

Abstract
Antiarrhythmic agents are traditionally classified according to Vaughan Williams into four classes of action. Class I antiarrhythmic agents include most of the drugs traditionally thought of as antiarrhythmics, and have as a common action, blockade of the fast-inward sodium channel on myocardium. These agents have a very significant toxicity, and while they are being used less, therapeutic drug monitoring (TDM) does significantly increase the safety with which they can be administered. Class II agents are antisympathetic drugs, particularly the β-adrenoceptor blockers. These are generally safe agents which do not normally require TDM. Class III antiarrhythmic agents include sotalol and amiodarone. TDM can be useful in the case of amiodarone to monitor compliance and toxicity but is generally of little value for sotalol. Class IV antiarrhythmic drugs are the calcium channel blockers verapamil and diltiazem. These are normally monitored by haemodynamic effects, rather than using TDM. Other agents which do not fall neatly into the Vaughan Williams classification include digoxin and perhexiline. TDM is very useful for monitoring the administration (and particularly the safety) of both of these agents.
Keywords: antiarrhythmic drugs, therapeutic drug monitoring
Introduction
This article will discuss the role of Therapeutic Drug Monitoring (TDM) in the clinical use of a large group of chemically disparate compounds. The unifying theme is that most or all of these compounds have been at times used to treat cardiac dysrhythmias. For many of these drugs (such as the classical ‘Class I’ antiarrhythmic agents), this has been the major clinical application. For others (e.g. β-adrenoceptor blockers, calcium channel antagonists, perhexilene), the major therapeutic importance lies elsewhere. Many of these drugs have undergone a decline in usage over recent years. This applies particularly to the Class I antiarrhythmic agents, many of which may well disappear from use over the next decade. Nonetheless this is a group of compounds with well-defined ‘therapeutic ranges’ and generally with dose-dependent toxicity, where TDM continues to play an important role. Increasingly, Class II and III agents are replacing the older antiarrhythmics. The value of TDM is less clear-cut with many of these new drugs.
Determination of plasma concentrations of drugs for TDM
Reliable, sensitive and specific assays for the drug of interest are clearly a requirement to establish concentration-efficacy-toxicity relationships. Assays need to be free from interference, not only from endogenous substances and from other concomitantly administered drugs, but also must distinguish between parent drug and any metabolites whether they be active or inactive. Examples of drugs with active (e.g. N-acetylprocainamide, (NAPA), the metabolite of procainamide) and inactive (e.g. mexiletine) metabolites are represented in this review.
Historically, assays were based on colorimetric, spectrophotometric and fluorometric techniques which exhibited decreasing degrees of specificity, respectively. Attempts to establish concentration-effect relationships were therefore generally unsatisfactory or of questionable value. However, the combination of these detection modalities with chromatographic resolution, has allowed the firm establishment of TDM as a valuable adjunct to appropriate drug therapy.
In the early phase of investigation, the majority of drug assays are based on high performance liquid chromatography (h.p.l.c.) or on gas liquid chromatography (g.l.c.). In experienced hands these are specific, accurate and reproducible methods. Even greater specificity may be obtained using gas chromatography mass spectrometry (GCMS). However, the potentially greater accuracy and reproducibility tends not to be realised once introduced into laboratories conducting assays on large numbers of specimens and commonly involving less experienced analysts. The most commonly used routine techniques are those based on commercial immunoassay kits [1], such as enzyme immunoassay (EIA), or fluorescence polarization immunoassay (FPIA). The commercial methods may be relatively expensive, and there is a potential for cross-reactivity of the antibody with related drugs or metabolites. However, they provide rapid, reproducible and generally accurate results, require less skilled personnel, and are amenable to a high degree of automation. In practice they commonly provide quicker and better results than the intrinsically more specific chromatographic techniques.
Enantiomers
Most commonly used routine assays, whether chromatographic or immunoassay based, are not stereospecific. Indeed while the chromatographic assays give concentrations which are the sum of the concentrations of the individual enantiomers, immunoassays may be selective or non-selective for the enantiomers and, generally speaking, there is no information available on their relative stereoselectivities. The drugs used as racemates and which are of relevance to the present review are, disopyramide, mexiletine, flecainide, propafenone, sotalol, perhexiline, propranolol, and verapamil. The ability to establish clear therapeutic ranges is made more difficult and data more questionable because of the lack of information on the disposition of the individual enantiomers which are commonly very different for these cardiac drugs. Nevertheless, despite the intrinsic variability introduced, nonstereoselective assays have been used successfully and are the basis for the vast majority of TDM services. There may be a redefining of some current therapeutic ranges if stereospecific assays are introduced into routine services.
All but two of the agents to be discussed below have traditionally been classified as possessing one or more of the four classes of antiarrhythmic action originally described by Vaughan Williams (Table 1). While increasingly under challenge, this classification and the further subclassification of the Class I agents into three subgroups ([2, 3] Table 2), remains in widespread clinical use. It is far less complicated than the more recently described ‘Sicilian Gambit’ [4]. Further argument as to the relative merits of the various classification systems is beyond the scope of this article, (for reviews see [5–7]) and for reasons of familiarity and pragmatism, the Vaughan Williams classification will be followed below.
Table 1.
Vaughan Williams’ classification of antiarrhythmic actions.
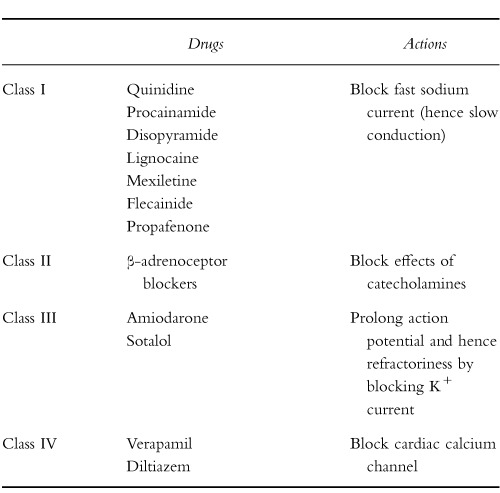 |
Table 2.
Subgroups of Class I drugs.
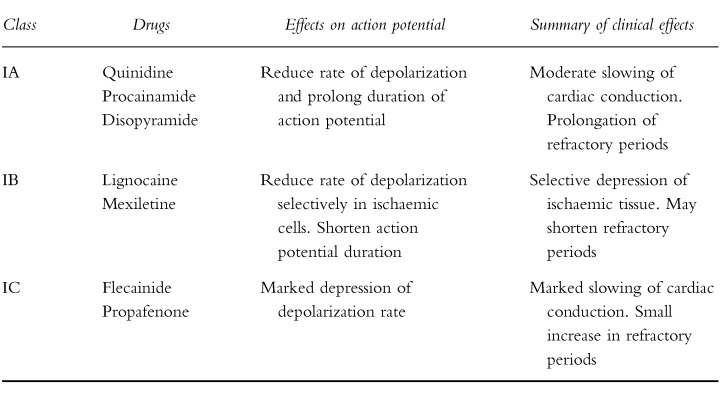 |
Class I antiarrhythmic agents
This is the oldest and by far the largest group and includes most of the classical antiarrhythmic agents. These compounds share the ability to block the fast inward sodium current responsible for the upstroke and rapid conduction of the cardiac action potential. Hence they are capable of slowing and even blocking intracardiac conduction. For this reason it came as little surprise to many clinicians when recent large-scale mortality trials confirmed that many of these drugs are capable, not only of preventing dysrhythmias, but also of causing them [8–18]. This phenomenon of ‘prodysrhythmia’ has become widely recognized, and has led to a dramatic reduction in the usage of these agents, particularly in Europe and Australasia.
Class IA drugs (quinidine, disopyramide and procainamide)
These drugs of which quinidine is the most widely used, have very similar electrophysiological properties both in vitro and vivo. They block both the inward sodium currents (an action common to all Class I agents), and the outward potassium currents responsible for repolarization of the cardiac action potential at concentrations in or near the therapeutic range [3, 19]. For this reason they are capable of causing proarrhythmic complications both via conduction slowing and via the promotion of oscillatory behaviour of the action potential associated with delayed repolarization, giving rise to a form of polymorphic ventricular tachycardia often referred to as ‘torsades de pointes’ [5, 20–23]. This is particularly a concern with quinidine and disopyramide. These drugs also share the unfortunate property that while their conduction-blocking actions are directly dose-dependent, their action potential prolonging effects and tendency to produce torsades de pointes may be more marked at lower concentrations than at higher concentrations [19]. Indeed many clinical reports of torsades due to quinidine and disopyramide have occurred with plasma concentrations at the lower end of (or even below) the therapeutic range [21, 22]. The reasons behind this paradox are well described [19], and unfortunately complicate the interpretation of TDM data with these compounds.
Quinidine
Quinidine is usually administered orally as the sulphate or gluconate or in various long-acting forms. The elimination half-life for quinidine sulphate or gluconate is 5–8 h, but the sustained release formulations which are almost universally used produce adequate plasma concentrations for at least 8 h [24]. A new steady state is not achieved for at least 24–36 h after a change in dosage following the initiation of therapy with such a sustained release formulation. Accordingly TDM and dosage adjustments should take this into account and should be based on trough levels sampled 8–12 h after the previous dose. Dosage adjustments should preferably not be made more frequently than every 2–3 days. (This general principle of using trough levels and only altering doses after allowing 3–5 half-lives to achieve steady state, applies to all drugs discussed below and will not be repeated under each new agent).
Plasma concentrations of quinidine are now most commonly determined by FPIA or EIA. In early development fluorometric assays were used because of the intrinsic high fluorescence of this drug [25], but these lack specificity [26]. While apparently producing relatively reliable data [27], inherently greater specificity was obtained with the introduction of h.p.l.c. assays [28, 29]. However, although quinidine is a single, stable isomer (note that a commonly promulgated fallacy is that quinidine and quinine are enantiomers [30]), it may contain dihydroquinidine (which is active), as a contaminant. Furthermore, several of the metabolites of quinidine are active, and accumulate to clinically significant concentrations during chronic therapy [31]. Earlier fluorescence assays were unreliable in these respects. Moderate cross reactivity of the antibodies in the commonly used FPIA assay [32] occurs with 3-hydroxyquinidine whose activity is ≈20% of that of the parent. This is one of the metabolites for which a correlation was identified between concentration and electrophysiological responses in human subjects [31]. The antibodies also do not distinguish between quinidine and the dihydroquinidine contaminant.
Therapeutic plasma levels are generally quoted as 3–8 μg ml−1 [33]. As referred to above, the dose-response curve for a particular form of quinidine toxicity, torsades de pointes, does not correlate well with this range, which largely refers to the efficacy of the compound in suppressing ectopic activity. Action potential prolongation and hazard for torsades de pointes are actually maximal at the lower end of the therapeutic range, and may occur at concentrations below this range [19]. This helps to explain the fact that torsades de pointes frequently occurs either early in a course of quinidine therapy or after the quinidine has been ceased and the blood levels are falling. Little can be done about this, other than pursuing a high level of clinical awareness. This is the rationale for the increasingly common practice of admitting patients to hospital for 2–3 days at the initiation of quinidine therapy.
Protein binding is 70–80%, and 80% of the drug is metabolised in the liver. The remainder is excreted unchanged in the urine. Renal excretion occurs by glomerular filtration and is pH dependent. Renal clearance of quinidine may diminish with increased urine pH [34], and reduced creatinine clearance, and hence is decreased in the elderly. Quinidine is not susceptible to peritoneal or haemodialysis. There is significant interpatient variability in bioavailability with a wide range of final daily dose (400–1200 mg day−1 ) being required to achieve therapeutic plasma concentrations. For these reasons therapeutic drug monitoring is definitely indicated.
Quinidine is implicated in a number of clinically significant drug interactions, which may increase requirements for monitoring. Inhibition of hepatic metabolism of quinidine occurs with drugs such as cimetidine and ketoconazole, and enhancement of metabolism by phenytoin and rifampicin. Macrolide antibiotics, particularly erythromycin, can both interfere with quinidine metabolism and produce additive potassium channel blockade. This form of interaction has been reported in association with life-threatening torsades de pointes [21]. Quinidine reduces the renal and nonrenal clearance of digoxin [35–37] and displaces digoxin from tissue binding sites. This leads to a reduction in the volume of distribution of digoxin of 30–40% and a reduction in digoxin clearance of 30–50%. Serum digoxin concentrations rise rapidly but do not plateau for up to 5 days or more. Most recommend halving the dose of digoxin on addition of quinidine and then monitoring digoxin levels closely.
Disopyramide
Disopyramide may be given orally or intravenously. Oral bioavailability is about 80% and peak plasma levels occur at 1–2 h [38]. The usual oral dose is 300–600 mg d−1.
Disopyramide has a half-life of 4–6 h in healthy volunteers. Elimination is largely renal, and half-life rises with falling creatinine clearance.
50–80% of the drug is normally excreted unchanged in the urine. There are several metabolites that probably do not contribute to the antiarrhythmic effect, but one has 24 times the anticholinergic potency of the parent, and may contribute to anticholinergic side-effects [39]. Long-acting formulations of disopyramide are available and effective [40].
Most commonly, disopyramide is assayed by EIA or FPIA. There are apparently no data on the relative specificities of the antibodies for the enantiomers of this racemic drug, which exhibits stereoselectivity with respect to metabolism, renal clearance and protein binding [41]. Interestingly, total clearance of each of the isolated enantiomers is similar but the S-enantiomer is cleared significantly more slowly when the racemate is given [42]. R-disopyramide and S-disopyramide have very similar activities with respect to prolonging the effective refractory period [43]. Consequently, while there are assays available to determine the enantiomers of disopyramide [44], the lack of stereospecificity of commonly used TDM assays would not appear to be a major concern S-disopyramide is 3–4 times more potent than R-disopyramide with respect to anticholinergic activity [45].
The therapeutic range for disopyramide concentration is ≈2.8–7.5 μg ml−1 [46]. Unlike most other antiarrhythmic drugs, protein binding of disopyramide shows nonlinear, saturable characteristics [47, 48]. This is of clinical importance since apparently small increases in total plasma concentration of disopyramide may mask larger rises in free (active) drug concentration [47]. There appears to be a better correlation between the overall change in QTc interval or ventricular tachycardia cycle length and free concentrations than with total concentrations of disopyramide [49, 50]. Routinely, however, total concentrations are reported and the quoted therapeutic range is that for total drug.
Disopyramide binds significantly to α1-acid glycoprotein, concentrations of which rise during many acute illnesses, including myocardial infarction [51]. In addition to these caveats, as with quinidine, there is correlation between relatively low plasma concentrations of disopyramide and high risk of torsades de pointes [19].
Interpretation of disopyramide assays is complicated not only by the issues outlined above but by a number of other matters. α1-acid glycoprotein levels are generally higher in elderly individuals, although the effect of this on decreasing amounts of free drug is probably balanced by the decreased renal function seen in elderly patients. Systemic clearance in children is at least twice as high as in normal adults although the compound does not commonly feature in paediatric practice.
Normally about 80% of disopyramide is excreted unchanged in the urine and although it is a basic compound, alterations in urine pH have little effect on the rate of renal excretion of disopyramide [52]. From a practical point of view it is important to either decrease the dose or more commonly increase the dosing interval of disopyramide as creatinine clearance falls [53].
Drugs which induce hepatic enzymes such as rifampicin and phenytoin increase conversion of disopyramide to its more anticholinergic metabolite and may enhance anticholinergic side-effects. Macrolide antibiotics, particularly erythromycin, can both interfere with disopyramide metabolism and produce additive potassium channel blockade. This form of interaction could lead to life-threatening torsades de pointes. Disopyramide does not appear to interact significantly with digoxin or warfarin.
Procainamide
Procainamide is usually given orally at a total dose of 3–6 g day−1. Bioavailability is high and peak plasma concentrations are achieved 1–2 h after tablet ingestion. Protein binding is only 10–20% and the elimination half-life is quite short (3–5 h). For this reason long acting formulations are commonly prescribed where this drug is to be used chronically. Approximately 40–70% of procainamide is excreted unchanged in the urine by glomerular filtration and active tubular secretion [54]. Approximately 16–30% of procainamide is acetylated by hepatic N-acetyltransferases, forming N-acetylprocainamide (NAPA). Proportions of metabolism to NAPA are about 16–20% in ‘slow acetylators’, and 25–30% in ‘rapid acetylators’ [55, 56]. NAPA is excreted via the kidneys and also undergoes limited metabolism. When monitoring procainamide therapy, it is customary to measure serum levels of both procainamide and NAPA which has significant antiarrhythmic action, particularly Class III activity (prolongation of the action potential via potassium channel blockade). NAPA (but not procainamide), can be removed by haemodialysis and haemoperfusion, but not by peritoneal dialysis [57]. Hepatic and/or renal disease will obviously interfere with excretion, and the dosage of procainamide should be reduced accordingly, based on TDM. Cardiac failure by interfering with hepatic metabolism and renal function may also necessitate dosage reduction.
The ratio between procainamide and NAPA in the plasma will depend on the acetylator status of the individual. Slow acetylators will have a low NAPA/procainamide ratio. The therapeutic plasma concentration of procainamide is generally thought to be in the range of 4–12 μg ml−1.
The FPIA and EIA assays are used most commonly for TDM of procainamide and of the active metabolite NAPA. The antibodies are, respectively, specific for both parent and metabolite [32]. Whether the therapeutic range should be based on the additive concentrations of the parent and metabolite (as is commonly the protocol) or on individually defined therapeutic ranges for each of procainamide and NAPA has not been established, but it is reasonable to monitor concentrations simultaneously. Addition of concentrations would appear to be an intrinsically flawed approach because it assumes equal activity and toxicity of parent and metabolite, and can only be done if both are expressed in molar units.
Class IB drugs (lignocaine and mexiletine)
Lignocaine
Lignocaine is widely used parenterally for the control of ventricular tachydysrhythmias. It is also finding an increasing role (along with orally active agents such as mexiletine and flecainide) in the management of various chronic pain syndromes particularly those thought to be neurogenic in origin [58–60].
Because of extensive hepatic first pass metabolism to potentially toxic metabolites [61, 62], lignocaine must be administrated parenterally. It has a volume of distribution at steady state of about 1.3 l kg−1, a distribution half-life of 8 min, and a plasma elimination half-life of ≈2 h. There are a number of effective dosage regimens in the literature designed to rapidly produce therapeutic blood levels without overshooting and causing toxicity. These usually involve a bolus or rapid infusion followed by a steady infusion to maintain plasma concentrations constant. Plasma concentrations should certainly be checked, to minimize toxicity particularly if the infusion is to be continued beyond 24 h. If inefficacy is an issue, then plasma levels should be checked when clinically indicated.
The usually quoted therapeutic plasma concentrations are approximately 2–6 μg ml−1. Toxicity is generally concentration-dependent, as is efficacy. There is no significant cross reactivity of the most commonly used EIA and FPIA assays [33, 63] with the primary metabolites, the concentrations of which are relatively low in plasma [63, 64]. Therapeutic ranges have not been established for treatment of chronic pain but the values quoted for antiarrhythmic effects appear clinically satisfactory.
Lignocaine has two active metabolites, monoethylglycine xylidide (MEGX) and glycine xylidide (GX) which also have short half-lives of 2 h and 1 h, respectively. Some of the central toxicity of lignocaine is attributed to central accumulation of the metabolites, particularly in patients with heart failure [62, 65].
Old age, and any drug or disease state which influences hepatic blood flow or metabolism will have significant effects on the pharmacokinetics of lignocaine. Common, clinically relevant examples include alcoholic liver disease, reduced hepatic blood flow due to heart failure or β-adrenoceptor blockers, and reduced metabolism due to cimetidine.
Mexiletine
Mexiletine is structurally very similar to lignocaine but is well absorbed after oral administration, with peak plasma concentrations occurring within 2–4 h. Bioavailability is about 80% [66–68]. For oral doses of 100–600 mg day−1 there is a linear relationship between plasma concentration and dose [69]. The therapeutic range is ≈0.6–1.7 μg ml−1 [68] for antiarrhythmic effects; the comment on lignocaine concentrations for chronic pain applies also to mexiletine (as it does also for flecainide).
The mean half-life of elimination after oral administration of a single dose is about 6–10 h but it may be higher (11–17 h) in patients with cardiac disease [66–68]. Mexiletine is eliminated largely by hepatic metabolism, with 85% being metabolised to inactive metabolites.
Approximately 15% is excreted unchanged in the urine, and as long as creatinine clearance is above 10 ml min−1, renal insufficiency has little or no relevance to plasma kinetics for mexiletine. Being a weak base, the proportion of drug excreted unchanged in urine is pH dependent. The half-life of mexiletine at pH 5.0 (2.8 h) is less than half of that at pH 8.0 (8.0 h) [70]. Mexiletine cannot be removed by dialysis.
There are variable and stereoselective differences in the disposition of the mexiletine enantiomers [71, 72]. The R-enantiomer has been reported to have greater antiarrhythmic activity [73]. Mexilitine is generally assayed using achiral GC or h.p.l.c. based assays, although stereoselective assays are available [74]. While intersubject variability in response may be explained in part by variable disposition of the enantiomers there are no studies which have investigated the correlation between concentration and effect using stereoselective assay.
Drugs which induce hepatic enzymes have been shown to enhance the hepatic elimination of mexiletine. These include rifampicin and phenytoin. By increasing the conjugation of mexiletine with glucuronic acid, cigarette smoking also enhances the elimination of mexiletine. Cimetidine, morphine and atropine can delay the absorption of mexiletine from the gut. Maximal blood concentrations and half-life are unchanged.
Class IC drugs (flecainide, propafenone)
Flecainide
Flecainide is very well absorbed orally with negligible hepatic first pass effect [75]. Elimination half-life ranges from 7 to 15 h in healthy volunteers and averages about 10 h in patients with cardiac disease [76]. There is considerable variability, however, and plasma concentration monitoring is recommended. Plasma flecainide levels are related linearly to dose over a wide range.
The therapeutic range is ≈200–1000 ng ml−1, with toxicity possible over 1000 ng ml−1 and quite likely over 1600 ng ml−1 [77, 78]. The drug is rapidly metabolized to compounds which are far less potent than the parent [79]. The depression of conduction velocity is quite strongly concentration-dependent, with no complicating factors such as exist with quinidine or disopyramide. The duration of the QRS interval on the surface ECG can be used as a crude indication of toxicity, with prolongation of the QRS interval signifying conduction slowing in normal myocardium. Nonetheless, this is not recommended as a substitute for plasma concentration monitoring.
There is some confusion arising from the use of the acetate salt of flecainide, rather than the free base, for the reference standards used in some FPIA kits and chromatographic procedures [80]. Indeed, the therapeutic range reported for flecainide (200–1000 ng ml−1 ) is that based on the acetate salt rather than the free base, flecainide; the corresponding range would be 175–870 ng ml−1 [81]. While it can be argued that use of molar units overcomes this difficulty, the range is probably not sufficiently well defined for clinical utility.
The immunoassays do not distinguish between the enantiomers of flecainide. Stereospecific h.p.l.c. assays are available [82–84]. However, there is little or no difference in the ability of the enantiomers of flecainide to depress sodium channels [85–87] and, consequently, it is unlikely that the relative disposition of the enantiomers is important.
In healthy subjects, 80–90% of oral flecainide is excreted in the urine either as unchanged drug or as relatively inactive metabolites. The presence of heart failure prolongs flecainide plasma half-life but does not affect urinary excretion after a single dose, and during long-term administration patients with heart failure may require a lower daily flecainide dose, (although it is generally recommended that flecainide is not given in the presence of clinical left ventricular dysfunction). Impairment of renal function prolongs plasma half-life and this effect correlates quite well with creatinine clearance. Flecainide is not significantly removed by haemodialysis.
Plasma protein binding of flecainide is ≈40%, and is relatively constant across the therapeutic concentration range and above.
Flecainide exhibits polymorphic metabolism, cosegregating with debrisoquine. Deficiency of the CYP2D6 isozyme (poor metabolisers) results in impaired metabolism but only of the R-enantiomer [88], the S-enantiomer presumably being metabolised by other pathways. Furthermore, quinidine which is a potent inhibitor of P4502D6, significantly reduces the clearance of R-flecainide [89]. The resulting increased concentrations of flecainide may also lead to increased QRS [89] and proarrythmic effects [90].
Propafenone
Propafenone is a Class IC agent but also exhibits some β-adrenoceptor blocking action [91, 92]. It is well absorbed orally, with maximum plasma concentrations occurring at 2–3 h [93]. Hepatic first-pass metabolism is extensive however, leading to a bioavailability of only 10–20%. Kinetics are nonlinear [94, 95]. Plasma half-life varies widely from about 2 to about 12 h.
Reported effective plasma concentrations range widely from 40 to over 3000 ng ml−1 [96–99]. In general, plasma concentration appears to correlate poorly with antiarrhythmic efficacy. Some of the reason for this may be genetic differences in metabolism discussed below. This has led to the recommendation that efficacy and toxicity should be monitored more by electrocardiographic parameters (such as QRS prolongation), and degree of suppression of overt dysrhythmias, rather than by plasma drug concentrations.
Propafenone is a racemic drug [100]. Plasma concentrations are generally determined by h.p.l.c. Stereospecific assays have been developed and applied to bioequivalence testing [101]. Interpretation of the plasma concentrations of propafenone is potentially complex because not only do the enantiomers have differing activities, but the metabolite, 5-hydroxypropafenone, is antiarrythmic [102] and achieves therapeutically relevant concentrations. While the enantiomers are similar with respect to antagonising sodium channels, S-propafenone is the enantiomer with β-antagonist activity [100]. Furthermore, R-propafenone impairs the metabolism of S-propafenone such that the actions of the racemic drug are not simply those predicted by summation of the effects of the individual enantiomers [103].
In more than 90% of patients, propafenone is metabolised rapidly and extensively in the liver [99, 100, 104]. In less than 10% of patients, the principal hepatic cytochrome P-450 enzyme responsible for propafenone metabolism, CYP2D6, appears to be either deficient or absent [99, 104, 105]. These patients demonstrate marked reduction in propafenone clearance, with long elimination half-life and high plasma concentrations relative to dose [99]. They are more susceptible to central nervous system side-effects and β-adrenoceptor blocking effects, owing to the high plasma concentration of the parent compound. However, the drug appears to be equally antiarrhythmic in both normal and slow metabolisers. As noted above the principal metabolite, 5-hydroxypropafenone, also possesses significant Class I antiarrhythmic actions [102].
Class II antiarrhythmic agents (β-adrenoceptor blockers)
The β-adrenoceptor blockers have a significant antiarrhythmic efficacy which is often discounted by clinicians [106, 107]. In addition they are the only class of antiarrhythmic agent to have demonstrated convincing mortality reduction post-myocardial infarction, an effect largely or entirely due to a 30% reduction in sudden death rates in these patients. They therefore represent an extremely valuable and somewhat under-rated component of the antiarrhythmic armamentarium of the clinician. On the other hand, their efficacy and side-effects correlate poorly with plasma concentrations, and unlike the Class I agents, there is little or no role for therapeutic drug monitoring with β-adrenoceptor blockers.
Class III antiarrhythmic agents
Sotalol
Sotalol is an increasingly popular antiarrhythmic agent which possesses both nonselective β-adrenoceptor blocking activity and Class III antiarrhythmic action [108–110]. This latter action is due to blockade of outward potassium currents, and produces a dose-dependent prolongation of cardiac action potential duration. It also produces a propensity to torsades de pointes [21, 111], which is generally associated with high blood levels of sotalol; this risk is exacerbated by hypokalaemia which may occur with concomitant high dose diuretics.
The oral bioavailability of sotalol is about 60%, and there is no significant hepatic first-pass metabolism. More than half the oral dose is recovered unchanged in the urine and there are no known active metabolites, nor is there any significant plasma protein binding. Hence fluctuations in serum concentration are small. Moreover, the lack of metabolic elimination means that this drug, unlike the structurally related β-adrenoceptor antagonists such as propranolol and metoprolol, does not exhibit polymorphic metabolism.
The plasma half-life is long, ranging from about 10–15 h and averaging ≈12 h. Plasma concentrations are linearly related to dose, and also vary directly in proportion to changes in creatinine clearance.
The usual oral dose of sotalol is 80 mg twice daily to 160 mg twice daily. Total daily doses above 320 mg−1 day lead to an increased incidence of torsades de pointes. In the presence of diminished renal function, the first step is usually to prolong the dosing interval to once daily. With creatinine clearance below 10–30 ml min−1, dosing every second day may be sufficient. Patients with more severe degrees of renal failure than this need careful blood concentration monitoring.
There are no major pharmacokinetic drug interactions commonly associated with sotalol usage. Sotalol, however, causes all of the side-effects predictable from its β-adrenoceptor blocking action including a number of pharmacodynamic interactions with other cardioactive compounds such as calcium channel blockers and other antiadrenergic drugs with which it may produce additive negative inotropic or chronotropic effects. It also may produce additive effects in terms of slowing cardiac repolarization, leading to enhanced prodysrhythmia with agents which also block outward potassium currents including amiodarone, tricyclic antidepressants, phenothiazines, terfenadine, astemizole and macrolide antibiotics, particularly erythromycin.
Therapeutic drug monitoring is not commonly carried out in association with sotalol usage. It may be of value in difficult situations, particularly in association with severe renal dysfunction or where there is a question concerning compliance. Plasma concentrations of sotalol during chronic oral therapy generally range from ≈1–3 μg ml−1 [109, 112]. Much more commonly however, the dosage is monitored according to effects on the QT interval, with prolongations of more than 15–20% being regarded as an indication to reduce the level or prolong the dosage interval.
Sotalol concentrations in plasma are generally determined by h.p.l.c. [113–115, 116], and stereospecific assays have been developed to monitor the disposition of the enantiomers [117]. There are relatively minor differences in the disposition of the enantiomers following administration of the racemate, the form used therapeutically. This is attributed to stereoselective differences in plasma protein binding. Their half-lives are similar [118]. The activity of the racemate is primarily attributable to the (−)-enantiomer [119].
Amiodarone
This very widely used drug is commonly classified as a Class III agent and certainly prolongs cardiac action potential in chronic dosing. Nonetheless it has a number of other actions which may well contribute both to its antiarrhythmic and proarrhythmic potential. These include significant sodium channel blocking (Class I action), significant antisympathetic action of a non competitive kind [120] and some degree of calcium channel blockade. It has been widely used since the 1960s, initially as a vascular smooth muscle relaxant for angina and subsequently as an antiarrhythmic agent. In many countries including Australia it is now the most commonly used antiarrhythmic.
The use of amiodarone is complicated by its very unusual pharmacokinetics and unwanted side-effects. These aspects are both well covered by recent reviews [120, 121], and will only be briefly outlined here. The administration of amiodarone (normally by mouth) is complicated by variable bioavailability (20–80%), and a terminal half-life of elimination which is usually 35–40 days, but may exceed 100 days. The major metabolite (desethylamiodarone) accumulates in high concentration in plasma and tissues, and possesses very similar electrophysiological properties to the parent [122, 123]. Dosage regimen varies very widely but most clinicians use a loading dose of 600 mg to 2000 mg day−1 for 1–8 weeks, followed by reduction to a maintenance dose of the order of 200–400 mg day−1. Where rapid loading is desirable, amiodarone maybe given intravenously (via a central vein) but the Class III action does not generally appear in the first few hours or even days of administration.
Many of the side-effects appear to be dose-dependent but blood concentration monitoring, other than to monitor compliance, is of limited benefit. The drug and its metabolite are found in tissues at very much higher concentrations than in plasma [123, 124]. There is some evidence that plasma concentrations above 0.5 μg ml−1 seem to be required for efficacy, but there are no convincing data showing a correlation between actual plasma concntrations and antiarrhythmic effect [125]. Similarly while serious toxicity seems to be more likely at concentrations above 2.5 μg ml−1 [126, 127], its incidence is more reliably correlated with measures of total drug usage, suggesting the importance of accumulation in target tissues over time.
Amiodarone concentrations are determined by h.p.l.c. which can separate parent from the active metabolite, desethylamiodarone [128, 129]. Amiodarone and desethylamiodarone are unstable and should be protected from light [130]. The assays available have been recently reviewed with respect to suitability of internal standards [131]. Although the metabolite is active the therapeutic range is based on reporting of parent drug concentrations only. This practice is intrinsically flawed, since ignoring the metabolite concentrations which may be greater than the parent makes definition of a clear therapeutic range unlikely.
There are a number of potentially significant drug interactions associated with amiodarone. In particular it may elevate serum digoxin levels and potentiate the actions of warfarin. When amiodarone is added to a maintenance digoxin regimen, the serum digoxin concentration rises linearly for up to a week until a new plateau is reached [127, 132, 133], and this may result in significant digitalis toxicity. The mechanism for this interaction is still unclear. The appropriate action is generally to halve the dose of digoxin and check plasma digoxin concentrations.
In patients taking warfarin, the INR is prolonged by the administration of amiodarone [134]. The mechanism of this interaction is also unknown. Additionally, amiodarone will produce additive cardiac depression with β-adrenoceptor blockers and calcium channel blockers.
Perhexiline
Perhexiline maleate has been used to treat angina pectoris for some 25 years without ever achieving wide popularity. It lacks significant negative inotropy or haemodynamic effects, but its widespread use has been significantly hampered by what are perceived to be unpredictable serious hepatic and neurological side-effects [135–139]. It was originally labelled as a calcium antagonist and subsequently shown to block at least some outward potassium channels [140]. There is considerable doubt, however, as to whether either of these actions is of relevance at therapeutic blood concentrations and recent evidence suggests that its anti-ischaemic actions may relate to its inhibition of carnitine palmitoyltransferase-1 [141].
Whatever its mechanism of action however, it has become evident that toxicity is directly related to perhexiline blood concentration, and that the drug has a saturable rate of hepatic metabolism that is genetically determined [142, 143]; see below). With the advent of a reliable assay for quantifying blood concentrations of perhexiline there has been a pronounced decrease in the incidence of serious side-effects with careful TDM.
The concentrations are generally reported as those of the racemic drug which is the form used clinically. There are no commercially available immunoassays and concentrations of perhexiline are measured routinely in relatively few laboratories. Direct assay by h.p.l.c. with fluorescence detection [144], or with derivatization which allows determination of the plasma concentrations of the cis-and trans-monohydroxy metabolites [145] are described. GC with electron capture detection following derivatization also has been used to quantify perhexiline and metabolites in plasma [146]. However, given the very high and stereoselective disposition of the enantiomers (2.5 l min−1 (+)-enantiomer, 1.0 l min−1, (−)-enantiomer [147]) one could question the likely value of such nonstereoselective assays, and the likely association between concentration of racemic drug and effect.
Plasma perhexiline concentration does not correlate with dose, as metabolism is saturable within the usual clinical dose range [144]. The major metabolic pathway of perhexiline is hepatic metabolism via CYP-2D6 and it is subject to genetic polymorphism, with up to 10% of the Caucasian population being ‘slow metabolizers’ [148]. Thus the dose range associated with ‘therapeutic’ plasma concentrations is very wide and it may range from 50 mg once per week to 600 mg day−1 [149]. The usual maintenance dose range is 100–400 mg day−1, aiming to achieve a plasma concentration in the range of 0.15–0.6 μg ml−1 (0.38–1.5 μm ). Patients who remain symptomatic with concentrations in this range may achieve additional benefit by cautious dose increases to achieve a concentration in the range of 0.6–1.2 μg ml−1. Significant hepatotoxicity and peripheral neuropathy is usually only observed with chronic plasma concentrations above 1.2 μg ml−1 (3 μm ). The most practical way of identifying slow metabolizers is to commence patients on 300 mg once daily for 1 week with a subsequent reduction to 100 mg once daily. A blood level performed after the initial week of therapy will be very high in the 10% of the population who are slow metabolizers, and these patients should be reduced to a very low maintenance dose of 50–100 mg once weekly monitored by further TDM. The remaining patients should be maintained initially on 100 mg once daily after the first week of therapy with further dose increments of 50–100 mg daily at 2–4 week intervals based on plasma concentration measurements and clinical efficacy.
Digoxin
Digoxin is by far the most widely prescribed cardiac glycoside. It may be administered intravenously, intramuscularly or orally. Oral bioavailability is about 75%, and the half-life is 40–150 h. It is not metabolized to a significant degree although metabolic clearance becomes more significant as renal function declines. It is excreted unchanged in the urine and for this reason dosage has to be adjusted carefully in patients with renal disease.
Digoxin was one of the first agents for which routine TDM was introduced. The fall in the prevalence of clinically significant digitalis toxicity since plasma concentration assays became readily available in the 1970s has been attributed to TDM.
Digoxin is commonly given to adults in a dose of 0.25 mg once daily if renal function is normal. The usually accepted therapeutic plasma concentration range is between 0.5 and 2.0 ng ml−1. The therapeutic and toxic concentration ranges overlap and toxicity may occur even within the therapeutic range. There is some clinical trial evidence that efficacy is concentration-dependent within the therapeutic range, and considerable evidence of concentration-dependence toxicity.
Digoxin is almost exclusively assayed by EIA and FPIA for the purpose of TDM. The commonly used immunoassays may not distinguish digoxin from other drugs e.g. spironolactone and its metabolites [150] and there is substantial cross reactivity with digoxin metabolites. The assays for digoxin are also unreliable for 10 days or more following administration of digoxin antibodies (e.g. ‘Digibind’) for the treatment of digoxin toxicity because of competition for binding of digoxin between the digoxin antibodies in the assay kit and the circulating Fab fragments [151–153]. Furthermore, endogenous substances [154], generally referred to as digoxin-like immunoreactive substances/factors (DLIS/DLIF) may also elicit a ‘digoxin-like’ response. Commercial kits demonstrate variable specificity for DLIF [155]. DLIF may be more problematic in renal failure [156], hepatic dysfunction, and neonates with attempts to reduce the interference by ultrafiltration giving improved specificity, but not in all specimens [157]. A more recent monoclonal antibody assay has demonstrated improved specificity both with respect to interference by DLIF and digoxin metabolites, and may also give reliable unbound concentrations of digoxin in the presence of ‘Digibind’ [153]. Thus digoxin concentrations need qualified interpretation in the light of these possible interferences and cross reactivities and may explain, in part, the variable response of patients even within the ‘therapeutic’ range of digoxin concentrations.
Since digoxin acts by binding to and blocking the sodium-potassium pump, there is significant potentiation of digoxin toxicity in the presence of hypokalaemia which in itself reduces pump activity significantly. Every effort should be made to maintain normal plasma potassium concentrations in patients taking digoxin. Similarly, digoxin is to an extent potentiated by elevated serum calcium concentration and vice versa.
Serum concentrations of digoxin may be dramatically influenced by other medications. Interaction of digoxin with quinidine is now well documented with the administration of quinidine to a patient already on a stable digoxin regimen leading to an increase in the plasma concentration of digoxin of 50–150% [158]. This increase begins to appear within hours. It is partly due to displacement of digoxin from binding sites, and maintained by a reduction in renal clearance of digoxin [159]. Inhibition of the ‘drug-pump’ P-glycoprotein, by quinidine may be the major mechanism [160, 161]. A number of other cardioactive agents, including verapamil, amiodarone, propafenone and diltiazem also commonly produce dramatic increases in digoxin concentrations. Anti-adrenergic agents do not affect serum digoxin levels but may produce additive negative chronotropic actions.
Conclusions
Therapeutic drug monitoring has played an extremely important role in the development and clinical application of many agents used to treat cardiac rhythm disturbances. This is particularly true of the Class I agents and of perhexiline and digoxin. TDM has been less important in monitoring the use of β-adrenoceptor blockers and Class IV drugs and has played only a limited role in the use of amiodarone, where there is some value in monitoring for compliance and toxicity.
The world wide reduction in the use of Class I antiarrhythmic agents will somewhat reduce the relevance of TDM to the practising cardiologist however, although its role in monitoring digoxin and perhexiline will remain. There are a number of novel, pure Class III antiarrhythmic agents in various stages of development, some of which will definitely require TDM if they come into wide-spread clinical use.
References
- 1.Goldberger BA, Jenkins AJ. Testing of abused drugs in urine by immunological techniques. AACC In-service Training Continuing Ed. 1992;13:7–18. [Google Scholar]
- 2.Vaughan Williams. EM A classification of antiarrhythmic actions reassessed after a decade of new drugs. J Cardiovasc Pharmacol. 1984;24:129–147. doi: 10.1002/j.1552-4604.1984.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 3.Campbell TJ. Subclassification of class I antiarrhythmic drugs: Enhanced relevance after CAST. Cardiovasc Drugs Ther. 1992;6:519–528. doi: 10.1007/BF00055611. [DOI] [PubMed] [Google Scholar]
- 4.Rosen MR, Schwartz PJ. The Sicilian Gambit. A new approach to the classification of antiarhythmic drugs based on their actions on arrhythmogenic mechanisms. Circulation. 1991;84:1831–1851. doi: 10.1161/01.cir.84.4.1831. [DOI] [PubMed] [Google Scholar]
- 5.Campbell TJ. Antiarrhythmic drugs. In: Thompson P, Churchill Livingstone, editors. Manual of Coronary Care. London: 1997. pp. 225–244. [Google Scholar]
- 6.Grant AO. Mechanisms of action of antiarrhythmic drugs: From ion channel blockage to arrhythmia termination. Pacing Clin Electrophysiol. 1997;20:432–444. doi: 10.1111/j.1540-8159.1997.tb06202.x. [DOI] [PubMed] [Google Scholar]
- 7.Katz AM. Cardiac ion channels. N Engl J Med. 1993;328:1244–1251. doi: 10.1056/NEJM199304293281707. [DOI] [PubMed] [Google Scholar]
- 8.Velebit V, Podrid PJ, Lown B, Cohen BH, Graboys TB. Aggravation and provocation of ventricular arrhythmias by antiarrhythmic drugs. Circulation. 1982;65:886–894. doi: 10.1161/01.cir.65.5.886. [DOI] [PubMed] [Google Scholar]
- 9.Ruskin JN, McGovern B, Garan H, Dimarco JP, Kelly E. Antiarrhythmic drugs: a possible cause of out-of-hospital cardiac arrest. N Engl J Med. 1983;309:1302–1306. doi: 10.1056/NEJM198311243092107. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman BF, Dangman JH. The role of antiarrhythmic drugs in sudden cardiac death. J Am Coll Cardiol. 1986;8:104A–109A. doi: 10.1016/s0735-1097(86)80036-5. [DOI] [PubMed] [Google Scholar]
- 11.Rae AP, Kay HR, Horowitz LN, Spielman SR, Greenspan AM. Proarrhythmic effects of antiarhythmic drugs with malignant ventricular arrhythmias evaluated by electrophsiologic testing. J Am Coll Cardiol. 1988;12:131–139. doi: 10.1016/0735-1097(88)90366-x. [DOI] [PubMed] [Google Scholar]
- 12.The Cardiac Arrhythmia Suppression Trial Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- 13.Cardiac Arrhythmia Suppression Trial II. Investigators. Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. N Engl J Med. 1992;327:227–233. doi: 10.1056/NEJM199207233270403. [DOI] [PubMed] [Google Scholar]
- 14.IMPACT Research Group. International mexiletine and placebo antiarrhythmic coronary trial I. Report on arrhythmia and other findings. J Am Coll Cardiol. 1984;4:1148. doi: 10.1016/s0735-1097(84)80133-3. [DOI] [PubMed] [Google Scholar]
- 15.Morganroth J. Early and late proarrhythmia from antiarrhythmic drug therapy. Cardiovasc Drugs Ther. 1992;6:11. doi: 10.1007/BF00050910. [DOI] [PubMed] [Google Scholar]
- 16.Coplen SE, Antman EM, Berlin JA, et al . Efficacy and safety of quinidine therapy for maintenance of sinus rhythm after cardioversion: a meta-analysis of randomized controlled trials. Circulation. 1990;82:1106–1116. doi: 10.1161/01.cir.82.4.1106. [DOI] [PubMed] [Google Scholar]
- 17.Morganroth J, Goin JE. Quinidine-related mortality in the short-to-medium-term treatment of ventricular arrhythmias: a meta-analysis. Circulation. 1991;84:1977–1983. doi: 10.1161/01.cir.84.5.1977. [DOI] [PubMed] [Google Scholar]
- 18.Teo KK, Yusuf S, Fuberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction. JAMA. 1993;270:1589–1595. [PubMed] [Google Scholar]
- 19.Wyse KR, Ye V, Campbell TJ. Action potential prolongation exhibits simple dose-dependence for sotalol, but reverse dose-dependence for quinidine and disopyramide: Implications for proarrhythmia due to triggered activity. J Cardiovasc Pharmacol. 1993;21:316–322. doi: 10.1097/00005344-199302000-00019. [DOI] [PubMed] [Google Scholar]
- 20.Campbell TJ. Pro-arrhythmic action of antiarrhythmic drugs: A review. Aust NZ J Med. 1990;20:275–282. doi: 10.1111/j.1445-5994.1990.tb01039.x. [DOI] [PubMed] [Google Scholar]
- 21.Jackman WM, Friday KJ, Anderson JL, et al . The long QT syndromes: a critical review, new clinical observations and a unifying hypothesis. Prog Cardiovasc Dis. 1988;31:115–172. doi: 10.1016/0033-0620(88)90014-x. [DOI] [PubMed] [Google Scholar]
- 22.Roden DM, Hoffman BF. Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Circ Res. 1985;56:857–867. doi: 10.1161/01.res.56.6.857. [DOI] [PubMed] [Google Scholar]
- 23.Dessertenne F. La tachycardie ventriculaire a deux foyers oppose variables. Arch Mal Coeur. 1966;59:263–272. [PubMed] [Google Scholar]
- 24.Taggart W, Holyoak W. Steady-state bioavailability of two sustained-release quinidine preparations. Clin Ther. 1983;5:357–361. [PubMed] [Google Scholar]
- 25.Huynh-ngoc T, Sirois G. Comparison of two spectroflurometric procedures for quinidine determination in biological fluids. Pharm Sci. 1977;66:591–592. doi: 10.1002/jps.2600660436. [DOI] [PubMed] [Google Scholar]
- 26.Guentert TW, Upton RA, Holford NH, Riegelman S. Divergence in pharmacokinetic parameters of quinidine obtained by specific and nonspecific assay methods. J Pharmacokinet Biopharmaceut. 1979;7:303–311. doi: 10.1007/BF01060020. [DOI] [PubMed] [Google Scholar]
- 27.Smith GH, Levy RH. Serum quinidine determination: comparison of mass-spectrometric and extraction-fluorescence methods. Drug Clin Pharmacy. 1982;16:693–695. doi: 10.1177/106002808201600909. [DOI] [PubMed] [Google Scholar]
- 28.MacKichan JJ, Shields BJ. Specific high performance liquid chromatograqphic determination of quinidine in serum, blood, and urine. Ther Drug Monit. 1987;9:104–112. doi: 10.1097/00007691-198703000-00018. [DOI] [PubMed] [Google Scholar]
- 29.Ahokas JT, Davies C, Ravenscroft PJ. Simultaneous analysis of disopyramide and quinidine in plasma by high-performance liquid chromatography. J Chromatogr. 1980;183:65–71. doi: 10.1016/s0378-4347(00)81399-7. [DOI] [PubMed] [Google Scholar]
- 30.Williams KM. Molecular asymmetry and its pharmacological consequences. Adv Pharmacol. 1991;22:57–135. doi: 10.1016/s1054-3589(08)60033-2. [DOI] [PubMed] [Google Scholar]
- 31.Kavanagh KM, Wyse G, Mitchell LB, Gilhooly T, Gillis AM, Duff HJ. Contribution of quinidine metabolites to electrophysiologic responses in human subjects. Clin Pharmacol Ther. 1989;46:352–358. doi: 10.1038/clpt.1989.150. [DOI] [PubMed] [Google Scholar]
- 32.TDx/TDx, FLx Systems Assays Manual. Abbott Park, Illinois: Abbott Laboratories; 1994. [Google Scholar]
- 33.Sokolow M, Ball RE. Factors influencing conversion of chronic atrial fibrillation with special reference to serum quinidine concentration. Circulation. 1956;14:568–583. doi: 10.1161/01.cir.14.4.568. [DOI] [PubMed] [Google Scholar]
- 34.Gerhardt RE, Knouss RF, Thyrum PT, et al . Quinidine excretion in aciduria and alkaluria. Ann Intern Med. 1969;71:927. doi: 10.7326/0003-4819-71-5-927. [DOI] [PubMed] [Google Scholar]
- 35.Leahey EB, Reiffel JA, Drusin RE, et al . Interaction between quinidine and digoxin. JAMA. 1978;240:533. [PubMed] [Google Scholar]
- 36.Bussey HI. The influence of quinidine and other agents on digitalis glycosides. Am Heart J. 1982;104:289. doi: 10.1016/0002-8703(82)90205-8. [DOI] [PubMed] [Google Scholar]
- 37.Bussey HI. Update on the influence of quinidine and other agents on digitalis glycosides. Am Heart J. 1984;107:143. doi: 10.1016/0002-8703(84)90148-0. [DOI] [PubMed] [Google Scholar]
- 38.Hinderling PH, Garrett ER. Pharmacokinetics of the antiarrhythmic disopyramide in healthy humans. J Pharmacokin Biopharmaceut. 1976;4:199–230. doi: 10.1007/BF01063614. [DOI] [PubMed] [Google Scholar]
- 39.Baines MW, Davies JE, Keilett DN, Munt PL. Some pharmacological effects of disopyramide and a metabolite. J Int Med Res. 1976;4(Suppl 1):5–7. [PubMed] [Google Scholar]
- 40.Zema M. Serum drug concentrations and adverse effects in cardiac patients after administration of a new controlled-release disopyramide preparation. Ther Drug Monit. 1984;6:192–198. doi: 10.1097/00007691-198406000-00011. [DOI] [PubMed] [Google Scholar]
- 41.Le Corre P, Gibassier D, Sado P, Le Verge R. Stereoselective metabolism and pharmacokinetics of disopyramide enantiomers in humans. Drug Metab Dis. 1988;16:858–864. [PubMed] [Google Scholar]
- 42.Giacomini KM, Nelson WL, Pershe RA, Valdivieso L, Turner-Tamiyasu Blaschke TF. In vivo interaction of the enantiomers of disopyramide in human subjects. J Pharmacokinet Biopharmaceut. 1986;14:335–338. doi: 10.1007/BF01059195. [DOI] [PubMed] [Google Scholar]
- 43.Vurke TR, Jr, Nelson WL. Resolution, absolute configuration, and antiarrhythmic properties of the enantiomers of disopyramide. J Med Chem. 1980;23:1044–1048. doi: 10.1021/jm00183a015. [DOI] [PubMed] [Google Scholar]
- 44.Le Corre P, Gibassier D, Sado P, Le Verge R. Simultaneous assay of disopyramide and monodesisopropyldisopyramide enantiomers in biological samples by liquid chromatography. J Chromatogr. 1988;424:424–429. doi: 10.1016/s0378-4347(00)81123-8. [DOI] [PubMed] [Google Scholar]
- 45.Giacomini KM, Cox BM, Blaschke TF. Comparative anticholinergic potencies of R-and S-disopyramide in longitudinal muscle strips from guinea pig ileum. Life Sci. 1980;27:1191–1197. doi: 10.1016/0024-3205(80)90471-3. [DOI] [PubMed] [Google Scholar]
- 46.Niarchos AP. Disopyramide: serum level and arrhythmia conversion. Am Heart J. 1976;92:57–64. doi: 10.1016/s0002-8703(76)80403-6. [DOI] [PubMed] [Google Scholar]
- 47.Meffin PJ, Robert EW, Winkle RA, et al . Role of concentration-dependent plasma protein binding in disopyramide disposition. J Pharmacokin Biopharmaceut. 1979;7:29–46. doi: 10.1007/BF01059439. [DOI] [PubMed] [Google Scholar]
- 48.David BM, Madsen BW, Ilett KF. Plasma bin Pharmacol. 1980;9:614–618. doi: 10.1111/j.1365-2125.1980.tb01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thibonniew M, Holford NHG, Upton RA, Blume CD, Williams RL. Pharmacokinetic–Pharmacodynamic analysis of unbound disopyramide directly measured in serial plasma samples in man. J Pharmacokinet Biopharmaceut. 1984;12:559–573. doi: 10.1007/BF01059552. [DOI] [PubMed] [Google Scholar]
- 50.Duff HJ, Mitchell LB, Nath CF, Manyari DE, Baynton R, Wyse DG. Concentration-response relationships of disopyramide in patients with ventricular tachycardia. Clin Pharmacol Ther. 1989;45:542–547. doi: 10.1038/clpt.1989.70. [DOI] [PubMed] [Google Scholar]
- 51.Routledge PA, Stargel WW, Wagner GS, Shand DG. Increased alpha-1-acid glycoprotein and lidocaine disposition in myocardial infarction. Ann Int Med. 1980;93:701–704. doi: 10.7326/0003-4819-93-5-701. [DOI] [PubMed] [Google Scholar]
- 52.Cunningham JL, Shen DD, Shudo I, et al . The effect of urine pH and plasma protein binding on renal clearance of disopyramide. Clin Pharmacokinet. 1977;2:373–383. doi: 10.2165/00003088-197702050-00004. [DOI] [PubMed] [Google Scholar]
- 53.Darim A, Nissen C, Azarnoff DL. Clinical pharmacokinetics of disopyramide. J Pharmacokinet Biopharm. 1982;10:465. doi: 10.1007/BF01059032. [DOI] [PubMed] [Google Scholar]
- 54.Giardina EGV, Dreyfuss J, Bigger JT, et al . Metabolism of procainamide in normal and cardiac subjects. Clin Pharmacol Ther. 1976;19:339–351. doi: 10.1002/cpt1976193339. [DOI] [PubMed] [Google Scholar]
- 55.Roden DM, Reele SB, Higgins SB, et al . Antiarrhythmic efficacy, pharmacokinetics and safety of N-acetyl-procainamide in human subjects: comparison with procainamide. Am J Cardiol. 1980;46:463–468. doi: 10.1016/0002-9149(80)90016-8. [DOI] [PubMed] [Google Scholar]
- 56.Reidenberg MM, Drayer DE, Levy M, Warner H. Polymorphic acetylation of procainamide in man. Clin Pharmacol Ther. 1975;17:722–30. doi: 10.1002/cpt1975176722. [DOI] [PubMed] [Google Scholar]
- 57.Braden GL, Fitzgibbons JP, Germain MJ, Ledewitz HM. Hemoperfusion for treatment of N-acetylprocainamide intoxication. Ann Intern Med. 1986;105:64–65. doi: 10.7326/0003-4819-105-1-64. [DOI] [PubMed] [Google Scholar]
- 58.Kastrup J, Petersen P, Dejgard A, Angelo HR, Hilsted J. Intravenous lidocaine infusion—a new treatment of chronic painful diabetic neuropathy? Pain. 1987;28:69–75. doi: 10.1016/0304-3959(87)91061-X. [DOI] [PubMed] [Google Scholar]
- 59.Swerdlow M. The use of local anaesthetics for the relief of chronic pain. Pain Clinic. 1988;2:3–6. [Google Scholar]
- 60.Glazer S, Portenoy RK. Systemic local anesthetics in pain control. J Pain Sympt. 1991;6:30–39. doi: 10.1016/0885-3924(91)90069-g. [DOI] [PubMed] [Google Scholar]
- 61.Stenson RE, Constantino RT, Harrison DC. Interrelationships of hepatic blood flow, cardiac output, and blood levels of lidocaine in man. Circulation. 1971;43:205–211. doi: 10.1161/01.cir.43.2.205. [DOI] [PubMed] [Google Scholar]
- 62.Blumer J, Strong JM, Atkinson AJ. The convulsant potency of lidocaine and its N-dealkylated metabolites. J Pharmacol Exp Ther. 1978;180:31–36. [PubMed] [Google Scholar]
- 63.Chen Y, Potter JM. Fluorescence polarization immunoassay and HPLC assays compared for measuring monoethylglycinexylidide in liver-transplant patients. Clin Chem. 1992;38:2426–2430. [PubMed] [Google Scholar]
- 64.Thomson AH, Kelman AW, DeVane PJ, Hillis WS, Whiting B. Changes in lignocaine disposition during long-term infusion in patients with acute ventricular arrrythmias. Ther Drug Monit. 1987;9:283–291. doi: 10.1097/00007691-198709000-00006. [DOI] [PubMed] [Google Scholar]
- 65.Halkin H, Meffin P, Melmon KL, Rowland M. Influence of congestive heart failure on blood levels of lidocaine and its active mono-de-ethylated metabolite. Clin Pharmacol Ther. 1975;17:669–676. doi: 10.1002/cpt1975176669. [DOI] [PubMed] [Google Scholar]
- 66.Prescott LF, Pottage A, Clements JA. Absorption, distribution and elimination of mexiletine. Postgrad Med J. 1977;53:50–55. [PubMed] [Google Scholar]
- 67.Haselbarth V, Doevendans J, Wolf M. Kinetics and bioavailability of mexiletine in healthy subjects. Clin Pharmacol Ther. 1981;29:729–736. doi: 10.1038/clpt.1981.103. [DOI] [PubMed] [Google Scholar]
- 68.Campbell NPS, Pantridge JF, Adgey AAJ. Long-term oral antiarrhythmic therapy with mexiletine. Br Heart J. 1978;40:796–801. doi: 10.1136/hrt.40.7.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pringle T, Fox J, McNeill JA, et al . Dose independent pharmacokinetics of mexiletine in healthy volunteers. Br J Clin Pharmacol. 1986;21:319–321. doi: 10.1111/j.1365-2125.1986.tb05196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jarzabek JI. Analyte of the month. AACC In-Service Training Continuing Ed. 1991;12:3–4. [Google Scholar]
- 71.Grech-Belanger O, Turgeon J, Gilbert M. Steroselective disposition of mexiletine in man. Br J Clin Pharmacol. 1986;21:481–487. doi: 10.1111/j.1365-2125.1986.tb02829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Igwemezie L, Kerr CR, McErlane KM. The pharmacokinetics of the enantiomers of mexiletine in humans. Xenobiotica. 1989;19:677–682. doi: 10.3109/00498258909042305. [DOI] [PubMed] [Google Scholar]
- 73.Turgeon J, Uprichard ACG, Belanger PM, Harron DQG, Grech-Belanger O. Resolution and electrophysiological effects of mexiletine enantiomers. J Pharm Pharmacol. 1991;43:630–635. doi: 10.1111/j.2042-7158.1991.tb03552.x. [DOI] [PubMed] [Google Scholar]
- 74.McErlane KM, Igwemezie L. Stereoselective analysis of the enantiomers of mexiletine by high-performance liquid chromatography using fluorescence detection and study of their stereoselective disposition in man. J Chromatogr. 1987;415:335–346. doi: 10.1016/s0378-4347(00)83225-9. [DOI] [PubMed] [Google Scholar]
- 75.Hodges M, Haugland JM, Conard GJ, Carlson GL, Frost JW, Ober RE. Human plasma pharmacokinetics of flecainide acetate (R-818), a new antiarrhythmic, following single oral and intravenous doses. Clin Pharmacol Ther. 1979;25:218. [Google Scholar]
- 76.Hodges M, Haugland JM, Granrud G, et al . Suppression of ventricular ectopic depolarizations by flecainide acetate, a new antiarrhythmic drug. Circulation. 1982;65:879–885. doi: 10.1161/01.cir.65.5.879. [DOI] [PubMed] [Google Scholar]
- 77.Lui HK, Lee G, Dietrich P, et al . Flecainide-induced QT prolongation and ventricular tachycardia. Am Heart J. 1982;103:567–569. doi: 10.1016/0002-8703(82)90346-5. [DOI] [PubMed] [Google Scholar]
- 78.Spivack C, Gottlieb S, Miura DS, Somberg JC. Flecainide toxicity. Am J Cardiol. 1984;53:329–330. doi: 10.1016/0002-9149(84)90449-1. [DOI] [PubMed] [Google Scholar]
- 79.Guehler J, Gornick CC, Tobler HG, et al . Electrophysiologic effects of flecainide acetate and its major metabolites in the canine heart. Am J Cardiol. 1985;55:807–812. doi: 10.1016/0002-9149(85)90161-4. [DOI] [PubMed] [Google Scholar]
- 80.Johnson JD, Carlson GL, Fox JM, Miller AM, Chang Chang. SF, Conard GJ. Quantitation of flecainide acetate, a new antiarrhythmic agent, in biological fluids by gas chromatography with electron-capture detection. J Pharmaceut Sci. 1984;73:1469–1471. doi: 10.1002/jps.2600731037. [DOI] [PubMed] [Google Scholar]
- 81.Ray J. Flecainide: What are we measuring? Ther Drug Monit. 1991;13:86. [PubMed] [Google Scholar]
- 82.Lie-A, Huen L, Stuurman RM, Ijdenberg FN, Dingma JH, Meijer DKF. High-performance liquid chromatographic assay of flecainide and its enantiomers in serum. Ther Drug Monit. 1989;11:708–711. doi: 10.1097/00007691-198911000-00017. [DOI] [PubMed] [Google Scholar]
- 83.Alessi-Severini S, Jamali F, Pasutto FM, Coutts RT, Gulamhusein S. High-Performance liquid chromatographic determination of the enantiomers of flecainide in human plasma and urine. J Pharm Sci. 1990;79:257–260. doi: 10.1002/jps.2600790316. [DOI] [PubMed] [Google Scholar]
- 84.Turgeon J, Kroemer HK, Prakash C, Blair IA. Stereoselective determination of flecainide in human plasma by high-performance liquid chromatography with florescence detection. J Pharm Sci. 1990;79:91–95. doi: 10.1002/jps.2600790202. [DOI] [PubMed] [Google Scholar]
- 85.Kroemer HK, Turgeon J, Parker RA, Roden DM. Flecainide enantiomers: Disposition in human subjects and electrophysiologic actions in vitro. Clin Pharmacol Ther. 1989;46:584–590. doi: 10.1038/clpt.1989.189. [DOI] [PubMed] [Google Scholar]
- 86.Lie-A-, Huen L, Van den Akker J, Den Hertog A, Meijer DKF. The action of flecainide acetate and its enantiomers on mammalian non-myelinated nerve fibres. Pharm Wkblt (Sci ) 1989;11:92–94. doi: 10.1007/BF02110256. [DOI] [PubMed] [Google Scholar]
- 87.Smallwood JK, Robertson DW, Steinberg MI. Electrophysiological effects of flecainide enantiomers in canine Purkinje fibres. Naunyn-Schmiedeberg’s Arch Pharmacol. 1989;339:625–629. doi: 10.1007/BF00168654. [DOI] [PubMed] [Google Scholar]
- 88.Gross AS, Mikus G, FisherC FisherC, Hertrampf R, Gundert-Remy U, Eichelbaum M. Stereoselective disposition of flecainide in relation to the sparteine/debrisoquine metaboliser phenotype. Br J Clin Pharmacol. 1989;28:555–556. doi: 10.1111/j.1365-2125.1989.tb03542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brigerdotter UM, Wong W, Turgeon J, Roden DM. Stereoselective genetically–determined interaction between chronic flecainide and quinidine in patients with arrhythmias. Br J Clin Pharmacol. 1992;33:275–280. doi: 10.1111/j.1365-2125.1992.tb04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mikus G, Gross AS, Beckmann J, Gertrampf R, Gundert-Remy U, Eichelbaum M. The influence of the sparteine/debrisoquin phenotype on the disposition of flecainide. Clin Pharmacol Ther. 1989;45:562–567. doi: 10.1038/clpt.1989.73. [DOI] [PubMed] [Google Scholar]
- 91.Dukes ID, Vaughan Williams EM. The multiple modes of action of propafenone. Eur Heart J. 1984;5:115–125. doi: 10.1093/oxfordjournals.eurheartj.a061621. [DOI] [PubMed] [Google Scholar]
- 92.Somberg JC, Tepper D, Landau S. Propafenone: a new antiarrhythmic agent. Am Heart J. 1988;115:1274–1279. doi: 10.1016/0002-8703(88)90021-x. [DOI] [PubMed] [Google Scholar]
- 93.Hollman M, Hege H, Brode E, et al. Pharmacokinetic and metabolic studies of propafenone in volunteers. In: Schlepper M, Molsson B, editors. Cardiac arrhythmias. Springer, Berlin Heidelberg New York: Proc First Int Rythmonorm Congress; 1983. pp. 125–132. [Google Scholar]
- 94.Connolly SJ, Kates RE, Lebsack CS, et al . Clinical efficacy and electrophysiology of oral propafenone for ventricular tachycardia. Am J Cardiol. 1983;52:1208–1213. doi: 10.1016/0002-9149(83)90575-1. [DOI] [PubMed] [Google Scholar]
- 95.Salerno DM, Granrud G, Sharkey P, et al . A controlled trial of propafenone for treatment of frequent and repetitive ventricular premature complexes. Am J Cardiol. 1984;53:77–83. doi: 10.1016/0002-9149(84)90687-8. [DOI] [PubMed] [Google Scholar]
- 96.Somberg JC. Vaughan Williams EM, Campbell TJ. Antiarrhythmic Drugs. Berlin: Springer-Verlag; 1989. Clinical use of Class Ic antiarrhythmic drugs; pp. 235–279. [Google Scholar]
- 97.Hiji JT, Duff JH, Burgess ED. Clinical pharmacokinetics of propafenone. Clin Pharmacokinet. 1991;21:1–10. doi: 10.2165/00003088-199121010-00001. [DOI] [PubMed] [Google Scholar]
- 98.Siddoway LA, Roden DM, Woosley RL. Clinical pharmacology of propafenone. Pharmacokinetics, metabolism and concentration-response relations. Am J Cardiol. 1984;54:9D–12D. doi: 10.1016/s0002-9149(84)80278-7. [DOI] [PubMed] [Google Scholar]
- 99.Siddoway LA, Thompson KA, McAllister CB, et al . Polymorphism of propafenone metabolism and disposition in man: Clinical and pharmocokinetic consequences. Circulation. 1987;75:785–791. doi: 10.1161/01.cir.75.4.785. [DOI] [PubMed] [Google Scholar]
- 100.Stoschietzky K, Klein W, Stark G, et al . Different stereoselective effects of R-and S-propafenone: Clinical pharmacology, electrophysiologic and radioligand binding studies. Clin Pharmacol Ther. 1990;47:740–744. doi: 10.1038/clpt.1990.102. [DOI] [PubMed] [Google Scholar]
- 101.Koytchev R, Alken RG, Mayer O, Bohm R, Ellrich A, Waldner-Kolblin RG. The bioequivalence of two oral propafenone preparations. Arzneimittel Forsch. 1995;45:542–545. [PubMed] [Google Scholar]
- 102.Oti-Amoako K, Vozeh S, Ha Huy-Riem Follath F. The relative potency of major metabolites and enantiomers of propafenone in an experimental reperfusion arrhythmia model. J Cardiovasc Pharmacol. 1990;15:75–81. doi: 10.1097/00005344-199001000-00012. [DOI] [PubMed] [Google Scholar]
- 103.Kroemer HK, Fromm MF, Buhl K, Terefe H, Blaschke G, Eichelbaum M. An enantiomer–enantiomer interaction of (S)-and (R)-propafenone modifies the effect of racemic drug therapy. Circulation. 1994;89:2396–2400. doi: 10.1161/01.cir.89.5.2396. [DOI] [PubMed] [Google Scholar]
- 104.Siddoway LA, McAlliser CB, Want T, et al . Polymorphic oxidative metabolism of propafenone in man. Circulation. 1983;68(Suppl: (III)):64–66. [Google Scholar]
- 105.Haefeli EW, Vozeh S, Ha HR, Fallath F. Comparison of the pharmacodynamic effects of intravenous and oral propafenone. Clin Pharmacol Ther. 1990;48:245–254. doi: 10.1038/clpt.1990.146. [DOI] [PubMed] [Google Scholar]
- 106.Held PH, Yusuf S. Effects of beta-blockers and calcium channel blockers in acute myocardial infarction. Eur Heart J. 1993;14((Supp F)):18–25. doi: 10.1093/eurheartj/14.suppl_f.18. [DOI] [PubMed] [Google Scholar]
- 107.Campbell TJ. Beta-blockers for ventricular arrhythmia: Have we underestimated their value? Aust N Z J Med. 1996;26:689–696. doi: 10.1111/j.1445-5994.1996.tb02941.x. [DOI] [PubMed] [Google Scholar]
- 108.Nademanee K, Field G, Hendrickson J, Singh PN, Singh BN. Electrophysiologic and antiarrhythmic effects of sotalol in patients with life-threatening ventricular tachyarrhythmias. Circulation. 1985;72:555–564. doi: 10.1161/01.cir.72.3.555. [DOI] [PubMed] [Google Scholar]
- 109.Cobbe SM. Sotalol. In: Vaughan Williams EM, Campbell TJ, editors. Antiarrhythmic drugs. Berlin: Springer-Verlag; 1989. pp. 365–387. [Google Scholar]
- 110.Campbell TJ. Class III antiarrhythmic action: the way forward. Med J Aust. 1993;158:732–733. doi: 10.5694/j.1326-5377.1993.tb121952.x. [DOI] [PubMed] [Google Scholar]
- 111.Soyka L, Witz C, Spangenbert R. Clinical safety profile of sotalol in patients with arrhythmias. Am J Cardiol. 1990;65:74A–81A. doi: 10.1016/0002-9149(90)90207-h. [DOI] [PubMed] [Google Scholar]
- 112.Neuvonen P, Elonen E, Tarssanen L. Sotalol intoxication, two patients with concentration-effect relationships. Acta Pharmacol Toxicol. 1979;45:52–57. doi: 10.1111/j.1600-0773.1979.tb02360.x. [DOI] [PubMed] [Google Scholar]
- 113.Boutagy J, Shenfield GM. Simplified procedure for the determination of sotalol in plasma by high-performance liquid chromatography. J Chromatogr. 1991;565:523–528. doi: 10.1016/0378-4347(91)80420-h. [DOI] [PubMed] [Google Scholar]
- 114.Gluth WP, Sorgel F, Gluth B, Braun J, Geldmacher von Mallinckrodt M. Determination of sotalol in human body fluids for pharmacokinetic and toxicokinetic studies using high-performance liquid chromatography. Arzneimittel Forsch. 1988;38:408–411. [PubMed] [Google Scholar]
- 115.Barek MJ, Vekshteyn M, Boarman MP, Gallo DG. Liquid chromatographic determination of sotalol in plasma and urine employing solid-phase extraction and fluorescence detection. J Chromatogr. 1987;421:309–318. doi: 10.1016/0378-4347(87)80410-3. [DOI] [PubMed] [Google Scholar]
- 116.Karkkainen S. High-performance liquid chromatographic determination of sotalol in biological fluids. J Chromatogr. 1984;336:313–319. [PubMed] [Google Scholar]
- 117.Fiset C, Philippon F, Gilbert M, Turgeon J. Stereoselective disposition of (±)-sotalol at steady-state conditions. Br J Clin Pharmac. 1993;36:75–77. doi: 10.1111/j.1365-2125.1993.tb05896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Poirier JM, Jaillon P, Lecocq B, Lecocq V, Ferry A, Ghevmol G. The pharmacokinetics of d-sotalol and d,l-sotalol in healthy volunteers. Eur J Clin Pharmacol. 1990;38:579–582. doi: 10.1007/BF00278585. [DOI] [PubMed] [Google Scholar]
- 119.Somani P, Watson DL. Antiarrhythmic activity of the dextro-and levo-rotatory isomers of 4-(2-isopropylamino-1-hydroxyethyl) methanesulphonamide. J Pharmacol Exp Ther. 1968;164:317–325. [PubMed] [Google Scholar]
- 120.Singh BN, Sarma JSM. Amiodarone and amiodarone derivatives. In: Singh BN, Dzau VJ, Vanhoutte PlM, Woosley RL, editors. Cardiovascular Pharmacology and Therapeutics. New York: Churchill Livingstone: 1994. pp. 689–710. [Google Scholar]
- 121.Bauman JL, Berk SI, Hariman RJ, et al . Amiodarone for sustained ventricular tachycardia: efficacy, safety and factors influencing long-term outcome. Am Heart J. 1987;114:436–444. doi: 10.1016/0002-8703(87)90549-7. [DOI] [PubMed] [Google Scholar]
- 122.Pallandi RT, Campbell TJ. Resting and rate-dependent depression of Vmax of guinea-pig ventricular action potentials by amiodarone and desethyl-amiodarone. Br J Pharmacol. 1987;92:97–103. doi: 10.1111/j.1476-5381.1987.tb11300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Holt DW, Tucker GT, Jackson PR, Storey GCA. Amiodarone pharmacokinetics. Am Heart J. 1983;106:840–847. doi: 10.1016/0002-8703(83)90006-6. [DOI] [PubMed] [Google Scholar]
- 124.Mitchell LB, Wyse G, Gillis AM, Duff HJ. Electropharmacology of amiodarone therapy initiation. Time courses of onset of electrophysiologic and antiarrhythmic effects. Circulation. 1989;80:34–42. doi: 10.1161/01.cir.80.1.34. [DOI] [PubMed] [Google Scholar]
- 125.Rotmensch HH, Belhassen B, Swanson BN, et al . Steady-state serum amiodarone concentrations: relationships with antiarrhythmic efficacy and toxicity. Ann Int Med. 1984;101:462–469. doi: 10.7326/0003-4819-101-4-462. [DOI] [PubMed] [Google Scholar]
- 126.Counihan PJ, McKenna WJ. Low-dose amiodarone for the treatment of arrhythmias in hypertrophic cardiomyopathy. J Clin Pharm. 1989;29:436–438. doi: 10.1002/j.1552-4604.1989.tb03357.x. [DOI] [PubMed] [Google Scholar]
- 127.Moysey JO, Jaggarao NSV, Grundy EN, et al . Amiodarone increases plasma digoxin concentration. Br Med J. 1981;282:272–275. doi: 10.1136/bmj.282.6260.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weir SJ, Ueda CT. Rapid liquid chromatographic assay for the determination of amiodarone and its N-deethyl metabolite in plasma, urine and bile. J Pharm Sci. 1985;74:460–465. doi: 10.1002/jps.2600740418. [DOI] [PubMed] [Google Scholar]
- 129.Plomp TA, Engels M, Robles Medina EO, Maes RA. Simultaneous determination of amiodarone and its major metabolite desethylamiodarone in plasma, urine and tissues by high-performance liquid chromatography. J Chromatogr. 1983;273:379–392. doi: 10.1016/s0378-4347(00)80958-5. [DOI] [PubMed] [Google Scholar]
- 130.Kannan R, Miller S, Perez V, Singh BN. Sensitive method for the measurement of amiodarone and desethylamiodarone in serum and tissue and its application to disposition studies. J Chromatogr. 1987;385:225–232. doi: 10.1016/s0021-9673(01)94634-5. [DOI] [PubMed] [Google Scholar]
- 131.Pollak PT. A systematic review and critical comparison of internal standards for the routine liquid chromatographic assay of amiodarone and desethylamiodarone. Ther Drug Monit. 1996;18:168–178. doi: 10.1097/00007691-199604000-00011. [DOI] [PubMed] [Google Scholar]
- 132.Nademanee K, Kannan R, Hendrickson J, et al . Amiodarone-digoxin interactions: Clinical significance, time course of development, potential pharmacokinetic mechanisms and therapeutic implications. J Am Coll Cardiol. 1984;4:111–116. doi: 10.1016/s0735-1097(84)80327-7. [DOI] [PubMed] [Google Scholar]
- 133.Denster PE, White NW Jr, Hanson CD. Pharmacokinetic evaluation of the digoxin–amiodarone interaction. J Am Coll Cardiol. 1985;5:108–112. doi: 10.1016/s0735-1097(85)80091-7. [DOI] [PubMed] [Google Scholar]
- 134.Serlin MJ, Sibeon RG, Green GJ. Dangers of amiodarone and anticoagulant treatment. Br Med J. 1981;283:57–58. doi: 10.1136/bmj.283.6283.58-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pexid Product Summary for Physicians. Cincinnati, Ohio: Merrel Dow Pharmaceuticals Inc.; 1982. [Google Scholar]
- 136.Lockhart JDF, Masheter HC. Report of a multicentre monitored release study of perhexiline maleate in the prevention of angina pectoris. Br J Clin Pract. 1976;30:172. [PubMed] [Google Scholar]
- 137.Horowitz JD, Morris PM, Drummer O, Goble AJ, Louis WJ. Saturable metabolism of perhexiline maleate: Correlations with long term toxicity. Am J Cardiol. 1981;47:399. [Google Scholar]
- 138.Singlas E, Goufet MAS, Imon P. Pharmacokinetics of perhexiline maleate in angina patients with and without peripheral neuropathy. Eur J Clin Pharmacol. 1978;14:195–201. doi: 10.1007/BF02089960. [DOI] [PubMed] [Google Scholar]
- 139.Laplane D, Bousser MG, Bouche P, Touboul PJ. Perhexiline maleate. 1978. Peripheral neuropathies caused by perhexiline maleate; pp. 89–96. Proceedings of a Symposium. Amsterdam, Excerpta Medica.; pp. . [PubMed] [Google Scholar]
- 140.Vaughan Williams EM. London: Academic Press; 1980. Antiarrhythmic action and the puzzle of perhexiline. [Google Scholar]
- 141.Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 142.Horowitz JK, Sia STB, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate treatment for severe angina pectoris — correlations with pharmacokinetics. Int J Cardiol. 1986;13:219–229. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 143.Cole PL, Beamer AD, McGowan N, et al. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990;81:1260–1270. doi: 10.1161/01.cir.81.4.1260. [DOI] [PubMed] [Google Scholar]
- 144.Horowitz JD, Morris PM, Drummer OH, Goble AJ, Louis WJ. High pressure liquid chromatographic assay of perhexiline maleate in plasma. J Pharmacol Sci. 1981;70:320–322. doi: 10.1002/jps.2600700325. [DOI] [PubMed] [Google Scholar]
- 145.Amoh AG, Gould BJ, Parke DV. Single-dose pharmacokinetics of perhexiline administered orally to humans. J Chromatogr. 1984;305:401–409. doi: 10.1016/s0378-4347(00)83354-x. [DOI] [PubMed] [Google Scholar]
- 146.Cooper RG, Harper G, Price AH, Evans DA, Lockhart JD. Simultaneous determination of perhexiline and its monohydroxy metabolites in biological fluids by gas chromatography–electron-capture detection. J Chromatogr. 1986;381:305–314. doi: 10.1016/s0378-4347(00)83596-3. [DOI] [PubMed] [Google Scholar]
- 147.Gould BJ, Amoah AGB, Parke DV. Stereoselective pharmacokinetics of perhixiline. Xenobiotica. 1986;16:491–502. doi: 10.3109/00498258609050254. [DOI] [PubMed] [Google Scholar]
- 148.Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JD. Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. Br Med J. 1982;284:295–299. doi: 10.1136/bmj.284.6312.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Siebert W. Perhexiline—stable plasma concentrations with formulation change. Aust NZ J Med. 1996;26:707–708. doi: 10.1111/j.1445-5994.1996.tb02945.x. [DOI] [PubMed] [Google Scholar]
- 150.Morris RG, Frewin DB, Taylor WB, Glistak Ml, Lehmann DR. The effect of renal and hepatic impairment and of spironolactone on digoxin immunoassays. Eur J Clin Pharmacol. 1988;34:233–239. doi: 10.1007/BF00540949. [DOI] [PubMed] [Google Scholar]
- 151.Natowicz M, Shaw L. Digoxin assay anomalies due to digoxin-specific fab immunotherapy. Ann Pharmacother. 1991;25:739–741. doi: 10.1177/106002809102500707. [DOI] [PubMed] [Google Scholar]
- 152.Kampa IS. The effect of digibind on various digoxin immunoassays. AACC In-Service Training Continuing Ed. 1993;14:183–186. [Google Scholar]
- 153.Miller JJ, Straub RW Valdes R Jr Analytical performance of a monoclonal digoxin assay with increased specificity on the ACS. 180. Ther Drug Monit. 1996;18:65–72. doi: 10.1097/00007691-199602000-00011. [DOI] [PubMed] [Google Scholar]
- 154.Goto A, Yamada K, Yagi N, Yoshioka M, Sugimoto T. Physiology and pharmacology of endogenous digitalis-like factors. Pharmacol Rev. 1992;44:377–399. [PubMed] [Google Scholar]
- 155.Loucari-Yiannakou E, Yiannakou L, Souvatzooglou A, Diamandis EP. Radioimmunoassay of digoxin in serum using monoclonal antibodies and assessment of interference by digoxin-like immunoreactive substances. Ther Drug Monit. 1990;12:195–200. doi: 10.1097/00007691-199003000-00015. [DOI] [PubMed] [Google Scholar]
- 156.Gault H, Vasdev S, Vlasses P, Longerich L, Dawe M. Interpretation of serum digoxin values in renal failure. Clin Pharmacol Ther. 1986;39:530–536. [PubMed] [Google Scholar]
- 157.Ray JE, Crisan D, Howrie D. Digoxin-like immunoreactivity in serum from neonates and infants reduced by centrifugal ultrafiltration and fluorescence polarization immunoassay. Clin Chem. 1991;37:94–98. [PubMed] [Google Scholar]
- 158.Woodcock BG, Rietbrock N. Digitalis—quinidine interactions. TIPS. 1997;3:118–122. [Google Scholar]
- 159.Su SF, Huang JD. Inhibition of the intestinal digoxin absorption and exsorption by quinidine. Drug Metab Disp. 1996;24:142–147. [PubMed] [Google Scholar]
- 160.Charuk JH, Loo TW, Clarke DM, Reithmeier RA. Interaction of rat kidney P-glycoprotein with a urinary component and various drugs including cyclosporin A. Am J Physiol. 1994;266:75. doi: 10.1152/ajprenal.1994.266.1.F66. F 66-F. [DOI] [PubMed] [Google Scholar]
- 161.Hori R, Okamuri N, Aiba T, Tanigawara Y. Role of P-glycoprotein in renal tubular secretion of digoxin in the isolated perfused rat kidney. J Pharmacol Exp Ther. 1993;266:1620–1625. [PubMed] [Google Scholar]