

Abstract
We have investigated the requirements for CRM1-mediated nuclear export and SUMO1 conjugation of the adenovirus E1B-55K protein during productive infection. Our data show that CRM1 is the major export receptor for E1B-55K in infected cells. Functional inactivation of the E1B-55K CRM1-dependent nuclear export signal (NES) or leptomycin B treatment causes an almost complete redistribution of the viral protein from the cytoplasm to the nucleus and its accumulation at the periphery of the viral replication centers. Interestingly, however, this nuclear restriction imposed on the wild type and the NES mutant protein is fully compensated by concurrent inactivation of the adjacent SUMO1 conjugation site. Moreover, the same mutation fully reverses defects of the NES mutant in the nucleocytoplasmic transport of Mre11 and proteasomal degradation of p53. These results show that nuclear export of E1B-55K in infected cells occurs via CRM1-dependent and -independent pathways and suggest that SUMO1 conjugation and deconjugation provide a molecular switch that commits E1B-55K to a CRM1-independent export pathway.
Keywords: CRM1, Mre11, nucleocytoplasmic transport, p53, SUMOylation
The 55K product from subgroup C adenovirus type 5 (Ad5) early region 1B (E1B-55K) belongs to a group of adenoviral regulatory proteins required for maximal virus production in a number of different normal human cell strains and human tumor cell lines (reviewed in ref. 1). In wild-type (WT) Ad5-infected cells, E1B-55K controls several processes, including selective nuclear export of viral late RNA transcripts, inhibition of cellular mRNA transport, and proteasomal degradation of the tumor suppressor protein p53 and Mre11, a subunit of the Mre11/Rad50/Nbs1 (MRN) DNA double-strand break repair complex (reviewed in ref. 2). Collectively available data suggest that these multiple lytic activities result from oligomerization, posttranslational modifications such as phosphorylation, continuous nucleocytoplasmic shuttling, and interactions with a variety of cellular and viral factors, most importantly the protein product from early region 4 ORF 6 (E4orf6) (reviewed in ref. 3 and references therein).
Over the past years, it has been well established that complex formation with E4orf6 increases the multifunctionality of the E1B protein. Several studies have shown that E4orf6 alters the intracellular distribution of E1B-55K in virus-infected cells directing the E1B protein to the nuclear matrix compartment (4) and the sites of viral RNA transcription and processing (5, 6). In addition, a substantial amount of novel information demonstrates that E4orf6 connects E1B-55K to components of a cellular E3 ubiquitin ligase, thereby allowing the proteasomal degradation of p53, Mre11, and Rad50 (reviewed in ref. 7). It appears that the latter activity also involves active nuclear export and cytoplasmic deposition of MRN subunits into aggresomes (8). Finally, several lines of evidence suggest that the E1B-55K/E4orf6 complex directly participates in the selective nuclear export of late viral mRNAs through active nucleocytoplasmic shuttling (3) and presumably through its ubiquitin-protein ligase activity (9).
Efforts to identify cellular targets that link the E1B-55K/E4orf6 complex to nuclear export pathways revealed that both viral proteins possess a single leucine-rich nuclear export signal (NES) of the HIV-1 Rev-type (10, 11) and can individually shuttle between the nuclear and cytoplasmic compartments via the export receptor CRM1 (reviewed in ref. 3). In the case of E1B-55K, nucleocytoplasmic trafficking may also involve covalent conjugation of the small ubiquitin-related modifier protein 1 (SUMO1), which appears to facilitate efficient nuclear import and/or subnuclear targeting of the Ad protein (12). Remarkably, in Ad5 E1B-55K, the lysine residue that serves as the SUMO1 conjugation site (K104) is situated in close proximity to the E1B NES both of which are separated by just 10 amino acids in the E1B-55K primary sequence (Fig. 1). It is, therefore, tempting to speculate that SUMOylation of E1B-55K regulates the NES-dependent nuclear export of the Ad protein. To evaluate this model, we examined the effect of amino acid substitution mutations in the NES, SUMO1 conjugation site (SCS) and both motifs on the nucleocytoplasmic export function of E1B-55K in the normal context of Ad-infected cells by generating and testing the appropriate virus mutants.
Fig. 1.

Effect of amino acid changes on stability and SUMOylation of E1B-55K mutant proteins. (A) Amino acid substitution mutations in E1B-55K mutant viruses. The leucine residues in the 55K NES known to be critical for CRM1-mediated nuclear export (11, 29) and the lysine residue that serves as the SUMO acceptor site (12) are indicated by triangles. Numbers refer to amino acid residues in the WT E1B-55K protein from H5pg4100. Amino acid changes in the E1B proteins from H5pm4101, H5pm4102, and H5pm4103 are indicated below. (B) Steady-state expression levels of E1B-55K proteins in WT and mutant virus-infected cells. A549 cells were infected with WT and mutant viruses at a multiplicity of 20 ffu per cell. Cells were harvested at the indicated times postinfection (p.i.), and total-cell extracts were prepared and subjected to immunoblotting by using anti-E1B-55K mouse monoclonal antibody (mab) 2A6. (C) Effect of mutations on SUMO1 conjugation of E1B-55K. Steady-state concentration of E1B-55K was detected by immunoblotting of total-cell lysates by using mab 2A6 (α-E1B; Left). The same extracts were subjected to immunoprecipitation with mab 2A6, and E1B/SUMO1 conjugates were visualized by SDS/PAGE and immunoblotting with anti-SUMO1 mouse mab 21C7 (α-SUMO1; Right). The bands representing the Ig heavy chain (IgH) and E1B-SUMO1 conjugate are indicated on Right.
Results
SUMOylation of E1B-55K Is Enhanced in the Absence of a Functional NES.
To analyze the role of SUMO1 conjugation and NES-dependent nuclear export on E1B-55K nucleocytoplasmic trafficking, we generated three virus mutants containing defined amino acid substitution mutations (Fig. 1A). In mutant virus H5pm4101, three leucine residues in the E1B NES, known to be critical for CRM1-mediated nuclear export (11) were substituted by alanines (L83A/L87A/L91A), whereas H5pm4102 carries a single amino acid substitution (K104R) previously shown to abolish SUMO1 modification of Ad5 E1B-55K (12). The double mutant H5pm4103 contains identical amino acid exchanges in both the E1B NES and E1B SCS. To test whether the virus mutants produce a stable protein, the steady-state levels of E1B-55K were determined by immunoblot analysis of total-cell lysates from infected cells (Fig. 1B). All three mutant viruses accumulated E1B-55K protein to levels comparable to WT H5dl309 or WT H5pg4100 through the course of the infection. No signal was obtained with total extracts from A549 cells infected with the Ad2/5 chimeric virus dl1520, which cannot express the E1B-55K coding sequence (13).
To confirm that substitution K104R eliminates E1B-55K SUMOylation, total infected-cell lysates were assayed for the presence of E1B-SUMO1 conjugates by combined immunoprecipitation and immunoblotting (Fig. 1C). Consistent with our previous work (12), only a minor proportion of E1B-55K present in WT Ad-infected cells was found conjugated to SUMO1 at steady state (Fig. 1C, lane 8). Interestingly, however, the amount of the ≈75-kDa SUMO1-modified form was greatly increased in H5pm4101-infected cells (Fig. 1C, lane 10), demonstrating that covalent binding of SUMO1 to E1B-55K is enhanced in the absence of a functional NES. This effect was not observed with other E1B-55K virus mutants containing an intact SCS (data not shown), indicating that elevated SUMO1 conjugation is a specific property of the NES mutant protein. Also, no E1B-SUMO1 conjugates were detected with the anti-SUMO1 mab in the immunoprecipitates from dl1520- (Fig. 1C, lane 9), H5pm4102-, or H5pm4103-infected cells extracts (Fig. 1C, lanes 11 and 12), verifying that the K104R substitution eliminates E1B-55K SUMOylation.
SUMOylation Regulates the Intranuclear Targeting of E1B-55K.
To assess the consequence of amino acid substitution mutations in E1B-55K on its intracellular distribution, the steady-state localization of WT and E1B mutant proteins was determined in infected A549 cells and compared with the viral E2A-72K DNA-binding protein (DBP) by double-label immunofluorescence. This protein is directly involved in viral DNA synthesis (14) and is firmly associated with the viral replication centers (15). Consistent with previous reports (5, 6), the E1B-55K protein present in WT H5pg4100-infected cells demonstrated a complex intracellular distribution (Fig. 2Ab). Within the cytoplasm, it was found in a diffusely distributed pattern together with a few intensely stained bodies adjacent to the nuclear membrane. Within the nucleus, the WT protein was mostly seen in a granular diffuse distribution. In some cells, the WT E1B protein localized in a limited number of globular condensations, which mostly coincided with globular or ring-like structures containing the DBP protein (Fig. 2 Aa–Ac). The intensity of diffuse nuclear staining seen with WT E1B-55K was greatly reduced in H5pm4101-infected cells (Fig. 2 Ag–Ai), and all of the cells examined (n > 100) demonstrated a close association of the E1B NES mutant protein with the viral replication centers. More specifically, the E1B-55K protein appeared to be concentrated in clusters of brightly stained smaller bodies organized about the periphery of a more uniformly stained DBP-positive spherical condensation. Fig. 2B shows a 5-fold enlargement of the boxed areas from Fig. 2 Ag–Ai to illustrate the distribution of the E1B-containing aggregates at the periphery of the replication centers. As opposed to the WT and the NES mutant protein, no colocalization of E1B-55K and DBP was evident in the nuclei of H5pm4102-infected cells through the course of the infection (Fig. 2 Aj–Al). Much of the E1B K104R mutant protein was seen diffusely distributed throughout the nucleoplasm (but excluded from the nucleoli) and concentrated in several cytoplasmic aggregates often located in close proximity to the nuclear membrane (Fig. 2 Aj–Al). Unexpectedly, a similar staining pattern was also observed in the cytoplasm and in the nucleus of H5pm4103-infected cells producing the NES/K104R mutant protein (Fig. 2 Am–Ao). As opposed to H5pm4101-infected cells, all of these cells examined (n > 100) contained numerous cytoplasmic inclusions, demonstrating that a large portion of the mutant E1B protein accumulates in the cytoplasm in the absence of a functional NES and SCS. The formation of K104R and NES/K104R cytoplasmic aggregates was not attributable to impaired nuclear import because both mutant proteins accumulated in the nucleus as efficiently as the WT 55K protein [supporting information (SI) Fig. 5]. Also, none of the cells contained the characteristic E1B-positive filamentous or globular aggregates at the periphery of the viral transcription centers invariably present in the nuclei of cells infected with H5pm4101 (compare Fig. 2 Ag–Ai). Rather, by focusing through the depth of the nucleus, it appeared that nearly all of the NES/K104R protein was uniformly distributed throughout the nucleoplasm. These findings indicate that accumulation of the E1B-NES mutant protein in globular or filamentous nuclear aggregates is dependent on the presence of a functional SCS.
Fig. 2.

Steady-state localization of E1B-55K and E2A-72K in WT and mutant virus-infected cells. (A) A549 cells were infected with WT and E1B mutant viruses at a multiplicity of 20 ffu per cell. Cells were fixed at 20 h p.i. and double-labeled in situ with anti-E2A-72K mouse mab B6–8 (α-DBP) and anti-E1B-55K rat mab 7C11 (α-E1B), and FITC- and Texas red-conjugated secondary antibodies, respectively. Representative anti-E2A (green; a, d, g, j, and m) and anti-E1B (red; b, e, h, k, and n) staining patterns are shown. The overlays of the green and red images are shown in c, f, i, l, and o (merge). In all panels, nuclei are indicated by a dotted line. (Magnification: ×7600.) (B) The boxed areas in g–i were enlarged 5-fold to illustrate the organization of the E1B NES mutant protein at defined sites around the periphery of the viral replication centers. (C) Subcellular distribution of WT and mutant E1B proteins in the presence of the CRM1 inhibitor LMB. A549 cells were infected at a multiplicity of 20 ffu per cell. At 21 h p.i., LMB was added to the medium. Three hours later, cells were fixed, and steady-state localization of E1B-55K was examined by indirect immunofluorescence as described above. In all panels, nuclei are indicated by a dotted line. (Magnification: ×7600.)
A nearly identical pattern of localization was observed when we tested the effect of the CRM1 inhibitor LMB on the subcellular distribution of the WT and the mutant E1B proteins in infected cells (Fig. 2C). Consistent with the results described above, inactivation of CRM1 caused the nuclear retention and the accumulation of the WT protein with the subnuclear structures (Fig. 2Cb). As before, the nuclear restriction and subnuclear localization imposed on the WT protein was fully compensated by the K104R mutation in H5pm4102- (Fig. 2Ce) and, significantly, H5pm4103-infected cells (Fig. 2Cf). Taken together, these results along with the data described above indicate that inactivation of NES-mediated nuclear export and/or SUMO1 conjugation substantially affects the subnuclear distribution of the Ad protein and its partition between the nucleus and cytoplasm in Ad-infected cells. Furthermore, they suggest that SUMOylation may play a role in the targeting of the Ad protein to the viral replication centers and indicate that SUMO1 conjugation and deconjugation regulate nuclear export of E1B-55K probably through one or more CRM1-independent pathways.
Cytoplasmic Sequestration of Mre11 by E1B-55K Involves a CRM1-Independent Export Pathway.
In addition to its important role in stimulating nuclear export of viral late mRNAs, recent studies indicate that E1B-55K further contributes to efficient viral replication by inactivating p53 and two components of the MRN complex (Mre11 and Rad50) through degradation by the ubiquitin-proteasome system in combination with E4orf6 (16, 17). In addition, inactivation of MRN functions involves deposition of nuclear MRN subunits into cytoplasmic inclusion bodies, which fit the criteria for aggresomes (8). It appears that this activity requires active CRM1 (8). These observations prompted us to investigate the effect of mutations in E1B-55K on the intracellular distribution of MRN in WT and mutant virus-infected A549 cells by double-label immunofluorescence. Representative samples of these experiments are shown in Fig. 3.
Fig. 3.

Effect of amino acid substitution mutations on steady-state localization of Mre11 and E1B-55K. A549 cells were mock-infected with WT or E1B mutant viruses at a multiplicity of 20 ffu per cell. Cells were fixed at 20 h p.i. and double-labeled in situ with anti-Mre11 mouse monoclonal antibody 12D7 (α-Mre11) and anti-E1B-55K rat mab 7C11 (α-E1B), and FITC- and Texas red-conjugated secondary antibodies, respectively. Representative anti-Mre11 (green; a, d, g, j, m, and p) and anti-E1B (red; b, e, h, k, n, and q) staining patterns are shown. The overlays of the green and red images are shown in c, f, i, l, o, and r (merge). Nuclei are indicated by a dotted line. Arrowheads in the merged images indicate Mre11 and/or E1B/Mre11-positive cytoplasmic inclusions. (Magnification: ×7600.)
In mock-infected cells, Mre11, the marker protein for the MRN complex, was almost entirely restricted to the nucleus and diffusely distributed throughout the nucleoplasm, but excluded from the nucleoli (Fig. 3 a–c). Immunofluorescence staining of E1B-55K and Mre11 in WT virus-infected cells revealed a variable number of E1B-55K-positive cytoplasmic aggregates some of which also contained Mre11 (indicated by arrowheads in Fig. 3 d–f). In nearly all dl1520-infected cells, Mre11 was mostly restricted to the nucleus and found concentrated in several short filaments (Fig. 3 g–i), which most likely correspond to reorganized PML-NB structures (18). Additionally, a portion of Mre11 appeared in small cytoplasmic inclusions of some infected cells in a perinuclear fashion; ≈10% of the H5pm4101-infected cells (Fig. 3 j–l) examined (n > 100) exhibited a few small Mre11-positive cytoplasmic aggregates, which, however, rarely coincided with the few E1B-55K-containing condensations present in a small fraction (5%) of H5pm4101-infected cells. By contrast, several large E1B-55K/Mre11-positive cytoplasmic aggregates were consistently observed in H5pm4102- (Fig. 3 m–o) and, significantly in H5pm4103-infected cells (Fig. 3 p–r). These results demonstrate that E1B-55K-mediated relocalization of nuclear Mre11 into cytoplasmic inclusions in late Ad5-infected A549 cells is largely dependent on CRM1-mediated nuclear export of E1B-55K in the presence of a functional SCS. Again, however, consistent with data described before, this requirement for CRM1 is fully compensated by mutational inactivation of the adjacent SCS. The latter result provides further support for the idea that conjugation and deconjugation of SUMO1 regulate CRM1-independent nuclear export of E1B-55K.
CRM1 Is Not Required for Proteasomal Degradation of Mre11 by E1B-55K and Maximal Virus Growth.
To test the effect of mutations in E1B-55K on the proteasomal degradation of p53 and Mre11, we determined the steady-state concentrations of Mre11 and p53 by immunoblot assays of total-cell lysates from WT and mutant virus-infected A549 cells (Fig. 4A). As expected, p53 and Mre11 were degraded in WT but not in dl1520 virus-infected cells. In the latter, Mre11 expression levels remained constant through the course of the infection, whereas p53 started to accumulate to high levels at 16 h p.i. Mre11 steady-state levels also gradually decreased in H5pm4101-, H5pm4102-, and H5pm4103-infected cells, although the degradation process was delayed in the presence of the mutant E1B proteins. This delay was not attributable to changes in steady-state concentrations of the E1B mutants or WT E4orf6 because the viral proteins accumulated to levels comparable to WT virus-infected cells. Furthermore, all three mutant proteins efficiently coprecipitated Mre11 at earlier times in the infection (16 h), suggesting that the amino acid substitutions do not interfere with Mre11-binding (Fig. 4B). In contrast to Mre11, degradation of p53 in H5pm4102- and H5pm4103-infected cells was as efficient as in WT virus-infected cells. Interestingly, however, p53 steady-state concentrations progressively increased at later times (48–72 h) in H5pm4101-infected cells, albeit to lower levels as compared with dl1520-infected cells. Thus, the absence of a functional NES in E1B-55K interferes with complete degradation of p53 at late times in the infection. Again, this defect is fully compensated by simultaneous inactivation of the adjacent SCS consistent with the concept that reversible SUMO modification may allow E1B-55K to access a CRM1-independent export pathway, which is apparently required for complete proteasomal degradation of p53.
Fig. 4.

Effect of amino acid substitutions on stability of p53 and Mre11. (A) Steady-state levels of Mre11 and p53 in WT and mutant virus-infected cells. A549 cells were infected with WT and E1B mutant viruses at a multiplicity of 20 ffu per cell. Cells were harvested at the indicated times p.i., and whole-cell extracts were prepared. Proteins from each time point were separated on SDS/PAGE and subjected to immunoblotting by using anti-Mre11 rabbit polyclonal antibody pNB-100–142, anti-p53 mouse mab DO-1, anti-E1B-55K mouse mab 2A6, and anti-E4orf6 rabbit polyclonal antibody 1807. (B) Coimmunoprecipitation of Mre11 with E1B-55K. Whole-cell extracts from infected A549 cells were prepared at 16 h after infection, coprecipitated (Ip) with mab 2A6 (α-E1B), and separated on SDS/PAGE followed by immunoblotting with antibody pNB-100–142.
To verify these data, we monitored degradation of ectopically expressed p53 and endogenous Mre11 in plasmid-transfected p53-negative H1299 cells. Steady-state expression levels of viral and cellular proteins were determined from total-cell lysates by immunoblotting (SI Fig. 6). Interestingly, in these assays, all E1B mutant proteins behaved like WT E1B-55K, demonstrating that complete degradation of p53 is independent from CRM1-dependent nuclear export of E1B-55K outside of the context of a normal Ad infection.
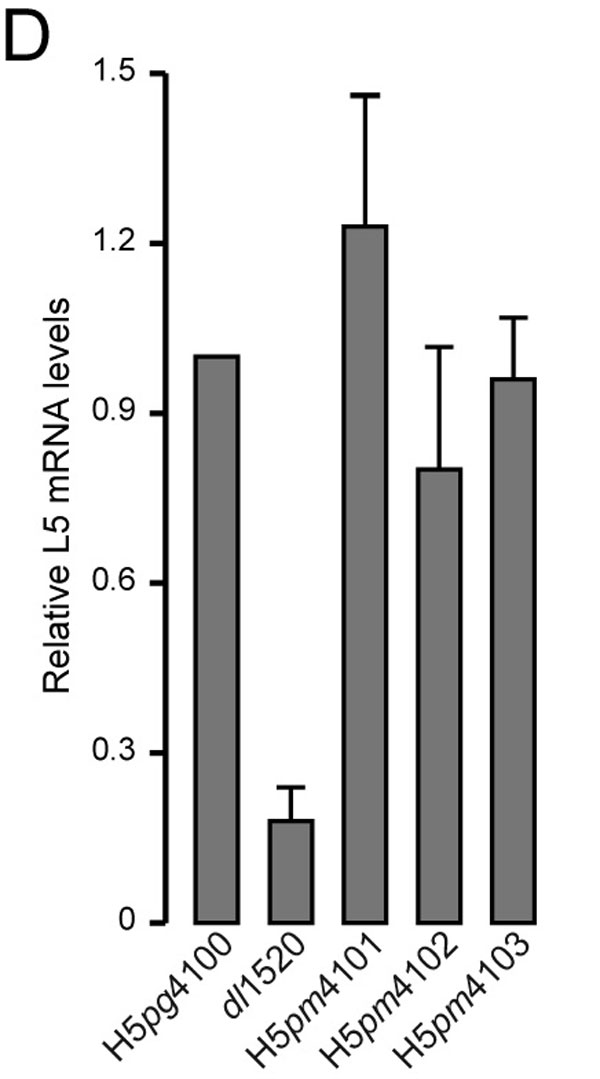
Finally, we assessed the effect of mutations on virus yield, viral DNA synthesis, late viral protein expression, and cytoplasmic accumulation of the viral late L5 fiber mRNA (SI Fig. 7). With the exception of dl1520, replication of the other mutant viruses was comparable to that of H5pg4100 (SI Fig. 7A). Also, in line with this result, no differences were observed when we monitored viral DNA replication (SI Fig. 7B) and late protein synthesis in WT and mutant virus-infected A549 cells (SI Fig. 7C). More significantly, neither of the amino acid changes in E1B-55K substantially affected the cytoplasmic accumulation (SI Fig. 7D) and the cytoplasmic-to-nuclear ratio of the L5 fiber mRNA (SI Fig. 7E), which serves as an indirect measure for the efficiency of nuclear mRNA export (3). Overall these findings are entirely consistent with recently published reports that the E1B NES is not required to promote late gene expression (19) and late viral mRNA transport (20) in Ad5-infected HeLa cells and the observation that stimulation of viral late mRNA export requires an active E1B-55K/E4orf6 ubiquitin ligase complex (9). Furthermore, they provide further support for the model (20) that E1B-55K can exit the nucleus through a CRM1-independent export pathway, which likely contributes to maximal virus production.
Discussion
Over the past years, a large number of proteins have been reported to be covalently modified by SUMO. Substantial evidence suggests that this posttranslational modification regulates protein–protein interactions and subcellular localization (reviewed in ref. 21). Until recently, direct evidence for links between SUMO and nuclear transport were only available for import. However, several recent studies have provided evidence for the requirement of SUMO in nuclear export (22, 23, 24). Collectively, these studies indicate that SUMO negatively or positively regulates nuclear export of proteins via CRM1-dependent and -independent pathways. Consistent with this model, studies in this report demonstrate that conjugation of SUMO1 to Ad5 E1B-55K regulates intranuclear targeting and nuclear export independent from CRM1 during productive infection.
Evidence for this concept originates from our previous observation that overexpression of SUMO1 in transformed rat cells stably expressing WT E1B-55K causes the accumulation of the Ad protein at defined subnuclear structures (12). Similar to this, the increase of the SUMO1-conjugated form of the E1B NES mutant (Fig. 1C) correlates with the localization of a large fraction of the viral protein to the periphery of the viral replication centers (Fig. 2). More significantly, the redistribution of the Ad protein observed in the nucleus of transfected and Ad-infected cells is eliminated by mutational inactivation of the E1B SCS (12) (Figs. 2 and 3), demonstrating that attachment of SUMO1 regulates intranuclear targeting of the Ad protein. Although there is no direct experimental evidence, it seems likely that SUMO1 targets E1B-55K to subnuclear sites, which are associated with the nuclear matrix (4, 25, 26). It is, therefore, tempting to speculate that SUMO1 conjugation facilitates the nuclear retention of E1B-55K, and thus may negatively regulate its nuclear export function. Consistent with this idea, mutational inactivation of the SCS compensates the nuclear retention of the NES mutant (Fig. 2) or the WT protein in LMB-treated cells (Fig. 2C) and completely restores the defect of the NES mutant in the deposition of Mre11 into cytoplasmic inclusion bodies (Fig. 3). These findings together with the observation that the NES/K104R and NES mutants fully support late viral mRNA export (SI Fig. 6 D and E), demonstrates that both mutant proteins can efficiently exit the nucleus in the absence of a functional NES and suggests that deconjugation of SUMO1 allows nuclear export of WT E1B-55K through one or more CRM1-independent pathways.
The involvement of SUMO1 in regulating subnuclear targeting and nuclear export of E1B-55K is surprising in light of the fact that the consequences of this modification on the subcellular distribution of the Ad protein are not proportionate to the small fraction of WT E1B-55K that is SUMO1-modified in infected cells at steady state (Fig. 1C). Similar scenarios have been described for many substrates of the SUMO conjugation machinery and have led to the model that SUMOylation may alter the long-term fate of the modified protein even though SUMO may be rapidly deconjugated (reviewed in ref. 27). Thus, SUMO1 modification and deconjugation of E1B-55K may be part of a highly dynamic process where E1B-55K undergoes rapid SUMO1 modification followed by equally rapid deconjugation. Interestingly, the latter process may be further augmented by E4orf6, which negatively regulates the association of E1B-55K with the matrix and presumably SUMO1 conjugation (4). Additionally and/or alternatively, the recent demonstration of noncovalent binding of SUMO to substrates (28) and the existence of multiple SUMO isoforms in mammalian cells (reviewed in ref. 21) may further reconcile the low level of SUMO modification with the large effects of the modification on E1B-55K localization and function.
We currently favor a model that is adapted from one proposed by Hay (27). In this hypothesis, SUMO1 conjugation interferes with CRM1 binding at the adjacent NES, thereby targeting the Ad protein to matrix-associated subnuclear structures through interactions with proteins containing SUMO-binding domains. According to our data, this compartment primarily corresponds to the peripheral zones of the viral replication centers in virus-infected cells (Fig. 2) and PML-NBs in stably transformed rat cells (12, 29). Once the modified E1B protein is committed to these sites, SUMO1 is rapidly deconjugated through combinatorial mechanisms that involve SUMO-specific proteases and E4orf6, leaving the unmodified protein in a competent state to access CRM1-dependent and -independent export pathways. Subsequently, E1B-55K and/or the E1B-55K/E4orf6 complex exits the nucleus in a SUMO1-independent fashion probably through physical interactions with different cellular exports receptors (30–32).
Although this model is attractive and can explain most of the observations from this and our previous studies (12, 29), the requirement for SUMO1 conjugation of E1B-55K during productive infection is still unclear. One possibility is that this modification regulates export functions of E1B-55K during the early and late phases of the infectious cycle. Such a model would account for the result that mutational inactivation of the E1B NES initially delays the degradation of Mre11, but this activity fully recovers as the late phase progresses (Fig. 4). Moreover, it appears that deposition of Mre11 into cytoplasmic inclusion bodies (Fig. 3) and efficient proteasomal degradation of p53 by the E1B-55K/E4orf6/E3-ubiquitin ligase is dependent on the E1B NES in the presence of a functional SCS (Fig. 4). These data suggest that nuclear depletion of Mre11 and proteasomal degradation of p53 through the CRM1 pathway is part of the mechanism by which E1B-55K inactivates p53 and MRN functions and indicate that both processes are regulated by reversible attachment of SUMO1. Also, an aspect that has not been considered yet is the observation that SUMO1 conjugation of E1B-55K is required for repression of p53-stimulated transcription in plasmid-transfected cells (12). In fact, this activity may involve the interaction of the SUMO-conjugated form of E1B-55K with PML-NBs (29) and/or PML-NB-associated factors such as Daxx or PML (33). Thus, similar to SUMO1-modified transcription factors, coactivators and corepressor (reviewed in ref. 21) SUMO1 conjugation of E1B-55K may additionally regulate transcriptional functions of the Ad protein during infection. Furthermore, the demonstration that SUMO1 contributes to the association of E1B-55K with the peripheral zones of the viral replication centers (Fig. 2) indicates that it may play a role in the selective CRM1-independent export of viral late mRNA transcripts. It should be noted, however, that the K104R mutation has no negative effect on viral mRNA export (SI Fig. 7), although it substantially interferes with the accumulation of E1B-55K at these virus-induced subnuclear structures (Fig. 2). Although clearly speculative at this point, it is possible that SUMO1-mediated accumulation of E1B-55K at the viral replication centers and regulated viral mRNA export is cell-type-dependent and, by analogy to dl1520/ONYX-015, is compensated by some cellular activity in A549 tumor cells (reviewed in ref. 3). Therefore, it will be interesting to test the effect of the K104R and NES/K104R mutant viruses in the context of primary cells, in which viral replication is dependent on E1B-55K-mediated viral mRNA export (34, 35). Further studies are now underway to test these possibilities.
Materials and Methods
Construction of Ad5 Recombinants, Virus Growth, and Titration.
The construction of plasmid pH5pg4100 and the transfer vector pE1–1235 was described recently (36). The Ad5 genome in pH5pg4100 was inserted into the PacI site of the bacterial cloning vector pPG-S2 (34). It lacked nucleotides 28593–30471 (encompassing E3) and contained an additional unique endonuclease restriction site at nucleotide 30955 (BstBI) (nucleotide numbering is according to the published Ad5 sequence from GenBank, accession no. AY339865). To generate Ad5 mutants carrying defined amino acid changes in the NES and SUMO conjugation motif of Ad5 E1B-55K (Fig. 1A) point mutations were first introduced into the 55K gene in pE1–1235 by site-directed mutagenesis, resulting in pE1–1236 (NES), pE1–1237 (K104R), and pE1B-1238 (NES/K104R). The 7.7-kb SwaI/BstZ17I-fragment from pH5pg4100 was then replaced with the corresponding fragments from plasmids pE1–1236, pE1–1237, and pE1–1238 to generate adenoviral plasmids pH5pm4101, pH5pm4102, and pH5pm4103, respectively. Finally, the viral genomes were released from the recombinant plasmids by PacI digestion, and mutant viruses H5pm4101, H5pm4102, and H5pm4103 and their WT parent H5pg4100 (Fig. 1A) were generated and analyzed exactly as described (36).
The following viruses were additionally used in this study: H5pg4100 (36), H5dl309 (37), and dl1520 (13). The Ad5 derivatives H5pg4100 and H5dl309 display a WT phenotype. All viruses were propagated in 293 monolayer cultures. The titers of the viruses used in this study were determined by a fluorescent-focus assay as described (36).
Antibodies, Indirect Immunofluorescence, and Immunoblotting.
The following mabs were used in this study: E1B-55K mouse mab 2A6 (38), E1B-55K rat mab 7C11 (this work), E2A-72K (DBP) mouse mab B6–8 (39), and E4orf6 rabbit polyclonal antibody 1807 (40). Primary antibodies specific for cellular proteins included SUMO1 mouse mab 21C7 (Zymed Laboratories, South San Francisco, CA), p53 mouse mab DO-1 (Santa Cruz Biotechnology, Santa Cruz, CA), Mre11 rabbit polyclonal antibody pNB 100–142 (Novus Biologicals, Littleton, CO), and Mre11 mouse monoclonal antibody 12D7 (Abcam, Cambridge, MA). The E1B rat mab 7C11 was generated against the carboxy-terminal region of the Ad5 protein corresponding to amino acids 348–496. A fragment of the 55K ORF (Ad5 nucleotide 3060–3510) was amplified by PCR with oligonucleotide primers 687 (5′-G-C-C-A-G-G-A-T-C-C-T-G-T-G-G-C-A-A-C-T-G-C-G-A-GG-3′) and 688 (5′- G-C-C-A-G-A-A-T-T-C-T-C-A-A-T-C-T-G-T-A-T-C-T-T-C-3′) and cloned into the BamHI and EcoRI sites of pGEX-2T (Amersham Pharmacia, Piscataway, NJ). The GST-E1B fusion protein was expressed in Escherichia coli TOPP5 (Stratagene, Cambridge, U.K.) purified from inclusion bodies (41) and used to immunize LOU/C rats.
Indirect immunofluorescence analysis and protein analysis were performed as described previously (29). For immunoprecipitation, protein A-Sepharose (30 mg/ml) was incubated with 1 ml of hybridoma supernatant 2A6 for 2 h at room temperature and washed twice in lysis buffer. The antibodies bound to protein A-Sepharose were added to the protein A-Sepharose precleared extracts and rotated overnight at 4°C. The immune complexes were washed three times with lysis buffer, separated by SDS/PAGE, and analyzed by immunoblotting.
Supplementary Material
Acknowledgments
We thank S. Allmeier for technical assistance and Roger Grand for critically reading the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
Abbreviations
- NES
nuclear export signal
- SCS
SUMO1 conjugation site.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0702158104/DC1.
References
- 1.Dobner T, Kzhyshkowska J. Curr Top Microbiol Immunol. 2001;259:25–54. doi: 10.1007/978-3-642-56597-7_2. [DOI] [PubMed] [Google Scholar]
- 2.Berk AJ. Oncogene. 2005;24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 3.Flint SJ, Gonzalez RA. Curr Top Microbiol Immunol. 2003;272:287–330. doi: 10.1007/978-3-662-05597-7_10. [DOI] [PubMed] [Google Scholar]
- 4.Lethbridge KJ, Scott GE, Leppard KN. J Gen Virol. 2003;84:259–268. doi: 10.1099/vir.0.18820-0. [DOI] [PubMed] [Google Scholar]
- 5.Ornelles DA, Shenk T. J Virol. 1991;65:424–429. doi: 10.1128/jvi.65.1.424-429.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez RA, Flint SJ. J Virol. 2002;76:4507–4519. doi: 10.1128/JVI.76.9.4507-4519.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weitzman MD, Ornelles DA. Oncogene. 2005;24:7686–7696. doi: 10.1038/sj.onc.1209063. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Shevchenko A, Shevchenko A, Berk AJ. J Virol. 2005;79:14004–14016. doi: 10.1128/JVI.79.22.14004-14016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo JL, Berk AJ. J Virol. 2007;81:575–587. doi: 10.1128/JVI.01725-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobbelstein M, Roth J, Kimberly WT, Levine AJ, Shenk T. EMBO J. 1997;16:4276–4284. doi: 10.1093/emboj/16.14.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krätzer F, Rosorius O, Heger P, Hirschmann N, Dobner T, Hauber J, Stauber RH. Oncogene. 2000;19:850–857. doi: 10.1038/sj.onc.1203395. [DOI] [PubMed] [Google Scholar]
- 12.Endter C, Kzhyshkowska J, Stauber R, Dobner T. Proc Natl Acad Sci USA. 2001;98:11312–11317. doi: 10.1073/pnas.191361798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barker DD, Berk AJ. Virology. 1987;156:107–121. doi: 10.1016/0042-6822(87)90441-7. [DOI] [PubMed] [Google Scholar]
- 14.Horwitz MS. Proc Natl Acad Sci USA. 1978;75:4291–4295. doi: 10.1073/pnas.75.9.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Puvion Dutilleul F, Pedron J, Cajean Feroldi C. Eur J Cell Biol. 1984;34:313–322. [PubMed] [Google Scholar]
- 16.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. Genes Dev. 2001;15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stracker TH, Carson CT, Weitzman MD. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 18.Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. J Virol. 2005;79:11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carter CC, Izadpanah R, Bridge E. Virology. 2003;315:224–233. doi: 10.1016/s0042-6822(03)00526-9. [DOI] [PubMed] [Google Scholar]
- 20.Flint SJ, Huang W, Goodhouse J, Kyin S. Virology. 2005;337:7–17. doi: 10.1016/j.virol.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 21.Gill G. Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 22.Sobko A, Ma H, Firtel RA. Dev Cell. 2002;2:745–756. doi: 10.1016/s1534-5807(02)00186-7. [DOI] [PubMed] [Google Scholar]
- 23.Wood LD, Irvin BJ, Nucifora G, Luce KS, Hiebert SW. Proc Natl Acad Sci USA. 2003;100:3257–3262. doi: 10.1073/pnas.0637114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vassileva MT, Matunis MJ. Mol Cell Biol. 2004;24:3623–3632. doi: 10.1128/MCB.24.9.3623-3632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leppard KN, Shenk T. EMBO J. 1989;8:2329–2336. doi: 10.1002/j.1460-2075.1989.tb08360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leppard KN, Everett RD. J Gen Virol. 1999;80:997–1008. doi: 10.1099/0022-1317-80-4-997. [DOI] [PubMed] [Google Scholar]
- 27.Hay RT. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 28.Lee YS, Jang MS, Lee JS, Choi EJ, Kim E. EMBO Rep. 2005;6:949–955. doi: 10.1038/sj.embor.7400511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Endter C, Hartl B, Spruss T, Hauber J, Dobner T. Oncogene. 2005;24:55–64. doi: 10.1038/sj.onc.1208170. [DOI] [PubMed] [Google Scholar]
- 30.Gabler S, Schütt H, Groitl P, Wolf H, Shenk T, Dobner T. J Virol. 1998;72:7960–7971. doi: 10.1128/jvi.72.10.7960-7971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bachi A, Braun IC, Rodrigues JP, Panté N, Ribbeck K, von Kobbe C, Kutay U, Wilm M, Görlich D, Carmo-Fonseca M, et al. RNA. 2000;6:136–158. doi: 10.1017/s1355838200991994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Higashino F, Aoyagi M, Takahashi A, Ishino M, Taoka M, Isobe T, Kobayashi M, Totsuka Y, Kohgo T, Shindoh M. J Cell Biol. 2005;170:15–20. doi: 10.1083/jcb.200405112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao LY, Colosimo AL, Liu Y, Wan Y, Liao D. J Virol. 2003;77:11809–11821. doi: 10.1128/JVI.77.21.11809-11821.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, et al. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez R, Huang W, Finnen R, Bragg C, Flint SJ. J Virol. 2006;80:964–974. doi: 10.1128/JVI.80.2.964-974.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groitl P, Dobner T. In: Adenovirus Methods and Protocols. Wold WS, Tollefson AE, editors. Totowa, NJ: Humana; 2007. pp. 29–39. [Google Scholar]
- 37.Jones N, Shenk T. Cell. 1979;17:683–689. doi: 10.1016/0092-8674(79)90275-7. [DOI] [PubMed] [Google Scholar]
- 38.Sarnow P, Sullivan CA, Levine AJ. Virology. 1982;120:510–517. doi: 10.1016/0042-6822(82)90054-x. [DOI] [PubMed] [Google Scholar]
- 39.Reich NC, Sarnow P, Duprey E, Levine AJ. Virology. 1983;128:480–484. doi: 10.1016/0042-6822(83)90274-x. [DOI] [PubMed] [Google Scholar]
- 40.Boivin D, Morrison MR, Marcellus RC, Querido E, Branton PE. J Virol. 1999;73:1245–1253. doi: 10.1128/jvi.73.2.1245-1253.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudolph R, Böhm G, Lilie H, Jaenicke R. In: Protein Function: A Practical Approach. Creighton TE, editor. Oxford: Oxford Univ Press; 1997. pp. 57–99. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}