. Upon reaction with dioxygen, an oxo/peroxo link may be formed (34,36,37). It would be interesting to determine how a binuclear center in DOHH would be configured and to examine how it is related to other known diiron enzymes. Further site-directed mutagenesis of other conserved amino acid residues of DOHH is underway to identify additional amino acid residues that may be critical for iron binding and catalysis and to gain further insights into the reaction mechanism of DOHH.
Interestingly, the concentrated solutions of the wild type enzyme purified in the absence of EDTA displayed a characteristic blue color and a broad absorption peak centered at 628 nm was detected (Supplemental Fig.1). In contrast, neither blue color or nor any absorption peak above 300 nm was observed for the apoenzyme at the same protein concentration and in the same buffer. Thus, the blue color of DOHH solutions appears to be due to the diiron binding. Analogously, another blue non-heme iron protein, neelaredoxin (with absorption peak at 666 nm) also contains a biiron center, proposed to be coordinated by specific His and Glu residues (38).
In this report, we have identified Fe(II) as the required metal for DOHH. Whereas both Fe(II) and Fe(III) can bind to DOHH apoenzyme (Fig. 4C) to induce a conformational change, only Fe(II) could restore the activity of the apoenzyme at 1–10 μM (Fig. 7A). In this concentration range, Fe(III) was without effect. These findings suggest that the iron binding and the conformational change per se are necessary, but are not sufficient, for DOHH catalysis and that only Fe(II) can fulfill the catalytic function. Other metals including Cd, Co, Cr, Cu, Mg, Mn, Ni and Zn did not convert the apoenzyme (slow migration form, band 2) to the faster moving form (band 1); presumably these metals also do not bind to the active site of DOHH. Consistent with the lack of conversion of DOHH apoenzyme to holoenzyme monomer (band 1) by these other metals (Fig. 4C), is their failure to enhance the activities of the apoenzyme (Fig. 7B).
DOHH is predicted to fold into a HEAT-repeat structure completely unrelated to the double stranded beta helix (DSBH) structure of other protein hydroxylases that catalyze the hydroxylation of proline, lysine, aspartic acid or asparagine residues (25,26). Nonetheless, DOHH and DSBH enzymes share common features such as Fe(II)-dependent reaction mechanism. Like DSBH enzymes, DOHH is dependent on molecular oxygen for hydroxylation (data not shown). However, there seems to be a fine distinction between DOHH and DSBH enzymes in details of mechanism, mode of iron binding, the consensus sequence of the coordination sites and metal specificity. For example, DSBH enzymes contain a consensus ligand sequence, His1-X-Asp/Glu-Xn-His2, one site per catalytic subunit for coordination of one iron atom (25). On the other hand, in the case of DOHH monomer, two iron atoms likely form a binuclear center coordinated by a pair of His1-Glu1-Xn-His2-Glu2 sequence (n=32) (one from each dyad arm and in which His1, His2 and Glu2 are three chelating residues) (4). Furthermore, the two iron binding sites of the dyad arms seem to closely cooperate for binuclear iron binding and catalysis.
DOHH catalyzes the final step in the maturation of eIF5A, an essential protein in eukaryotic cell proliferation. DOHH as a key component of this maturation pathway and its product, eIF5A, have been implicated in the regulation of mammalian cell proliferation (1,6–11,23). The metal chelating compounds that effectively inhibit deoxyhypusine hydroxylation in eIF5A, arrest cell cycle progression at the boundary of G1/S in mammalian cells, including human cancer cells and HUVEC cells (23,24). Furthermore, these compounds caused a strong inhibition of angiogenesis in model assays, presumably by concomitant inhibition of DOHH and collagen prolyl-4 hydroxylase (24). eIF5A and DOHH have also been implicated in HIV-1 viral replication (39–43). Furthermore, metal chelating inhibitors of DOHH, e.g. deferiprone and ciclopirox olamine, have been reported to inhibit replication of HIV-1 (44) (H.M. Hanauske-Abel, personal communication). Taken together, the ability to limit eIF5A hypusination may provide a useful tool for targeted control of cell growth and expansion in disease states.
We are actively pursuing determination of the crystal structures of DOHH apo- and holoenzymes and a DOHH complex with its protein substrate. The crystal structures will offer the ultimate validation of DOHH model structure and the proposed iron binding mode and will provide further insights into the DOHH reaction mechanism and the specificity of enzyme-substrate interactions. Characterization of the reaction mechanism and determination of the crystal structure of DOHH will pave the way to the development of specific inhibitors of DOHH.
Supplementary Material
Acknowledgments
We thank Drs. Hans E. Johansson (Biosearch Technologies, Inc.) and Hartmut M. Hanauske-Abel (UMDNJ, New Jersey Medical School) for helpful discussions. This research was supported by the Intramural Research Program of the NIH (NIDCR, NIDDK).
Abbreviations
- eIF5A
eukaryotic initiation factor 5A
- eIF5A-1
primary isoform of eIF5A
- eIF5A(Lys)
eIF5A precursor
- eIF5A(Dhp)
eIF5A intermediate containing deoxyhypusine
- DOHH
deoxyhypusine hydroxylase
- DSBH
double stranded beta helix
- GST
glutathione-S-transferase
- GSH
glutathione
- DTT
dithiothreitol
- CD
circular dichroism
References
- 1.Park MH. J Biochem (Japan) 2006;139:161–169. doi: 10.1093/jb/mvj034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolff EC, Lee YB, Chung SI, Folk JE, Park MH. J Biol Chem. 1995;270:8660–8666. doi: 10.1074/jbc.270.15.8660. [DOI] [PubMed] [Google Scholar]
- 3.Joe YA, Wolff EC, Park MH. J Biol Chem. 1995;270:22386–22392. doi: 10.1074/jbc.270.38.22386. [DOI] [PubMed] [Google Scholar]
- 4.Park JH, Aravind L, Wolff EC, Kaevel J, Kim YS, Park MH. Proc Natl Acad Sci U S A. 2006;103:51–56. doi: 10.1073/pnas.0509348102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbruzzese A, Park MH, Folk JE. J Biol Chem. 1986;261:3085–3089. [PubMed] [Google Scholar]
- 6.Chen KY, Liu AY. Biol Signals. 1997;6:105–109. doi: 10.1159/000109115. [DOI] [PubMed] [Google Scholar]
- 7.Byers TL, Lakanen JR, Coward JK, Pegg AE. Biochem J. 1994;303:363–368. doi: 10.1042/bj3030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chattopadhyay MK, Tabor CW, Tabor H. Proc Natl Acad Sci U S A. 2003;100:13869–13874. doi: 10.1073/pnas.1835918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishimura K, Murozumi K, Shirahata A, Park MH, Kashiwagi K, Igarashi K. Biochem J. 2005;385:779–785. doi: 10.1042/BJ20041477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gerner EW, Mamont PS, Bernhardt A, Siat M. Biochem J. 1986;239:379–386. doi: 10.1042/bj2390379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caraglia M, Marra M, Giuberti G, D'Alessandro AM, Baldi A, Tassone P, Venuta S, Tagliaferri P, Abbruzzese A. J Biochem (Tokyo) 2003;133:757–765. doi: 10.1093/jb/mvg097. [DOI] [PubMed] [Google Scholar]
- 12.Bartig D, Schümann H, Klink F. System Appl Microbiol. 1990;13:112–116. [Google Scholar]
- 13.Glick BR, Ganoza MC. Proc Natl Acad Sci U S A. 1975;72:4257–4260. doi: 10.1073/pnas.72.11.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanawa-Suetsugu K, Sekine S, Sakai H, Hori-Takemoto C, Terada T, Unzai S, Tame JR, Kuramitsu S, Shirouzu M, Yokoyama S. Proc Natl Acad Sci USA. 2004;101:9595–9600. doi: 10.1073/pnas.0308667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schnier J, Schwelberger HG, Smit-McBride Z, Kang HA, Hershey JW. Mol Cell Biol. 1991;11:3105–3114. doi: 10.1128/mcb.11.6.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wöhl T, Klier H, Ammer H. Mol Gen Genet. 1993;241:305–311. doi: 10.1007/BF00284682. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki K, Abid MR, Miyazaki M. FEBS Lett. 1996;384:151–154. doi: 10.1016/0014-5793(96)00310-9. [DOI] [PubMed] [Google Scholar]
- 18.Park MH, Joe YA, Kang KR. J Biol Chem. 1998;273:1677–1683. doi: 10.1074/jbc.273.3.1677. [DOI] [PubMed] [Google Scholar]
- 19.Thompson GM, Cano VS, Valentini SR. FEBS Lett. 2003;555:464–468. doi: 10.1016/s0014-5793(03)01305-x. [DOI] [PubMed] [Google Scholar]
- 20.Sugimoto A. Differentiation. 2004;72:81–91. doi: 10.1111/j.1432-0436.2004.07202004.x. [DOI] [PubMed] [Google Scholar]
- 21.Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM. Genetics. 1999;153:135–177. doi: 10.1093/genetics/153.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Csonga R, Ettmayer P, Auer M, Eckerskorn C, Eder J, Klier H. FEBS Lett. 1996;380:209–214. doi: 10.1016/0014-5793(96)00020-8. [DOI] [PubMed] [Google Scholar]
- 23.Hanauske-Abel HM, Park MH, Hanauske AR, Popowicz AM, Lalande M, Folk JE. Biochim Biophys Acta. 1994;1221:115–124. doi: 10.1016/0167-4889(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 24.Clement PM, Hanauske-Abel HM, Wolff EC, Kleinman HK, Park MH. Int J Cancer. 2002;100:491–498. doi: 10.1002/ijc.10515. [DOI] [PubMed] [Google Scholar]
- 25.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 26.Aravind L, Koonin EV. Genome Biol. 2001;2:RESEARCH0007. doi: 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kivirikko KI, Pihlajaniemi T. Adv Enzymol Relat Areas Mol Biol. 1998;72:325–398. doi: 10.1002/9780470123188.ch9. [DOI] [PubMed] [Google Scholar]
- 28.Hanauske-Abel HM, Popowicz AM. Curr Med Chem. 2003;10:1005–1019. doi: 10.2174/0929867033457601. [DOI] [PubMed] [Google Scholar]
- 29.Phizicky EM, Martzen MR, McCraith SM, Spinelli SL, Xing F, Shull NP, Van Slyke C, Montagne RK, Torres FM, Fields S, Grayhack EJ. Methods Enzymol. 2002;350:546–559. doi: 10.1016/s0076-6879(02)50984-8. [DOI] [PubMed] [Google Scholar]
- 30.Andrade MA, Petosa C, O'Donoghue SI, Muller CW, Bork P. J Mol Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 31.Park JH, Wolff EC, Folk JE, Park MH. J Biol Chem. 2003;278:32683–32691. doi: 10.1074/jbc.M304247200. [DOI] [PubMed] [Google Scholar]
- 32.Provencher SW, Glockner J. Biochemistry. 1981;20:33–37. doi: 10.1021/bi00504a006. [DOI] [PubMed] [Google Scholar]
- 33.Park MH, Liberato DJ, Yergey AL, Folk JE. J Biol Chem. 1984;259:12123–12127. [PubMed] [Google Scholar]
- 34.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235–350. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 35.Tshuva EY, Lippard SJ. Chem Rev. 2004;104:987–1012. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 36.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 37.Wei PP, Skulan AJ, Wade H, DeGrado WF, Solomon EI. J Am Chem Soc. 2005;127:16098–16106. doi: 10.1021/ja053661a. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Sharma P, Le Gall J, Mariano AM, Teixeira M, Xavier AV. Eur J Biochem. 1994;226:613–618. doi: 10.1111/j.1432-1033.1994.tb20087.x. [DOI] [PubMed] [Google Scholar]
- 39.Bevec D, Jaksche H, Oft M, Wöhl T, Himmelspach M, Pacher A, Schebesta M, Koettnitz K, Dobrovnik M, Csonga R, Lottspeich F, Hauber J. Science. 1996;271:1858–1860. doi: 10.1126/science.271.5257.1858. [DOI] [PubMed] [Google Scholar]
- 40.Ruhl M, Himmelspach M, Bahr GM, Hammerschmid F, Jaksche H, Wolff B, Aschauer H, Farrington GK, Probst H, Bevec D, Hauber J. J Cell Biol. 1993;123:1309–1320. doi: 10.1083/jcb.123.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bevec D, Hauber J. Biol Signals. 1997;6:124–133. doi: 10.1159/000109118. [DOI] [PubMed] [Google Scholar]
- 42.Liu YP, Nemeroff M, Yan YP, Chen KY. Biol Signals. 1997;6:166–174. doi: 10.1159/000109123. [DOI] [PubMed] [Google Scholar]
- 43.Hofmann W, Reichart B, Ewald A, Müller E, Schmitt I, Stauber RH, Lottspeich F, Jockusch BM, Scheer U, Hauber J, Dabauvalle MC. J Cell Biol. 2001;152:895–910. doi: 10.1083/jcb.152.5.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrus L, Szabo P, Grady RW, Hanauske AR, Huima-Byron T, Slowinska B, Zagulska S, Hanauske-Abel HM. Biochemical Pharmacology. 1998;55:1807–1818. doi: 10.1016/s0006-2952(98)00053-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.