

Abstract
The Golgi ribbon is a complex structure of many stacks interconnected by tubules that undergo fragmentation during mitosis through a multistage process that allows correct Golgi inheritance. The fissioning protein CtBP1-S/BARS (BARS) is essential for this, and is itself required for mitotic entry: a block in Golgi fragmentation results in cell-cycle arrest in G2, defining the ‘Golgi mitotic checkpoint'. Here, we clarify the precise stage of Golgi fragmentation required for mitotic entry and the role of BARS in this process. Thus, during G2, the Golgi ribbon is converted into isolated stacks by fission of interstack connecting tubules. This requires BARS and is sufficient for G2/M transition. Cells without a Golgi ribbon are independent of BARS for Golgi fragmentation and mitotic entrance. Remarkably, fibroblasts from BARS-knockout embryos have their Golgi complex divided into isolated stacks at all cell-cycle stages, bypassing the need for BARS for Golgi fragmentation. This identifies the precise stage of Golgi fragmentation and the role of BARS in the Golgi mitotic checkpoint, setting the stage for molecular analysis of this process.
Keywords: BARS, fission, G2, Golgi complex, mitotic checkpoint
Introduction
The Golgi complex is the essential station for protein processing and sorting at the center of the secretory pathway. In mammalian cells, this organelle is structured in the form of numerous stacks (usually 70–100; Trucco et al, 2004) that are laterally connected by tubular bridges known as the ‘non-compact' zones. This generates a continuous membranous system, the ‘Golgi ribbon', that is located in a perinuclear position (Rambourg et al, 1987; Shorter and Warren, 2002). One intriguing aspect of the physiology of the Golgi complex is its mechanism of mitotic inheritance, which involves progressive and reversible disassembly of the ribbon into dispersed elements, allowing the correct partitioning of the Golgi membranes between the daughter cells (Shorter and Warren, 2002; Colanzi et al, 2003). This mitotic fragmentation takes place in two main sequential steps: the pericentriolar Golgi membranes are first converted to scattered tubulo-reticular elements and then these are further fragmented and dispersed throughout the cytoplasm, appearing as the Golgi ‘haze' (Colanzi et al, 2003; Axelsson and Warren, 2004; Altan-Bonnet et al, 2006). The molecular mechanisms involved here are partially understood, and through the development of in vitro Golgi fragmentation assays, some of the relevant components have been identified (Acharya et al, 1998; Colanzi et al, 2000, 2003; Sutterlin et al, 2001, 2002; Shorter and Warren, 2002). Among these, a key player is the protein CtBP1-S/BARS (BARS) (Hidalgo Carcedo et al, 2004), which acts by inducing fission of Golgi tubules during both normal trafficking and mitosis (Weigert et al, 1999).
A remarkable recent development in this area is that mitotic Golgi fragmentation is required not only for Golgi partitioning but also for entry into mitosis. This was established through a variety of approaches, which showed that inhibition of Golgi fragmentation in living cells results in arrest of the cell cycle at the G2 stage (Sutterlin et al, 2002; Hidalgo Carcedo et al, 2004). These approaches included treatments affecting the function of the Golgi matrix protein GRASP-65 (Sutterlin et al, 2002; Preisinger et al, 2005; Yoshimura et al, 2005) as well as the microinjection of an anti-BARS blocking antibody and BARS dominant-negative mutants, and BARS depletion using antisense oligonucleotides (Hidalgo Carcedo et al, 2004). These anti-BARS treatments blocked fragmentation of the Golgi complex and progression through the cell cycle, whereas readdition of recombinant BARS restored these processes (Hidalgo Carcedo et al, 2004). Based on these lines of evidence, the existence of a novel checkpoint that appears to sense the integrity of the Golgi complex, the ‘Golgi checkpoint', has been postulated (Sutterlin et al, 2002; Hidalgo Carcedo et al, 2004; Preisinger et al, 2005). The physiological relevance of this checkpoint is witnessed by its link with profound and prolonged inhibition of mitotic entrance (Sutterlin et al, 2002; Hidalgo Carcedo et al, 2004; Yoshimura et al, 2005).
Unfortunately, basic aspects of the relationship between Golgi fragmentation and mitotic ingression remain unclear, including the exact cell-cycle phase during which this Golgi checkpoint is triggered (see below) and the precise event in Golgi fragmentation that controls the checkpoint. This information is fundamental for identification of the mechanisms behind the Golgi checkpoint. In particular, a confusing feature here is that it is generally believed that Golgi fragmentation initiates during prophase (Shorter and Warren, 2002). This, however, would be incompatible with the demonstration that inhibition of Golgi fragmentation blocks cell-cycle progression in G2 (Sutterlin et al, 2002; Hidalgo Carcedo et al, 2004), an earlier cell-cycle phase. One possible explanation relates to the ability of mammalian cells to retreat from the earliest mitotic stage (prophase) in the presence of a ‘stress' (Mikhailov and Rieder, 2002); therefore, in principle, a cell could sense Golgi fragmentation in early mitosis, and if something is amiss, it would rapidly exit prophase, returning to the premitotic G2 phase. Alternatively, the alterations in the Golgi complex that are necessary for entry into mitosis could occur already during G2; in this case, however, they should be so subtle structurally as to have so far escaped detection.
Because BARS controls both Golgi fragmentation and the Golgi checkpoint, a potentially useful approach to address the above questions is likely to be the identification of the precise site of action of BARS in mitotic Golgi partitioning. A relevant hint as to the role of BARS derives from our previous work, which showed that it operates in the fission of Golgi tubules involved in a variety of processes (Weigert et al, 1999; Mironov et al, 2004). This suggests that the role of BARS in Golgi partitioning might be in the severing of the tubular non-compact zones of the Golgi ribbon.
Here, using BARS-related molecular tools and a variety of morphological and functional approaches, we clarify these issues and identify an early stage of the Golgi partitioning process that has not been fully characterized so far, namely, the severing of the Golgi ribbon into separate stacks or small groups of stacks. We find that this step occurs in G2 and is necessary to pass the G2/M Golgi checkpoint. Presumably, this is the minimal level of Golgi fragmentation needed to divide the Golgi between daughter cells. BARS is specifically required for severing of the non-compact zones in G2, resulting in ribbon breakdown. The further disassembly of the Golgi complex, which leads to complete fragmentation of this organelle, is BARS independent and takes place only after the cells have entered mitosis. These results set the stage for defining the molecular processes involved in the Golgi checkpoint.
Results
The Golgi ribbon is severed into isolated stacks during G2
As indicated above, inhibition of fragmentation of the Golgi complex leads to cell-cycle arrest in the G2 phase (Sutterlin et al, 2002; Hidalgo Carcedo et al, 2004), suggesting that Golgi partitioning initiates during G2 rather than, as generally believed, in prophase (Shorter and Warren, 2002). If this is the case, this partitioning most likely involves minor modifications of the general structure of the organelle that have gone unnoticed until now.
To explore this possibility, we first used confocal microscopy to investigate the general morphology of Golgi membranes in HeLa cells during G2, taking advantage of the combined staining patterns of anti-phosphorylated-H1 and anti-H3 antibodies to identify late-G2 cells (Hendzel et al, 1997; Hidalgo Carcedo et al, 2004). When labelled with antibodies against giantin, the Golgi ribbon in S phase appeared as a large continuous perinuclear formation, either compact or elongated, with alternating heavily and lightly stained areas that possibly represent stacks and non-compact zones (Figure 1A). A characteristic continuous ribbon was present in most of the cells found in S phase (Figure 1B). During late G2 (1–2% of the cells), the Golgi complex showed breaks that interrupted the continuity of the Golgi membranes (with a considerable increase in discrete Golgi objects) in more than 90% of the G2 cells (Figure 1A and B), indicating that some morphological modification was taking place. Then, in late prophase/prometaphase, the pericentriolar Golgi stacks broke down into smaller fragments, and finally, between prometaphase and early anaphase, the Golgi membranes underwent further fragmentation and were diffusely dispersed throughout the cytosol (mitotic Golgi haze) (Figure 1A), in agreement with previous observations (Shorter and Warren, 2002). Similar results were obtained when staining the Golgi with an anti-GM130 antibody (not shown).
Figure 1.

Morphology of Golgi membranes in HeLa cells through the cell cycle. (A) HeLa cells were grown on coverslips and fixed and labelled with anti-phosphohistone-H1 and -H3 polyclonal antibodies (pH1/pH3) as markers of different cell-cycle phases (Hidalgo Carcedo et al, 2004), and with a giantin antibody to label the Golgi complex. Images were acquired using a confocal microscope set at maximal resolution. For quantitative analysis of Golgi phenotypes, fixed imaging conditions were applied to all of the images. Scale bar, 5 μm. (B) Percent distribution of Golgi phenotypes as described in Materials and methods. Data shown are representative of a total of 70–120 cells for each experimental condition across three independent experiments.
To analyze the morphology of Golgi membranes during G2 at the ultrastructural level, HeLa cells were induced to accumulate in G2 with an 18-h incubation with bisbenzimide, a topoisomerase-I inhibitor that activates the DNA damage checkpoint and promotes the accumulation of cells in G2 (Supplementary Figure S1) (Dubey and Raman, 1983). A survey of the Golgi by electron microscopy showed that the ribbon in non-treated (interphase) cells consisted of stacks of variable sizes, which were often aligned along their main axis and appeared connected by tubular non-compact zones, often visible even in random thin sections (Figure 2A). G2-blocked cells did not show major differences either in the number of cisternae that composed the single stacks or in the average diameter of the stacks; however, when compared with non-treated cells, they showed stacks that were isolated (i.e. not interconnected by tubules) in most cases and were not longitudinally aligned (Figure 2A). To define more precisely the connectivity of the Golgi ribbon in G2, we used serial sections to examine the ribbon in three-dimensional space. This analysis showed that while nearly all of the control stacks were interconnected, the stacks of G2 cells were either isolated or connected in small groups of two to four stacks. The ribbon connectivity was also quantified in thin sections by assessing the fraction of the stacks that were isolated or connected in small groups. Although this method does not reveal connections outside the plane of the section, it is quite suitable to compare G2 with control conditions. Again, the differences indicated a large loss of ribbon connectivity during G2 (Figure 2B). Thus, these data are consistent with confocal microscopy observations and indicate that during G2, the alterations in the global structure of the Golgi complex consist of the disappearance of the interstack tubular connections.
Figure 2.
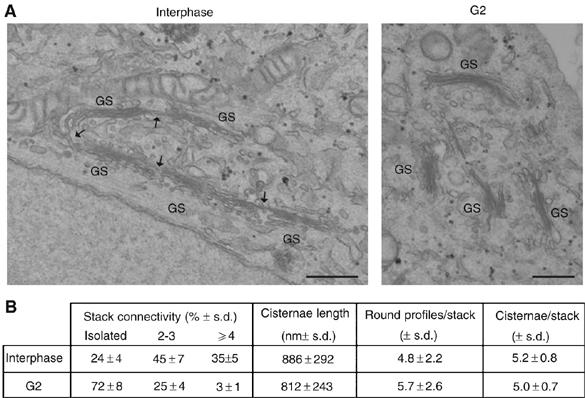
Ultrastructure of Golgi membranes during G2 and interphase. (A) Non-treated HeLa cells and HeLa cells induced to accumulate in G2 with bisbenzimide were processed for electron microscopy. Representative images of thin sections of non-treated (interphase) and G2-blocked (G2) cells are shown. Note that Golgi membranes are organized as stacks (GS) connected by non-compact zones (arrows) in non-synchronized HeLa cells, and as isolated stacks (GS) during G2. Scale bar, 0.5 μm. (B) Morphometric analysis of the ribbon extension in thin sections from non-treated (interphase) and G2-blocked HeLa cells (G2), as indicated. Stack connectivity represents the relative percent distribution of Golgi stacks found isolated, in groups of 2–3 or of at least four connected stacks. The data are representative of more than 80 Golgi containing sections for each experimental condition in three independent experiments.
Next, we further assessed the loss of ribbon integrity by a different quantitative approach based on analysis of the diffusion-mediated process of fluorescence recovery after photobleaching (FRAP) of Golgi-resident enzymes (Lippincott-Schwartz and Patterson, 2003). Golgi enzymes diffuse along the length of the Golgi ribbon, as revealed by their fast FRAP (Cole et al, 1996); thus, if the tubular connections between adjacent stacks are severed, there should be a block in diffusion of the enzymes between stacks, as has been previously reported (Mironov et al, 2004). HeLa cells were transfected with the medial-Golgi-resident enzyme galactosyltransferase fused to GFP (GalT-GFP; Cole et al, 1996) and induced to accumulate in G2 with bisbenzimide (Supplementary Figure S1). Then, a region corresponding to about half of the Golgi membranes was bleached by repeated laser illumination at high intensity and the FRAP of GalT-GFP was examined. Figure 3A and B illustrate the pattern and time course of the FRAP of the bleached area. In non-treated (interphase) cells, recovery of GalT-GFP fluorescence (normalized to the unbleached areas) was rapid, consistent with an intact Golgi ribbon (Figure 3A–C, interphase), and reached a plateau in 5 min. In contrast, in cells in G2, GalT-GFP FRAP reached a much lower plateau than that seen in control cells (Figure 3A–C, G2), suggesting that in G2, the continuity of the Golgi ribbon is interrupted.
Figure 3.

BARS mediates fission of the Golgi ribbon during G2. (A–C) HeLa cells were transfected with GalT-GFP and left non-treated (interphase/Int) or were induced to accumulate in G2 with bisbenzimide (C, G2 blocked). They were then mock microinjected (G2(−)) or microinjected with BARS-DN SBD (G2+SBD/SBD) and fluorescent dextran as microinjection marker and subjected to FRAP analysis after bleaching about 50% of the Golgi mass. (A) Representative images of GalT-GFP HeLa cells before bleaching (Pre-bleach), at the end of bleaching (Bleach) and 300 s after bleaching (300 s). The bleached areas are delineated by white-bordered rectangles. Scale bar, 5 μm. (B) Time courses of FRAP of interphase (top), G2 (middle) and BARS-DN-SBD-microinjected G2 (bottom) cells illustrated in (A). Fluorescence intensities in bleached areas were monitored every 5 s. Recovery curves of fluorescence intensities are normalized to non-bleached areas and corrected for background versus times. (C) Mean FRAP (±s.d.) 300 s after bleaching. Data are from analysis of 18–20 cells for each condition, and from four independent experiments performed as in (A). Statistical significances were evaluated by Student's t-test: (a) versus (b), P<0.0005; (b) versus (c), P<0.008 and (a) versus (c), P<0.03. (D) HeLa cells were transfected with GalT-GFP and left overnight non-treated (Int) or to accumulate in S phase with aphidicolin (G2 enriched). Eleven to thirteen hours after aphidicolin washout, the G2-enriched cells were microinjected with recombinant GST (8 mg/ml) (GST), recombinant BARS-DN SBD (8 mg/ml) (SBD) or the p50-2 anti-BARS antibody (3–6 mg/ml) (Ab), with fluorescent dextran as microinjection marker. Finally, the cells were subjected to FRAP after bleaching 15–20% of the Golgi mass. Mean FRAP (±s.d.) measured 300 s after bleaching and derived from 32–38 cells for each experimental condition, from five independent experiments. Statistical significances evaluated by Student's-t test: (a) versus (b), P<0.0001; (b) versus (c), P<0.0001; (b) vs (d), P<0.0001 and (a) versus (c), P<0.2. (E) HeLa cells arrested in S phase using aphidicolin (Hidalgo Carcedo et al, 2004). After 1 h, cells were microinjected with recombinant GST (8 mg/ml), generic IgG (3-6 mg/ml), recombinant BARS-DN SBD (8 mg/ml) (SBD) or the p50-2 anti-BARS antibody (3–6 mg/ml) (Ab), with FITC-conjugated dextran as microinjection marker; after 13 h, they were fixed and labelled for cell-cycle phase (Hoechst 33258; not shown), as detailed in (Hidalgo Carcedo et al, 2004). More than 200 cells were microinjected for each condition. The relative mitotic index was calculated after measuring the percentages of microinjected cells in mitosis normalized to non-microinjected cells on the same coverslip. Means (±s.d.) from three independent experiments.
To rule out the possibility that the reduced FRAP seen in G2 is due to nonspecific effects of bisbenzimide, we set up an alternative approach to assess FRAP in cells in G2. HeLa cells were transfected with GalT-GFP and induced to accumulate at the beginning of S phase by synchronization with aphidicolin, a DNA polymerase inhibitor (see Hidalgo Carcedo et al, 2004). After the aphidicolin washout, the number of cells in mitosis increased with time, peaking 13 h after removal of aphidicolin. GalT-GFP FRAP was then evaluated in interphase (non-synchronized) cells and in a G2-enriched cell population represented by the non-mitotic cells seen 11–13 h after aphidicolin washout, which is prevalently composed of cells in various stages of G2 (Supplementary Figure S1). Even under these conditions, the FRAP in G2-enriched cells measured 5 min after bleaching was significantly reduced compared with that of interphase cells (Figure 3D), in spite of the non-complete G2 synchrony. Indeed, a small fraction of the non-mitotic cell population had high FRAP values, most likely because this represented cells that were not in G2, in which, therefore, the ribbon had not yet undergone fragmentation.
Altogether, this demonstrates that during G2, the non-compact zones through which the Golgi enzymes diffuse along the ribbon (Cole et al, 1996) become fragmented, resulting in the production of isolated stacks or small groups of Golgi stacks. As this Golgi fragmentation step occurs in G2, it might be the Golgi-related event that is required for mitotic entry. Since BARS is essential for mitotic entry, to address this point, we analyzed its role in this early Golgi fragmentation.
BARS is required for fission-dependent cleavage of the non-compact zones in G2
We have previously reported that BARS promotes the fission of tubules originating from the rims of Golgi stacks under a variety of conditions (Weigert et al, 1999). It thus seems likely that the role of BARS in Golgi mitotic partitioning might primarily be the severing of the inter-stack tubular connections.
Although our previous investigations into the role of BARS in Golgi fragmentation were obtained in NRK cells (Hidalgo Carcedo et al, 2004), since our experiments on cell-cycle synchronization were performed in the HeLa cell system, we addressed this here by first also assessing the role of BARS on mitotic ingression in HeLa cells. HeLa cells were transfected with GalT-GFP and induced to accumulate at the beginning of S phase by aphidicolin treatment. After aphidicolin washout, the cells were microinjected with either the BARS substrate-binding domain (SBD), which acts as a dominant-negative BARS (BARS-DN SBD), or the p52 affinity-purified anti-BARS blocking antibody to block BARS function. The cells were then fixed 13 h after aphidicolin washout to evaluate the number of cells in mitosis, as previously described (Hidalgo Carcedo et al, 2004). In cells microinjected with BARS-DN SBD or with the anti-BARS blocking antibody, the number of cells undergoing mitosis was greatly reduced compared with control (GST- or IgG-microinjected) cells (Figure 3E), indicating that BARS controls mitotic entry also in HeLa cells.
Having verified this, we tested the effect of BARS blockers on G2-specific fission of the non-compact zones using the above-described FRAP assay in HeLa cells. We first tested the effects of inhibition of BARS activity in bisbenzamide-blocked G2 cells. As shown in Figure 3A–C, microinjection of BARS-DN SBD prevented the reduction in FRAP in G2-blocked cells, indicating that the G2 division of the ribbon into isolated stacks was inhibited. We also used FRAP with a G2-enriched cell population as described above. Under these conditions, in cells where BARS was inhibited by microinjection of BARS-DN SBD or the anti-BARS blocking antibody, the FRAP was markedly greater than that of control (GST-microinjected) G2-enriched cells; indeed, it was comparable with that seen in interphase cells (Figure 3D). Microinjection of recombinant BARS or generic IgGs in interphase cells did not have any effect on the FRAP (data not shown).
These data indicate that BARS is required for G2-specific severing of the Golgi ribbon into stacks. Since BARS also controls mitotic entrance (Hidalgo Carcedo et al, 2004), this indicates that ribbon fragmentation in G2 is the event that controls progression into mitosis.
BARS function is dispensable for G2/M transition if the Golgi complex is in the form of isolated stacks
An alternative possibility, however, is that BARS influences mitotic entrance in ways unrelated to Golgi fragmentation. Albeit unlikely, this possibility is not formally ruled out by the data above. To address this point, we reasoned that if BARS-dependent fission of non-compact tubular zones is the essential BARS effect for passing through the Golgi checkpoint, then BARS should be required for entry into mitosis only in cells that have a normal Golgi ribbon, and would instead be dispensable when the ribbon organization is lost. A previous indication of this comes from studies using the microtubule depolymerizing agent nocodazole, which divides the Golgi ribbon into isolated stacks (Hidalgo Carcedo et al, 2004). Nocodazole, however, might have severe side effects. Thus, to test this prediction, we used LdlG-CHO cells, which lack the Golgi protein GM130 (Vasile et al, 2003). When grown at 34°C, LdlG-CHO cells show an apparently normal Golgi structure as revealed by immunofluorescence microscopy (Figure 4A); however, since GM130 is required for maintenance of the Golgi ribbon (Puthenveedu et al, 2006), LdlG-CHO cells lack the ribbon organization and have Golgi membranes in the form of isolated but fully functional, ministacks that are clustered around the nucleus (Marra et al, 2007). Moreover, if LdlG-CHO cells are transfected with GM130, they reacquire normal Golgi ribbon structure (Marra et al, 2007). Based on these considerations, BARS should not be required for mitotic entry in LdlG-CHO cells, which possess no Golgi ribbon organization; in contrast, in GM130-transfected LdlG-CHO cells, where the Golgi ribbon can reform, BARS should be necessary for entrance into mitosis. This prediction was tested using the BARS blockers: microinjection of the anti-BARS blocking antibody into LdlG-CHO cells had no effect on entry into mitosis, whereas the same treatment in GM130-transfected LdlG-CHO cells blocked them in G2 (Figure 4B). An analogous case in a different cell line is described below. These results therefore establish a causal link between inhibition of severing of the Golgi ribbon in G2 by BARS blockers and inhibition of mitotic entrance. Notably, once the cells had entered mitosis, further fragmentation of the Golgi complex in cells microinjected with BARS blockers was essentially identical to that of control (IgG microinjected) cells, indicating that after entry into mitosis, BARS does not have an essential role in the subsequent stages of Golgi fragmentation (not shown).
Figure 4.
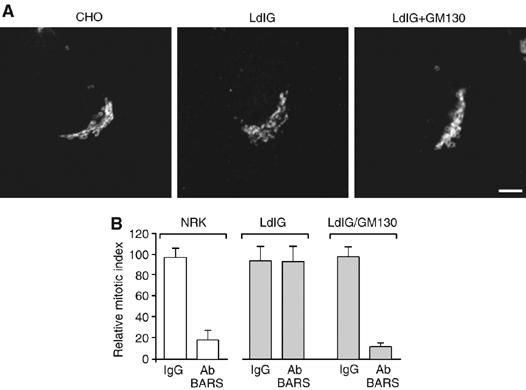
Inhibition of BARS does not affect progression into mitosis in cells that do not possess an intact Golgi ribbon. (A) CHO, LdlG (CHO cells defective in GM130 expression) and LdlG/GM130 (LdlG cells stably expressing GM130) cells (as indicated) were fixed and treated for immunofluorescence microscopy. The structure of the Golgi complex was monitored using an anti-giantin antibody. Scale bar, 5 μm. (B) NRK (normal rat kidney), LdlG and LdlG/GM130 cells (as indicated) arrested in S phase using aphidicolin (Hidalgo Carcedo et al, 2004). After 1 h, cells were microinjected with generic IgG (3–6 mg/ml; IgG) or the p50-2 BARS antibody (3–6 mg/ml; Ab). After a further 7 h (NRK) or 14 h (LdlG and LdlG/GM130), cells were fixed and labelled for microinjection (goat antibody to rabbit IgG; not shown) and cell-cycle phase (Hoechst 33258; not shown), as detailed in (Hidalgo Carcedo et al, 2004). More than 200 NRK and 600 LdlG and LdlG/GM130 cells were microinjected per sample. The relative mitotic index was calculated after measuring the percentage of microinjected cells in mitosis normalized to non-microinjected cells on the same coverslip. Means (±s.d.) from three independent experiments.
Therefore, these data identify the first stage of the partitioning process, the severing of the Golgi ribbon into separate stacks (which is necessary to pass the G2/M checkpoint), as the target of the action of BARS in the mitotic entry process.
Cells derived from BARS knockout embryos lose both Golgi ribbon organization and the Golgi mitotic checkpoint
Our previous (Hidalgo Carcedo et al, 2004) and present data all converge toward an essential role for BARS in Golgi fragmentation and entry into mitosis. Thus, an apparent discrepancy arises between these data and those based on a BARS knockout (KO) mouse (Hildebrand and Soriano, 2002). These KO data reveal that although this BARS KO is embryonically lethal, a cell line derived from immortalized fibroblasts obtained from 8-day embryos (CtBP90 mouse embryo fibroblasts (MEFs)) shows normal proliferation (Hildebrand and Soriano, 2002). Moreover, as shown in Figure 5, the mitotic cycle and the mode of Golgi complex partitioning during mitosis in CtBP90 MEFs appears normal, at least at the immunofluorescence level. We therefore examined how CtBP90 MEFs compensate for a lack of BARS with respect to these mitotic events.
Figure 5.
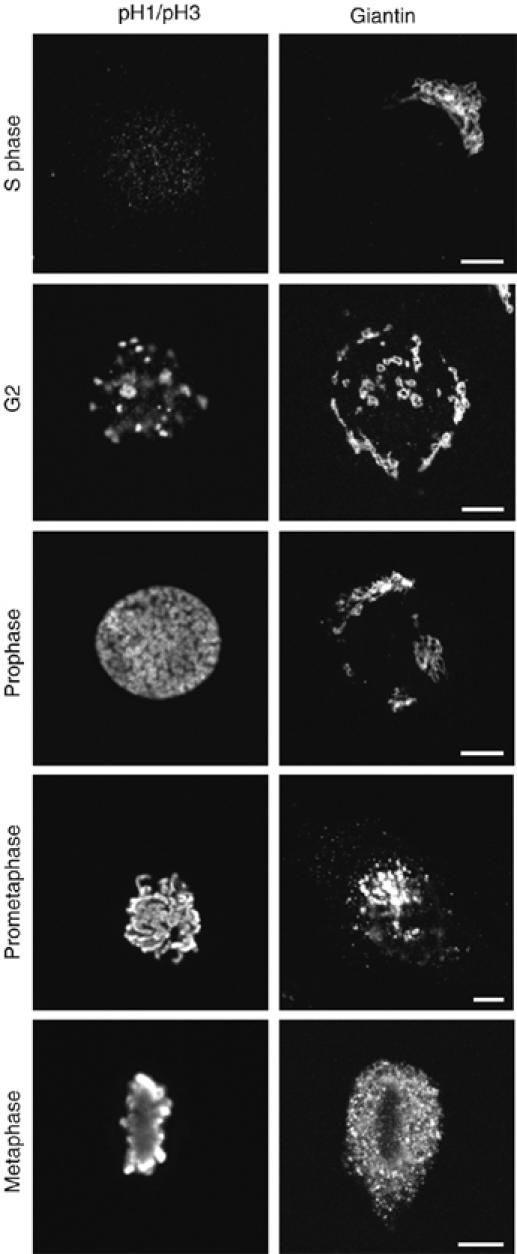
Morphology of Golgi membranes in CtBP90 cells through the cell cycle. CtBP90 cells were grown on coverslips and fixed and labelled with anti-phospho-histone-H1 and -H3 polyclonal antibodies (pH1/pH3) as markers of different cell-cycle phases (Hidalgo Carcedo et al, 2004), and with an anti-giantin antibody to label the Golgi complex. Images were acquired using confocal microscope set at maximal resolution. Images shown are representative of three independent experiments. Scale bar, 5 μm.
First, since the essential step for entry into mitosis is fragmentation of the Golgi ribbon by cleavage of the non-compact zones, we examined its organization in CtBP90 MEFs. Remarkably, in electron microscopy, the normal ribbon organization of the Golgi complex was absent in CtBP90 MEFs, a feature that was not evident by simple confocal analysis (Figure 6A and B). Although the stacks were morphologically normal and located near the cell center as usual, they were physically isolated from each other and were not aligned (Figure 6B). The absence of an extended Golgi ribbon was confirmed by quantitative electron microscopy, using the approaches described above for HeLa cells in G2 (the results were similar to HeLa cells in G2; see Figure 6D) and by FRAP: CtBP90 MEFs were transfected with GalT-GFP and half of the fluorescent Golgi complex was bleached. The bleached area did not show appreciable FRAP when monitored for 5 min (Figure 7A and B). These data thus indicate that BARS KO MEFs have a fragmented ribbon, which would explain their ability to bypass the Golgi checkpoint and enter mitosis despite their lack of BARS.
Figure 6.
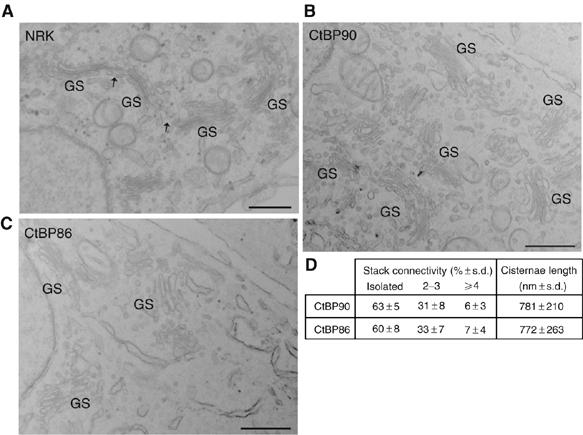
Ultrastructure of Golgi membranes in CtBP90 and CtBP86 cells. (A–C) NRK, CtBP90 and CtBP86 cells were processed for electron microscopy. Representative images of thin sections of NRK (A) CtBP90 (B) and CtBP86 (C) and cells are shown. Note that the Golgi membranes are organized as a continuous ribbon, composed of stacks (GS) and non-compact zones (arrows) in NRK cells, whereas they are organized as isolated and non-aligned stacks (GS) in CtBP90 and CtBP86 cells. (D) Morphometric analysis of the ribbon extension in thin sections from CtBP90 and CtBP86 cells, as indicated. Stack connectivity represents the relative percent distribution of Golgi stacks found isolated, in groups of 2–3 or of at least four connected stacks. Images are representative of more that 60 Golgi-containing sections from three independent experiments. Scale bar, 0.5 μm.
Figure 7.
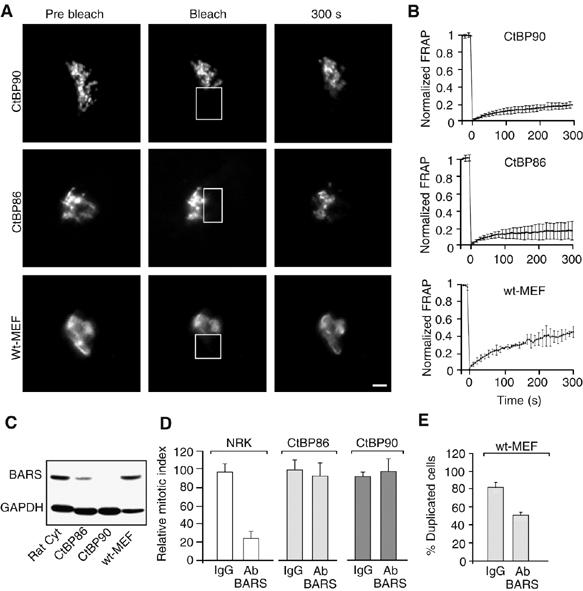
Inhibition of BARS does not affect entry into mitosis in CtBP90 cells, which are organized in the form of isolated stacks. Cells were transfected with GalT-GFP and subjected to FRAP analysis after bleaching 50% of the Golgi mass. (A) Representative images of GalT-GFP in CtBP86, CtBP90 and wild-type (wt)-MEF cells before bleaching (prebleach), immediately after bleaching (bleach) and 300 s after bleaching (300 s). Bleached areas are shown by white-bordered rectangles. Fluorescence intensities in bleached areas were monitored every 5 s. Recovery curves of fluorescence intensities were normalized to unbleached areas and corrected for background versus time. Scale bar, 5 μm. (B) Mean time courses of FRAP (±s.d.) from 20–32 cells from four independent experiments preformed as indicated in (A) with CtBP86 (top), CtBP90 (middle) and wild-type (bottom; wt) MEFs. Fluorescence intensities in bleached areas were monitored every 5 s. Recovery curves of fluorescence intensities are normalized to non-bleached areas and corrected for background versus times. (C) Cytosols prepared from rat brain cytosol (rat cyt.), CtBP86 and CtBP90 MEFs, and wt-MEFs (30 μg per lane) were analyzed by immunoblotting using the anti-BARS antibody (BARS) and an anti-GAPDH antibody (GAPDH) as reference. (D) Cells were arrested in S phase with 2 mM thymidine (see Materials and methods and (Hidalgo Carcedo et al, 2004)). After 1 h, cells were microinjected with generic IgG (3–6 mg/ml) or the p50-2 BARS antibody (3–6 mg/ml; Ab BARS). After a further 7 h (NRK) or 8 h (CtBP86 and CtBP90), cells were fixed and labelled for microinjected cells (goat antibody to rabbit IgG; not shown) and cell-cycle phase (Hoechst 33258; not shown) as detailed in (Hidalgo Carcedo et al, 2004). More than 200 NRK and 600 CtBP86 and CtBP90 MEFs were microinjected per sample. The relative mitotic index was calculated after measuring the percentage of microinjected cells in mitosis normalized to non-microinjected cells on the same coverslip. Means (±s.d.) from four independent experiments. (E) Unsynchronized wt-MEFs were microinjected with generic IgG (2–4 mg/ml) or the p50-2 anti-BARS antibody (2-4 mg/ml; Ab BARS) in the presence of fluorescent dextran as microinjection marker. Cells were fixed 12 h after injection and labelled for microinjected cells (goat antibody to rabbit IgG), cell-cycle phase (Hoechst 33258) and scored for the number of duplicated cells. The percentages of duplicated cells that were microinjected with IgG or the anti-BARS antibody are shown. More than 200 wt-MEF cells were microinjected per sample. Means (±s.d.) from four independent experiments.
We also wanted to know whether the fragmented ribbon found in CtBP90 MEFs results from a specific adaptation of these cells to the lack of BARS or if it is a feature common to all of the MEF lines. We first examined CtBP86 MEF cells that were derived from a partial BARS KO (i.e. from heterozygous embryos) and therefore had lower levels of BARS (about 30%) than other MEFs (Figure 7C). Interestingly, also in these cells, the extended Golgi ribbon organization was lost, as determined by electron microscopy (Figure 6C and D) and FRAP analyses (Figure 7A and B). In line with this, microinjection of the anti-BARS antibody had no effect on mitotic entry in both CtBP90 and CtBP86 MEFs (Figure 7D). In contrast, unrelated MEFs that had normal BARS levels showed FRAP values significantly greater than that of the CtBP MEFs. At the same time, however, this FRAP was somewhat lower than that seen in HeLa cells (compare Figure 7B, wt-MEF, with Figure 3C, Int.), suggesting that the Golgi membranes in wild-type MEFs are organized as a less extended ribbon than that in HeLa cells (potentially due to the accelerated duplication rate of MEF cells; Ciemerych and Sicinski, 2005). MEF cells should therefore have a Golgi checkpoint and require BARS for mitotic entry. To test this is not straightforward, because these cells do not synchronize easily and tend to undergo apoptosis when subject to drug-based sychronization protocols. We thus used an alternative scheme that did not require stressful synchronizing treatments. We microinjected a large number of unsynchronized cells with either the anti-BARS antibody or control IgGs and 12 h later analyzed the number of cells that had duplicated in the two groups (Figure 7E). With the anti-BARS antibody in wild-type MEFs, there was a significant, albeit not complete, inhibition of cell duplication (as compared to IgGs; Figure 7E). This incomplete effect might be due to either the lower basal degree of ribbon integrity in these MEF cells when compared with HeLa cells, as noted above, and/or by the fact that only very prolonged duplication blocks are detected by the protocol used here. In any case, these data provide an explanation of how BARS KO MEFs can bypass the need for BARS for cell duplication and they confirm the link between Golgi ribbon organization and the presence of the Golgi mitotic checkpoint.
Discussion
This study reports on three closely interrelated findings. First, mitotic Golgi fragmentation involves a very early and now fully characterized stage: the cleavage of the tubular non-compact zones that interconnect the Golgi stacks into a ribbon structure. This ribbon cleavage stage occurs in G2 and results in isolated stacks (or small groups of stacks) that remain structurally and functionally normal in their usual pericentriolar position. Second, although structurally minimal and, in fact, difficult to detect without specifically designed approaches, ribbon cleavage is of great functional significance, in that it controls the transition from G2 to M; namely, the Golgi checkpoint (see below). Third, ribbon cleavage requires the fission-inducing protein BARS. This first cleavage is followed by two previously characterized steps that are independent of BARS: the disassembly of the Golgi into scattered tubular elements in prometaphase, and the further fine dispersion of these elements into the Golgi haze in metaphase.
These conclusions rest on the following lines of evidence: (a) using electron microscopy and FRAP approaches, we have shown that during G2, the normal non-compact zones of the Golgi ribbon are mostly fragmented, although the stacks maintain an overall intact morphology and remain clustered in a perinuclear area; these data are consistent with the reduction in cisternal length previously observed in prophase (Misteli and Warren, 1995); (b) cleavage of the non-compact zones can be inhibited by a variety of treatments that block BARS activity; and (c) these same treatments, and other treatments that block early Golgi fragmentation (Sutterlin et al, 2002; Yoshimura et al, 2005) cause cell-cycle arrest in G2, that is, prevent mitotic entrance. Moreover, in cells that already have Golgi membranes that are organized as isolated stacks (either spontaneously or through experimental manipulation), microinjection of the BARS blockers does not affect G2/M transition, indicating that in the absence of a ribbon organization of the Golgi, BARS becomes superfluous for mitotic entry. Of note, this is also the case in CtBP90 BARS-KO MEFs, which have a Golgi complex that is permanently composed of isolated stacks or small groups of stacks, and thus do not need BARS for G2/M transition.
While this manuscript was under revision, it was reported that the inhibition of MEK1 kinase in HeLa cells caused a 2 h delay in mitotic entry. This delay was abrogated if the Golgi ribbon was broken down by depletion of the golgin GRASP65. Imaging revealed that this breakup of the ribbon begins before M phase and is reduced by inhibition of MEK1 activity (Feinstein and Linstedt, 2007). Thus, even if this mitosis-specific MEK1 function appears to facilitate rather than to be required for G2/M progression, which is instead the case for BARS, this independent observation is perfectly in line with our data and leads to the interesting speculation that the severing of the non-compact zones by BARS might be somehow coordinated with this newly identified role for MEK1.
The data presented here have several implications related to both the mechanisms of Golgi ribbon formation and the interplay between cell-cycle regulation and Golgi membrane structure/function.
The Golgi ribbon appears to be a dynamic entity that is regulated by complex and coordinated actions of several different factors, some that are devoted to its maintenance and some others to its breakdown. Among the maintenance factors, there are several Golgi-matrix proteins, possibly acting as membrane tethers, including Golgin-84, GM130, GRASP-65, Golgin-245, Golgin-45, p115 and Cog3 (Short et al, 2001; Allan et al, 2002; Diao et al, 2003; Rios and Bornens, 2003; Rios et al, 2004; Yoshino et al, 2005; Zolov and Lupashin, 2005; Puthenveedu et al, 2006); microtubule motors and various cytoskeleton components such as CD1 (cytoplasmic dynein 1), Hook3 (Golgi-localized microtubule-binding protein), CLASP (peripheral cis-localized protein; proposed to bind short-range detyrosinated microtubules), MACF1b (microtubule actin-crosslinking factor), GMAP-210 and AKAP-450 (Walenta et al, 2001; Sillibourne et al, 2002; Rios et al, 2004; Lin et al, 2005; Mimori-Kiyosue et al, 2005). Also, the lipid composition of Golgi membranes is important for ribbon maintenance; for instance, inhibition of phospholipase A2 and increased levels of sphingosine cause fragmentation of the Golgi complex (Chan et al, 2004; Hu et al, 2005). Interestingly, while all these factors are involved in maintaining the ribbon structure, only one player, BARS, is so far known to act by cleaving the ribbon into stacks. It is thus important to elucidate the interplay between BARS, the relevant maintenance factors and the regulatory kinases. Probably, the ribbon represents the ‘background state' of the mammalian Golgi, which must be actively and rapidly broken down by activating BARS and inactivating maintenance factors when needed at specific times, such as just before mitotic entry. The persistent inactivation of maintenance factors might represent a backup mechanism through which embryonic cells adapt to a lack of BARS activity. It will be of interest to determine the factor(s) involved in the permanent loss of the ribbon structure in BARS KO cells.
The Golgi complex is organized in the form of a ribbon in all mammalian and probably other animal cells. However, the reason of this peculiar organization is still an open question (Colanzi et al, 2003). The transport of cargo to, through and out of the Golgi complex in cultured cells can take place efficiently when the ribbon is interrupted (Cole et al, 1996; Trucco et al, 2004), and plant and insect cells do not have a ribbon (Colanzi et al, 2003), indicating that the ribbon provides some evolutionary advantage in certain (but not all) organisms that is not apparent in cultured cells. A role for the ribbon in the equal distribution of Golgi enzymes among the stacks and, consequently, for efficient glycosylation of secretory proteins has been proposed recently (Puthenveedu et al, 2006). However, these conclusions are not supported by a different study (Marra et al, 2007). This proposal, therefore, needs further analysis. It is tempting to speculate that the ribbon organization has functions that are not strictly related to membrane transport, but instead related to regulatory functions. In agreement with this, Golgi membranes are home to several signal transduction molecules (Rios and Bornens, 2003; Sallese et al, 2006) and are directly involved in the generation of the proapoptotic mediator ganglioside GD3 (Ferri and Kroemer, 2001). Moreover, the Golgi complex is reoriented towards the leading edge in migrating fibroblasts (Nobes and Hall, 1999; Palazzo et al, 2001), suggesting that the ribbon functions in strict coordination with the microtubule-organizing center and the cystoskeleton to accomplish the correctly targeted delivery of membranes to specific sites at the cell surface. In line with this, the fragmentation of the Golgi ribbon induced by overexpression of GRASP65 impairs polarized dendrite outgrowth in hippocampal neurons (Horton et al, 2005).
The mechanism of action of BARS and its regulation in this process are further important issues for future studies. The addition of recombinant BARS alone to permeabilized cells does not induce fragmentation of the Golgi (Hidalgo Carcedo et al, 2004), and the microinjection of recombinant BARS in interphase cells does not induce the breakup of the Golgi ribbon (this study, data not shown); moreover, in permeabilized cells, BARS can reconstitute Golgi fragmentation only in the presence of mitotic cytosol (previously depleted of BARS) and not of interphase cytosol (Hidalgo Carcedo et al, 2004). Collectively, these data are in line with specific mitotic activation of BARS in severing the non-compact zones. BARS is known to be a target for several kinases (Corda et al, 2006). Since PAK1 has been reported to be required for mitotic entry and is known to phosphorylate the BARS homologue CtBP1 (Barnes et al, 2003), it is a candidate activator of BARS in G2. BARS phosphorylation probably determines its molecular partners in the fission of the Golgi ribbon. Here, the availability of in vitro assays of mitotic Golgi fragmentation (Acharya et al, 1998) will provide experimental access to these questions.
Regarding the BARS-independent Golgi disassembly that occurs after cells enter mitosis, several golgins are phosphorylated by various kinases, including Plk1, the Raf1/MEK1/ERK1c cascade and Cdc2, cargo exit from the endoplasmic reticulum is blocked and Arf1 is released from Golgi membranes (Shorter and Warren, 2002; Altan-Bonnet et al, 2003). All these events may contribute to a loss of the stacked organization of the Golgi complex and transformation of the Golgi cisternae into the tubular network seen during the early stages of mitosis (Shorter and Warren, 2002). Moreover, these conditions might facilitate further fragmentation/fission by a variety of mechanisms including vesiculation and/or redistribution into the endoplasmic reticulum, all of which can act independently of BARS (Axelsson and Warren, 2004; Altan-Bonnet et al, 2006).
Why is the BARS-mediated severing of the Golgi ribbon into stacks required for mitotic ingression? The simplest explanation is that correct inheritance of Golgi membranes between the new cells that are formed is critical for cell survival, and therefore cells have evolved mechanisms to ensure and/or check the equal division of this organelle (McGowan and Russell, 2004). As the Golgi complex in mammalian cells has the properties of being virtually a ‘single-copy' organelle and of residing in an asymmetrical location, the formation of two equal and inheritable Golgi pools before the centrosomes start forming the mitotic spindle in late prophase should be required for entry into mitosis. To this end, the minimal essential Golgi fragmentation step is division of the ribbon into its constituent stacks. Our morphological analysis indicates that during late G2, the Golgi complex reorganizes from a large ribbon-like network (Trucco et al, 2004) into a mixed population of isolated stacks and small groups of two to four connected stacks. This level of breakdown, rather than a full Golgi disassembly, appears to be sufficient to enter mitosis. Evolutionary evidence supports this idea, in that many non-mammalian cell types that do not have Golgi stacks organized in a ribbon, such as plant cells and Drosophila embryo cells, divide well without the need for disassembling the stacks (Colanzi et al, 2003).
Finally, a key question is how ribbon breakdown controls the Golgi checkpoint. A series of possible mechanisms can be envisioned on the basis of our current observations. One intriguing possibility is related to the requirement for entry into mitosis of separation and ‘maturation' of the duplicated centrosomes in G2. This maturation begins in G2 and involves the sequential centrosomal recruitment and activation of PAK1 kinase, Aurora-A kinase and CDC25B phosphatase, which in turn are all essential for centrosomal recruitment and initial activation of the Cdk1/ cyclin B complex, the master regulator of mitotic entry (Jackman et al, 1995; Brittle and Ohkura, 2005; Zhao et al, 2005). Since the Golgi complex is tightly associated with microtubules and the centrosome (Shima et al, 1998), we can suppose that an undivided Golgi ribbon might mechanically prevent the correct separation and maturation of the centrosomes, which would in turn cause a block of entry into mitosis.
An alternative possibility is that Golgi fragmentation during G2 is essential for the exposure of proteins or lipids on, or release from, the severed membranes that trigger mitotic entrance. This is analogous to the observation that during prophase, a set of proteins including clathrin, Nir2, Sak1 and cullin-2 are released from Golgi membranes and acquire new functions that are related to the formation of the mitotic spindle, chromosome segregation and cytokinesis (Litvak et al, 2004; Royle et al, 2005; Altan-Bonnet et al, 2006).
The identification of this precise stage in Golgi partitioning that controls the Golgi mitotic checkpoint, and the availability of BARS-related molecular tools to manipulate it both in vivo and in vitro, provides the basis for defining the mechanisms by which the Golgi complex controls G2/M transition, with potentially important physiological and pharmacological consequences.
Materials and methods
Reagents
The following antibodies and constructs were used: generic IgGs (Jackson ImmunoResearch, West Grove, PA, USA), anti-phosphorylated-H1 and anti-phosphorylated-H3 antibodies (Upstate, Charlottesville, VA, USA) and anti-giantin monoclonal antibody (Dr H P Hauri, University of Basel, Basel, Switzerland); anti-BARS p52 antibody has been previously described (Hidalgo Carcedo et al, 2004), and cDNAs of GalT-GFP were from Dr J Lippincott-Schwartz (NIH, Bethesda, MD, USA). The CtBP90 and CtBP86 MEFs were from Dr J Hildebrand (University of Pittsburg, Pittsburgh, PA, USA). Unless otherwise stated, all other chemicals and reagents were obtained from previously indicated sources (Hidalgo Carcedo et al, 2004) or from Sigma (Milan, Italy). Growth of the HeLa, CHO, NRK and MEF cells were as previously described (Hildebrand and Soriano, 2002; Trucco et al, 2004). Poly-histidine-tagged CtBP/BARS and BARS mutants were expressed and purified as described previously (Nardini et al, 2003; Valente et al, 2005).
Microinjection and cell-cycle synchronization
The affinity-purified anti-BARS antibody and preimmune IgGs (both at 3–6 mg/ml) were microinjected into 200–600 aphidicolin-arrested (2.5 μg/ml aphidicolin) or thymidine-arrested (2 mM thymidine) cells 45 min after removal of this S-phase block. Cells were then incubated in complete medium for the appropriate times before fixing. To induce G2 block, cells were incubated for 18 h with bisbenzimide (1 μg/ml Hoechst 33342 or 40 μg/ml Hoechst 33258) (Dubey and Raman, 1983). Cells were stained with an anti-giantin antibody to visualize Golgi membranes, with TRITC-conjugated dextran to visualize microinjected cells, with Hoechst to visualize DNA organization and with anti-phosphorylated-H1 and anti-phosphorylated-H3 antibodies as markers of cell-cycle phases (Hendzel et al, 1997), according to the manufacturer's instructions (Upstate, Charlottesville, VA, USA). Cell-cycle synchrony under various conditions was evaluated by FACS analysis and/or imunofluorescence detection of the pH1/pH3 epitopes combined with Hoechst. Microinjection of recombinant protein was as for the antibody; SBD and GST were microinjected at 8 mg/ml.
Analysis of FRAP
Living cells were imaged at 37°C on the temperature-controlled stage of a Zeiss LSM 510 laser scanning confocal microscope using the 488-nm line of a 25-mW argon laser and an oil-immersion objective (× 63, 1.4 numerical aperture). GalT-GFP fluorescence was photobleached in a defined region of the Golgi mass with 10 iterations at 80% laser power/100% transmission. After bleaching, fluorescence intensity in the bleached area was monitored over time by scanning at 0.5% transmission every 5 s. GalT-GFP photobleaching was irreversible under these conditions, as judged by bleaching the entire Golgi complex. Data sets where the focal plane shifted were discarded by visual inspection of recorded images. To assess differences in GalT-GFP mobility in the Golgi mass under different conditions, we determined recovery curves of fluorescence intensities normalized to the unbleached areas and corrected for background versus time. To evaluate FRAP in microinjected cells, HeLa cells were first transfected with GalT-GFP, and at appropriate times randomly microinjected with various recombinant proteins or antibodies in the presence of TRITC-dextran as microinjection marker. Under confocal microscopy, cells that were both transfected and microinjected were further evaluated by phase contrast to determine if they had interphase or mitotic features (rounded cell shape, condensed chromosomes). Cells not in mitosis were further processed for FRAP analysis. Images were cropped with Adobe Photoshop and composed using Adobe Illustrator. Statistical significance of FRAP measurements was assessed by two-tailed Student's t-test.
Microscopy
Fluorescence and confocal microscopy were as described previously (Polishchuk et al, 2000). For quantitative analysis of Golgi phenotypes, the images were acquired using identical Confocal settings, analyzed using the Object Count tool and assigned to one of the three following classes: (a) intact ribbon (one major object, either compact or elongated, plus 0–3 minor objects); (b) partially fragmented (4–10 objects) and (c) fragmented (11 up to 30 objects). For electron microscopy, HeLa cells and MEFs were grown in Petri dishes, fixed with 2% glutaraldehyde and processed for electron microscopy, as described previously (Hidalgo Carcedo et al, 2004). Ultrathin sections (80 nm) were observed at 80 keV with a Teknai 12 electron microscope. Images were evaluated using Analysis software. In serial-sections analysis, adjacent aligned stacks with at least one contiguous cisterna were considered to be connected. The length of individual stacks and the part of the ribbon seen in the section were measured using the freehand line selection tool in analysis.
Supplementary Material
Supplementary Data
Acknowledgments
We thank R Polishchuk and H Gad for critical reading of the manuscript, MA De Matteis for sharing unpublished information and providing the LdlG cells, J Hildebrand (University of Pittsburg, Pittsburgh, PA, USA) for the generous gift of the CtBP90 MEFs, R Polishchuk, P Liberali and A Di Campli for help with the FRAP experiments, J Lippincott-Schwartz for supplying constructs, N Martelli for FACS analysis, CP Berrie for editorial assistance, Elena Fontana for preparing figures and the Italian Association for Cancer Research (AIRC, Milan, Italy), Telethon (Italy), and the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR, Italy) for financial support. AP and MB were fellows of the Italian Foundation for Cancer Research (FIRC, Milan, Italy) and CHC was a recipient of a fellowship of the Alfredo Leonardi Fund and GL Pfeiffer Foundation.
References
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V (1998) Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell 92: 183–192 [DOI] [PubMed] [Google Scholar]
- Allan VJ, Thompson HM, McNiven MA (2002) Motoring around the Golgi. Nat Cell Biol 4: E236–242 [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet N, Phair RD, Polishchuk RS, Weigert R, Lippincott-Schwartz J (2003) A role for Arf1 in mitotic Golgi disassembly, chromosome segregation, and cytokinesis. Proc Natl Acad Sci USA 100: 13314–13319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altan-Bonnet N, Sougrat R, Liu W, Snapp EL, Ward T, Lippincott-Schwartz J (2006) Golgi inheritance in mammalian cells is mediated through endoplasmic reticulum export activities. Mol Biol Cell 17: 990–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson MA, Warren G (2004) Rapid, endoplasmic reticulum-independent diffusion of the mitotic Golgi haze. Mol Biol Cell 15: 1843–1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CJ, Vadlamudi RK, Mishra SK, Jacobson RH, Li F, Kumar R (2003) Functional inactivation of a transcriptional corepressor by a signaling kinase. Nat Struct Biol 10: 622–628 [DOI] [PubMed] [Google Scholar]
- Brittle AL, Ohkura H (2005) Centrosome maturation: Aurora lights the way to the poles. Curr Biol 15: R880–R882 [DOI] [PubMed] [Google Scholar]
- Chan D, Strang M, Judson B, Brown WJ (2004) Inhibition of membrane tubule formation and trafficking by isotetrandrine, an antagonist of G-protein-regulated phospholipase A2 enzymes. Mol Biol Cell 15: 1871–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciemerych MA, Sicinski P (2005) Cell cycle in mouse development. Oncogene 24: 2877–2898 [DOI] [PubMed] [Google Scholar]
- Colanzi A, Deerinck TJ, Ellisman MH, Malhotra V (2000) A specific activation of the mitogen-activated protein kinase kinase 1 (MEK1) is required for Golgi fragmentation during mitosis. J Cell Biol 149: 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A, Suetterlin C, Malhotra V (2003) Cell-cycle-specific Golgi fragmentation: how and why? Curr Opin Cell Biol 15: 462–467 [DOI] [PubMed] [Google Scholar]
- Cole NB, Smith CL, Sciaky N, Terasaki M, Edidin M, Lippincott-Schwartz J (1996) Diffusional mobility of Golgi proteins in membranes of living cells. Science 273: 797–801 [DOI] [PubMed] [Google Scholar]
- Corda D, Colanzi A, Luini A (2006) The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol 16: 167–173 [DOI] [PubMed] [Google Scholar]
- Diao A, Rahman D, Pappin DJ, Lucocq J, Lowe M (2003) The coiled-coil membrane protein golgin-84 is a novel rab effector required for Golgi ribbon formation. J Cell Biol 160: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey DD, Raman R (1983) Effects of Hoechst 33258 on different cell cycle events. I. Inhibition of synthetic activities in bone-marrow cells of the mole rat Bandicota bengalensis. Exp Cell Res 149: 419–432 [DOI] [PubMed] [Google Scholar]
- Feinstein TN, Linstedt AD (2007) Mitogen-activated protein kinase kinase 1-dependent Golgi unlinking occurs in G2 phase and promotes the G2/M cell cycle transition. Mol Biol Cell 18: 594–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255–E263 [DOI] [PubMed] [Google Scholar]
- Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD (1997) Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106: 348–360 [DOI] [PubMed] [Google Scholar]
- Hidalgo Carcedo C, Bonazzi M, Spano S, Turacchio G, Colanzi A, Luini A, Corda D (2004) Mitotic Golgi partitioning is driven by the membrane-fissioning protein CtBP3/BARS. Science 305: 93–96 [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Soriano P (2002) Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 22: 5296–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton AC, Racz B, Monson EE, Lin AL, Weinberg RJ, Ehlers MD (2005) Polarized secretory trafficking directs cargo for asymmetric dendrite growth and morphogenesis. Neuron 48: 757–771 [DOI] [PubMed] [Google Scholar]
- Hu W, Xu R, Zhang G, Jin J, Szulc ZM, Bielawski J, Hannun YA, Obeid LM, Mao C (2005) Golgi fragmentation is associated with ceramide-induced cellular effects. Mol Biol Cell 16: 1555–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M, Firth M, Pines J (1995) Human cyclins B1 and B2 are localized to strikingly different structures: B1 to microtubules, B2 primarily to the Golgi apparatus. EMBO J 14: 1646–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CM, Chen HJ, Leung CL, Parry DA, Liem RK (2005) Microtubule actin crosslinking factor 1b: a novel plakin that localizes to the Golgi complex. J Cell Sci 118: 3727–3738 [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living cells. Science 300: 87–91 [DOI] [PubMed] [Google Scholar]
- Litvak V, Argov R, Dahan N, Ramachandran S, Amarilio R, Shainskaya A, Lev S (2004) Mitotic phosphorylation of the peripheral Golgi protein Nir2 by Cdk1 provides a docking mechanism for Plk1 and affects cytokinesis completion. Mol Cell 14: 319–330 [DOI] [PubMed] [Google Scholar]
- Marra P, Salvatore L, Mironov AJ, Di Campli A, Di Tullio G, Trucco A, Beznoussenko G, Mironov A, De Matteis MA (2007) The biogenesis of the Golgi ribbon: the roles of membrane input from the ER and of GM130. Mol Biol Cell [advance online publication 21 February 2007; doi:10.1091/mbc.E06-10-0886] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, Russell P (2004) The DNA damage response: sensing and signaling. Curr Opin Cell Biol 16: 629–633 [DOI] [PubMed] [Google Scholar]
- Mikhailov A, Rieder CL (2002) Cell cycle: stressed out of mitosis. Curr Biol 12: R331–R333 [DOI] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Grigoriev I, Lansbergen G, Sasaki H, Matsui C, Severin F, Galjart N, Grosveld F, Vorobjev I, Tsukita S, Akhmanova A (2005) CLASP1 and CLASP2 bind to EB1 and regulate microtubule plus-end dynamics at the cell cortex. J Cell Biol 168: 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov AA, Colanzi A, Polishchuk RS, Beznoussenko GV, Mironov AA Jr, Fusella A, Di Tullio G, Silletta MG, Corda D, De Matteis MA, Luini A (2004) Dicumarol, an inhibitor of ADP-ribosylation of CtBP3/BARS, fragments golgi non-compact tubular zones and inhibits intra-golgi transport. Eur J Cell Biol 83: 263–279 [DOI] [PubMed] [Google Scholar]
- Misteli T, Warren G (1995) Mitotic disassembly of the Golgi apparatus in vivo. J Cell Sci 108: 2715–2727 [DOI] [PubMed] [Google Scholar]
- Nardini M, Spano S, Cericola C, Pesce A, Massaro A, Millo E, Luini A, Corda D, Bolognesi M (2003) CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J 22: 3122–3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144: 1235–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo AF, Joseph HL, Chen YJ, Dujardin DL, Alberts AS, Pfister KK, Vallee RB, Gundersen GG (2001) Cdc42, dynein, and dynactin regulate MTOC reorientation independent of Rho-regulated microtubule stabilization. Curr Biol 11: 1536–1541 [DOI] [PubMed] [Google Scholar]
- Polishchuk RS, Polishchuk EV, Marra P, Alberti S, Buccione R, Luini A, Mironov AA (2000) Correlative light-electron microscopy reveals the tubular-saccular ultrastructure of carriers operating between Golgi apparatus and plasma membrane. J Cell Biol 148: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preisinger C, Korner R, Wind M, Lehmann WD, Kopajtich R, Barr FA (2005) Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J 24: 753–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthenveedu MA, Bachert C, Puri S, Lanni F, Linstedt AD (2006) GM130 and GRASP65-dependent lateral cisternal fusion allows uniform Golgi-enzyme distribution. Nat Cell Biol 8: 238–248 [DOI] [PubMed] [Google Scholar]
- Rambourg A, Clermont Y, Hermo L, Segretain D (1987) Tridimensional structure of the Golgi apparatus of nonciliated epithelial cells of the ductuli efferentes in rat: an electron microscope stereoscopic study. Biol Cell 60: 103–115 [DOI] [PubMed] [Google Scholar]
- Rios RM, Bornens M (2003) The Golgi apparatus at the cell centre. Curr Opin Cell Biol 15: 60–66 [DOI] [PubMed] [Google Scholar]
- Rios RM, Sanchis A, Tassin AM, Fedriani C, Bornens M (2004) GMAP-210 recruits gamma-tubulin complexes to cis-Golgi membranes and is required for Golgi ribbon formation. Cell 118: 323–335 [DOI] [PubMed] [Google Scholar]
- Royle SJ, Bright NA, Lagnado L (2005) Clathrin is required for the function of the mitotic spindle. Nature 434: 1152–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallese M, Pulvirenti T, Luini A (2006) The physiology of membrane transport and endomembrane-based signalling. EMBO J 25: 2663–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Cabrera-Poch N, Pepperkok R, Warren G (1998) An ordered inheritance strategy for the Golgi apparatus: visualization of mitotic disassembly reveals a role for the mitotic spindle. J Cell Biol 141: 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short B, Preisinger C, Korner R, Kopajtich R, Byron O, Barr FA (2001) A GRASP55–rab2 effector complex linking Golgi structure to membrane traffic. J Cell Biol 155: 877–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Warren G (2002) Golgi architecture and inheritance. Annu Rev Cell Dev Biol 18: 379–420 [DOI] [PubMed] [Google Scholar]
- Sillibourne JE, Milne DM, Takahashi M, Ono Y, Meek DW (2002) Centrosomal anchoring of the protein kinase CK1delta mediated by attachment to the large, coiled-coil scaffolding protein CG-NAP/AKAP450. J Mol Biol 322: 785–797 [DOI] [PubMed] [Google Scholar]
- Sutterlin C, Hsu P, Mallabiabarrena A, Malhotra V (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109: 359–369 [DOI] [PubMed] [Google Scholar]
- Sutterlin C, Lin CY, Feng Y, Ferris DK, Erikson RL, Malhotra V (2001) Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc Natl Acad Sci USA 98: 9128–9132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trucco A, Polishchuk RS, Martella O, Di Pentima A, Fusella A, Di Giandomenico D, San Pietro E, Beznoussenko GV, Polishchuk EV, Baldassarre M, Buccione R, Geerts WJ, Koster AJ, Burger KN, Mironov AA, Luini A (2004) Secretory traffic triggers the formation of tubular continuities across Golgi sub-compartments. Nat Cell Biol 6: 1071–1081 [DOI] [PubMed] [Google Scholar]
- Valente C, Spano S, Luini A, Corda D (2005) Purification and functional properties of the membrane fissioning protein CtBP3/BARS. Methods Enzymol 404: 296–316 [DOI] [PubMed] [Google Scholar]
- Vasile E, Perez T, Nakamura N, Krieger M (2003) Structural integrity of the Golgi is temperature sensitive in conditional-lethal mutants with no detectable GM130. Traffic 4: 254–272 [DOI] [PubMed] [Google Scholar]
- Walenta JH, Didier AJ, Liu X, Kramer H (2001) The Golgi-associated hook3 protein is a member of a novel family of microtubule-binding proteins. J Cell Biol 152: 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert R, Silletta MG, Spano S, Turacchio G, Cericola C, Colanzi A, Senatore S, Mancini R, Polishchuk EV, Salmona M, Facchiano F, Burger KN, Mironov A, Luini A, Corda D (1999) CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid. Nature 402: 429–433 [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Yoshioka K, Barr FA, Lowe M, Nakayama K, Ohkuma S, Nakamura N (2005) Convergence of cell cycle regulation and growth factor signals on GRASP65. J Biol Chem 280: 23048–23056 [DOI] [PubMed] [Google Scholar]
- Yoshino A, Setty SR, Poynton C, Whiteman EL, Saint-Pol A, Burd CG, Johannes L, Holzbaur EL, Koval M, McCaffery JM, Marks MS (2005) tGolgin-1 (p230, golgin-245) modulates Shiga toxin transport to the Golgi and Golgi motility towards the microtubule-organizing centre. J Cell Sci 118: 2279–2293 [DOI] [PubMed] [Google Scholar]
- Zhao ZS, Lim JP, Ng YW, Lim L, Manser E (2005) The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell 20: 237–249 [DOI] [PubMed] [Google Scholar]
- Zolov SN, Lupashin VV (2005) Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J Cell Biol 168: 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data