

Abstract
Tuberous sclerosis complex (TSC) is a genetic disorder caused by mutations in one of two tumor suppressor genes, TSC1 and TSC2. Here, we show that absence of Drosophila Tsc1/2 leads to constitutive dS6K activation and inhibition of dPKB, the latter effect being relieved by loss of dS6K. In contrast, the dPTEN tumor suppressor, a negative effector of PI3K, has little effect on dS6K, but negatively regulates dPKB. More importantly, we demonstrate that reducing dS6K signaling rescues early larval lethality associated with loss of dTsc1/2 function, arguing that the S6K pathway is a promising target for the treatment of TSC.
Keywords: growth, TSC, S6K, TOR, PTEN, PKB
Hereditary cancers have revealed the existence of a number of tumor suppressor genes that function to control cell proliferation and maintain normal tissue homeostasis (Macleod 2000). Two tumor suppressors have been implicated in the PI3K-signaling pathway, PTEN (Cantley and Neel 1999) and more recently, a complex composed of two proteins, hamartin (TSC1) and tuberin (TSC2; Montagne et al. 2001). The lipid phosphatase PTEN, which constrains PI3K signaling by dephosphorylating its product phosphatidylinositol 3,4,5-trisphosphate (PIP3), is found mutated in a number of cancers (Cantley and Neel 1999). Mutations in either TSC1 or TSC2 are associated with widespread medically distinct tumors of the brain, eyes, skin, heart, lungs, and kidneys (Young and Povey 1998). TSC1 and TSC2 contain putative coiled-coil domains, with TSC1 having a predicted transmembrane domain and TSC2 a region similar to that of small-GTPase-activating protein (GAP) domains (Montagne et al. 2001). Although little is known concerning the growth regulatory targets of TSC1 and TSC2, genetic studies in Drosophila (Potter et al. 2001; Tapon et al. 2001) have led recently to the hypothesis that dTsc1/2 acts as a negative effector of dS6K or of a dS6K target (Potter et al. 2001). In these models, dS6K was placed as a downstream effector of dPI3K, via dPKB (Potter et al. 2001). However, dS6K and dPI3K/dPKB appear to reside on parallel growth-promoting pathways rather than functioning in a linear-signaling cascade (Radimerski et al. 2002). Seemingly consistent with these findings, in Drosophila devoid of dPTEN, dPKB appears to be the sole critical target activated by elevated PIP3 levels (Stocker et al. 2002).
Here, we use double-stranded RNA mediated interference (dsRNAi) in Drosophila Kc167 cultured cells to demonstrate that dTsc1/2 acts to suppress dS6K activation, whereas dPTEN negatively regulates dPKB activation but has little effect on dS6K activity. Similar findings are obtained in second instar larvae deficient for either dTsc or dPTEN function, whereas overexpression of either dTsc1/2 or dPTEN in second instar larvae selectively inhibits dS6K or dPKB activity, respectively. In addition, loss of dTsc1/2 function in Kc167 cells or in larvae also leads to suppression of dPKB activity, an effect that is relieved by loss of dS6K. More strikingly, we demonstrate that a relative subtle pharmacological or genetic reduction in dS6K signaling is sufficient to rescue larval lethality associated with loss of dTsc function. These latter findings strongly suggest that the S6K pathway is a promising target for pharmaceutical intervention in tuberous sclerosis treatment.
Results and Discussion
To determine whether loss of dTsc1/2 or dPTEN directly affected dS6K activity, each was depleted in Drosophila Kc167 cells by dsRNAi (Clemens et al. 2000). Quantitative Real Time PCR showed that such treatment strongly reduced levels of both transcripts (Fig. 1A). Compared with control cells, depletion of dTsc1 increased dS6K activity and T398 phosphorylation, consistent with the reduced electrophoretic mobility of dS6K (Fig. 1B). These results are in agreement with recent findings in TSC1 null mammalian cells (Kwiatkowski et al. 2002). Insulin treatment of either control cells or dTsc1-depleted cells did not significantly increase these responses beyond that of dTsc1 depletion alone (Fig. 1B), indicating that loss of dTsc function leads to full dS6K activation. RAD001, a rapamycin derivative (Radimerski et al. 2002), blocked dS6K activity in both control and dTsc1-depleted cells treated with insulin (Fig. 1B). However, it was consistently noted that the RAD001 block of insulin-induced dS6K activation was not as strong in dTsc1-depleted cells (Fig. 1B), suggesting that not all the effects of dTsc on dS6K are dependent on the Drosophila target of rapamycin, dTOR. Similar results to those described here were obtained by dTsc2 depletion (Supplementary Fig. 1). In addition, the effects appeared specific, as dTsc1 depletion had no effect on the basal activity of other AGC-kinase family members, such as dPKB or Drosophila atypical PKC (Fig. 1C,D). However, insulin-induced dPKB activation and S505 phosphorylation were repressed in dTsc1-depleted cells as compared with control cells (Fig. 1C), consistent with dS6K acting in a negative feedback loop to dampen dPKB signaling (Haruta et al. 2000). In contrast to loss of dTsc1, depletion of dPTEN had little effect on dS6K activity and T398 phosphorylation, whereas it led to elevated levels of both basal and insulin-stimulated dPKB activity and S505 phosphorylation (Fig. 1, cf. E and F). Thus, loss of dTsc1/2, but not dPTEN, leads to constitutive dS6K activation.
Figure 1.

dS6K is constitutively activated in dTsc- but not dPTEN-depleted Drosophila Kc167 cells. (A) Real-time PCR analysis following dTsc1 and dPTEN dsRNAi. (B) Analysis of dS6K activity following dTsc1 dsRNAi. Co, untreated control; RNAi, dsRNAi targeting dTsc1; +ins, insulin (100 nM) stimulation for 30 min; RAD, 15 min RAD001 (20 nM) pretreatment. (Top panel) In vitro dS6K activity. (Bottom panels) Western blot analysis of dS6K T398 phosphorylation (top) and dS6K band-shift (bottom). (C) Analysis of dPKB activity following dTsc1 dsRNAi treatment. (Top panel) In vitro dPKB activity assay. CT, Crosstide. (Bottom panels) Western blot analysis of dPKB S505 phosphorylation (top) and total dPKB (bottom). (D) Analysis of atypical dPKC activity following dTsc1 dsRNAi treatment. (Top) In vitro dPKC activity assay using myelin basic protein as substrate (MBP). (Bottom) Western blot of total atypical dPKC. (E) Analysis of dPKB activity following dPTEN dsRNAi treatment. Panels are as in C. (F) Analysis of dS6K activity following dPTEN dsRNAi treatment. The top panel is as in B, the bottom panels are Western blot analyses of dS6K T398 phosphorylation (top) and total dS6K (bottom).
To determine whether the findings above could be corroborated in the animal, dS6K activity was measured in extracts of dTsc1, dPTEN, and dS6K null larvae. The results show that dS6K activity in extracts derived from dTsc1 null larvae was strongly increased over that of wild-type larvae, whereas it was slightly increased in larvae lacking dPTEN (Fig. 2A). The opposite was found for dPKB activity, which was strongly repressed in dTsc1 null larvae, and up-regulated in dPTEN-deficient larvae (Fig. 2B). Hence, it cannot be excluded that reduced dPKB activity contributes to larval lethality of dTsc mutants. Given that loss of dTsc function leads to increased dS6K activity, it was reasoned that ectopic expression of dTsc1/2, but not dPTEN, would inhibit dS6K activity. To test this hypothesis, both tumor suppressors were expressed ubiquitously in larvae using the GAL4/UAS system, such that the GAL4 promoter chosen in each case led to developmental arrest at late larval second instar. Extracts from larvae overexpressing dTsc1/2 display strongly reduced dS6K activity, whereas those from dPTEN overexpressing larvae have normal levels of dS6K activity (Fig. 2C). In contrast, dPKB activity is strongly suppressed in dPTEN overexpressing larvae and little affected in extracts from larvae overexpressing dTsc1/2 (Fig. 2D). These data corroborate previous findings that dS6K and dPKB act in parallel signal transduction pathways (Radimerski et al. 2002), and provide compelling evidence that they are negatively controlled by distinct tumor suppressor genes.
Figure 2.

dTsc controls dS6K activity in vivo. (A) Analysis of dS6K activity from second instar larvae lacking either dTsc1 (left) or dPTEN (right) compared with wild-type larvae (y w) and dS6K null larvae. (Top panels) In vitro dS6K kinase assay. (Bottom panels) Western blot detection of dS6K and eIF4E as loading control. (B) Analysis of dPKB activity from second instar dTsc1 or dPTEN null larvae or larvae harboring a kinase inactive dPKB. (Top) In vitro dPKB kinase assay. CT, Crosstide. (Bottom) Western blot detection of dPKB. (C) Analysis of dS6K activity following either ubiquitous dPTEN (da-GAL4>UAS-dPTEN) or dTsc1/2 overexpression (act5C-GAL4>UAS-dTsc1/2) in larvae. Panels are as in A. (D) Analysis of dPKB activity following either ubiquitous dPTEN (da-GAL4>UAS-dPTEN) or dTsc1/2 overexpression (act5C-GAL4>UAS-dTsc1/2) in larvae. Panels are as in B. (E) Analysis of dPKB activity in larvae null for dS6K, null for dTsc1, and null for both dS6K and dTsc1. Panels are as in B. (Bottom) Western blot detection of total dS6K.
Despite the fact that dS6K and dPKB act in parallel signaling pathways, loss of dTsc1/2 function leads to inhibition of dPKB activity (Figs. 1C, 2B; Supplementary Fig. 1B), suggesting cross-talk between the two pathways. Compatible with such a model, recent studies have shown that rapamycin treatment of adipocytes inhibits a negative feedback loop, which normally functions to dampen insulin-induced PKB activation (Haruta et al. 2000). As RAD001 inhibits dS6K activity (Fig. 1B; Radimerski et al. 2002) and increases dPKB activity (Radimerski et al. 2002), it raised the possibility that the effects of dTsc mutants on dPKB are mediated through dS6K. Consistent with this hypothesis, inhibition of dPKB activity due to loss of dTsc function was relieved in the absence of dS6K (Fig. 2E). Similar results were obtained by using dsRNAi in cell culture (see Supplementary Fig. 2). Thus, the suppression of dPKB by loss of dTsc function requires dS6K.
To genetically test the specificity of dTsc1/2 and dPTEN tumor suppressor function, either dTsc1 or dPTEN were removed in cells giving rise to the adult eye structure, by inducing mitotic recombination with the FLP/FRT system under the control of the eyeless promoter (Newsome et al. 2000). In a wild-type genetic background, loss of either dTsc1 or dPTEN within the developing eye causes strong overgrowth of the head (Fig. 3, cf. A–C; Gao et al. 2000; Gao and Pan 2001). Eye overgrowth by removal of dTsc1 is strongly suppressed in a genetic background null for dS6K, as is ommatidia size (Fig. 3, cf. D and E), in agreement with a previous report analyzing double mutant clones of dTsc2 and dS6K in the eye (Potter et al. 2001). In contrast, removal of dPTEN in the eyes of dS6K null flies still induces overgrowth of clones with enlarged ommatidia (Fig. 3, cf. D and F). These findings are supported by results showing that eye overgrowth by removal of dTsc1 is still observed in clones devoid of dPKB function (Potter et al. 2001) and overgrowth by removal of dPTEN is suppressed in a viable dPKB mutant genetic background (Stocker et al. 2002). Thus, dTsc1/2 appears to be specific for the dS6K-signaling pathway, whereas dPTEN antagonizes PI3K signaling to counteract dPKB activation by decreasing PIP3 levels (Stocker et al. 2002).
Figure 3.

Overgrowth in eye imaginal tissues mediated by loss of dTsc, but not by loss of dPTEN, requires dS6K. (A–F) Lateral views of eyes. Clusters of pigmented ommatidia correspond to heterozygous tissue. Absence of red pigment represents homozygous mutant tissue in which mitotic recombination occurred. Relative ommatidia size, X, was determined from either control or mutant tissues as described in Materials and Methods. The value in parenthesis represents relative ommatidia size in the dS6K null background. Genotypes: (A) ey-FLP; FRT82B 3R3.7 P(w+)/+, (B) ey-FLP; FRT82B dTsc1Q87X/FRT82B 3R3.7 P(w+), (C) ey-FLP; dPTEN117 FRT40/P(w+) l(2)2L-3.1 FRT40, (D) dS6Kl1 FRT82B dTsc1Q87X/dS6Kl1 FRT82B 3R3.7 P(w+), (E) eyFLP; dS6Kl1 FRT82B dTsc1Q87X/dS6Kl1 FRT82B 3R3.7 P(w+), (F) ey-FLP; dPTEN117 FRT40/P(w+) l(2)2L-3.1 FRT40; dS6Kl1/dS6Kl1. Bar, 0.25 mm.
As dTsc1/2 loss-of-function overgrowth in clones is suppressed by removing dS6K (Fig. 3E), it was reasoned that reducing increased dS6K activity in dTsc1 loss-of-function larvae (Fig. 2A) might rescue second larval instar lethality. Consistent with this, feeding dTsc1 null larvae low doses of RAD001, which induces a developmental delay of 3 d in wild-type larvae, allowed them to reach late wandering third larval instar (Fig. 4A). The dTsc1 null larvae died shortly after pupation, presumably because wandering third instar larvae stop feeding and thus fail to receive the drug during pupal stages. To circumvent the problem of feeding, we attempted to reduce dS6K signaling by reducing the dosage of the gene. Compared with wild-type pupae (Fig. 4B, pupae 1), dS6K null larvae are significantly reduced in size (Fig. 4B, pupae 2), and lack of one allele of dTsc1 in this background had no significant effect on the dS6K null phenotype (Fig. 4B, pupae 3). Strikingly, the second instar lethality caused by lack of both dTsc1 alleles was rescued to early pupal stages in the dS6K null background (Fig. 4B, pupae 4); however, these larvae were still small and severely delayed in development. In contrast, larvae in which one allele of dS6K had been removed in a dTsc1 null background developed to early pupal stages with little developmental delay, although they were now significantly larger than wild type (Fig. 4B, pupae 5). On the basis of these latter findings, it was reasoned that further reduction of dS6K signaling, but not its abolishment, may allow dTsc1 null animals to develop beyond early pupal stages. To test this in the dTsc1 null background, we used either one allele of dTOR bearing a mutation in the kinase domain alone or in combination with one null allele of dS6K. dTsc1 null larvae bearing one kinase mutant dTOR allele survived with higher frequency to pupae than animals with one null allele of dS6K, with a few emerging as adults (Table 1). However, genetically lowering dS6K signaling further by combining the dTOR and dS6K loss-of function alleles, resulted in more than 60% of animals surviving to the adult stage (Table 1). The rescued females and males were slightly larger than wild-type flies, with overall patterning appearing normal (Fig. 4C). Furthermore, the rescued females were semifertile when crossed to wild-type males, whereas the rescued males were fully fertile when crossed to wild-type females (data not shown). Similarly, animals lacking dTsc2 function (Canal et al. 1998; Ito and Rubin 1999) were rescued to viability (data not shown) by the same genetic approach applied above. Importantly, flies lacking one dS6K allele and bearing one kinase mutant dTOR allele display no obvious mutant phenotype (data not shown). Therefore, lowering but not abolishing dS6K signaling is sufficient to allow development of Drosophila lacking dTsc function.
Figure 4.

Reduction of dS6K signaling rescues lethality caused by loss of dTsc1 function. (A) From left to right: +/+, y w third instar wandering larvae; −/−, dTsc1Q87X/dTsc1Q87X larvae (lethality occurs at late second instar stages); −/−/+, dTsc1Q87X/dTsc1Q87X third instar wandering larvae fed 30 μM RAD001. (B) Dorsal view of pupal cases. Genotypes: (Pupae 1) y w; (pupae 2) dS6Kl1/dS6Kl1; (pupae 3) dS6Kl1/dS6Kl1 dTsc1Q87X; (pupae 4) dS6Kl1 dTsc1Q87X/dS6Kl1 dTsc1Q87X; (pupae 5) dTsc1Q87X/dS6Kl1 dTsc1Q87X. (C) Rescue of dTsc1 loss-of-function lethality by genetic reduction of dTOR/dS6K signaling. (Left) Upper fly, y w female; lower fly, dTOR2L1/+; dS6Kl1 dTsc1Q87X/dTsc1Q87X female. (Right) Upper fly, y w male; lower fly, dTOR2L1/+; dS6Kl1 dTsc1Q87X/dTsc1Q87X male. Bars, 1 mm.
Table 1.
Reduction of dS6K signaling rescues the lethality of the dTsc1 loss-of-function mutation
| Genotype
|
Animals surviving to pupal stage
|
Animals surviving to adult stage
|
|
|
|---|---|---|---|---|
|
0% | 0% | (n = 80) | |
|
45% | 0% | (n = 120) | |
|
82.5% | 18.5% | (n = 114) | |
|
93% | 62% | (n = 125) |
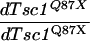
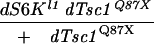
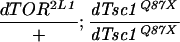
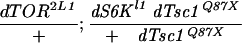
Taken together, our results demonstrate that the tumor suppressor dTsc1/2 is a critical component in controlling dS6K activation. Interestingly, this effect may be dTOR independent, as insulin-induced dS6K activation is more elevated in dTsc1/2-depleted cells pretreated with RAD001 than in control cells (Fig. 1B; Supplementary Fig. 1), and in preliminary studies, clonal overgrowth in the eye induced by loss of dTsc1 is not suppressed in a semiviable, heterorallelic dTOR mutant background. We further show that overexpression of dTsc1/2 selectively suppresses the dS6K-signaling pathway, whereas dPTEN operates on the dPI3K-signaling pathway (Fig. 2). It was shown recently that double mutations for dPTEN and dTsc1 are additive for clonal overgrowth (Gao and Pan 2001), compatible with dS6K and dPKB independently mediating growth. Nevertheless, inhibition of dPKB by loss of dTsc function shows that there is negative cross-talk between the two signaling pathways (Fig. 2). Given this negative cross-talk, the observation that in double mutant clones growth is additive, suggests that in the absence of dPTEN, inhibition of dPKB by loss of dTsc is circumvented. However, despite the observation that double mutations for dPTEN and dTsc1 are additive for clonal overgrowth, overgrowth induced by absence of dPTEN is suppressed in clones mutant for dTOR (Oldham et al. 2000; Zhang et al. 2000). As dS6K does not prevent such overgrowth (Fig. 3), it is possible that this suppression actually represents an intermediate phenotype, or that dPTEN negatively acts on a dTOR target distinct from dS6K. At this point, it is important to gain a deeper knowledge of the molecular mechanisms by which dTsc1/2 acts to suppress dS6K function and how the signaling components of these two pathways cross-talk with one another.
Recently, a successful Phase I clinical trial was completed for a rapamycin analog in the treatment of solid tumors. The results of the trial demonstrated that the drug was efficacious at subtoxic doses, and suggested that specific tumor types may be more sensitive to inhibition by rapamycin than others (Hidalgo and Rowinsky 2000). The question that arose from the trial is, which tumors would be susceptible to rapamycin treatment? Here, we demonstrate for the first time in vivo that a mild reduction in dS6K signaling, which alone has no blatant phenotype, is sufficient to restore viability of flies devoid of dTsc function. Thus, these findings imply that rapamycin or its derivatives might be very promising pharmaceutical agents in the treatment of tumors arising from TSC.
Materials and methods
Fly strains
The following null alleles were used: dS6Kl1 (Montagne et al. 1999), dTsc1Q87X (Tapon et al. 2001), and dPTEN117 (Stocker et al. 2002). dS6Kl1 and dTsc1Q87X alleles were combined on the same chromosome by recombination. dTOR2L1 (Oldham et al. 2000) and dPKB1 (Staveley et al. 1998) encode kinase mutant proteins. UAS-dPTEN and UAS-dTsc1 UAS-dTsc2 strains were described previously (Goberdhan et al. 1999; Tapon et al. 2001). For ubiquitous GAL4 expression, actin5C-GAL4 (act-GAL4) and daughterless-GAL4 (da-GAL4) lines were used. Experiments were performed in the y w genetic background, crosses were set at 25°C, and adult phenotype analysis was done in females. Mosaic clones within the developing eye were generated by mitotic recombination using the ey-FLP FRT system as described (Newsome et al. 2000). Relative size of ommatidia was determined by measuring the area inscribed within the black outlines of individual eyes with the Adobe Photoshop 4.0 histogram function and dividing this value by the number of ommatidia within each outlined area. At least 50 ommatidia were counted for each genotype.
Double-stranded RNA mediated interference
Double-stranded RNA (dsRNAi) was performed essentially as described (Clemens et al. 2000) with an incubation time of 1 wk. Cells were maintained and treated as reported (Radimerski et al. 2002). All primers were designed starting with the T7 RNA polymerase binding site as follows: 5′-TTAATACGACTCACTATAGGGAGA-3′. dTsc1; accession no. AF173560, sense-primer 436–453, antisense-primer 1081–1098, dTsc2; accession no. AF172995, sense primer 591–608, antisense primer 1371–1388, dPTEN; accession no. AF144232, sense primer 205–222, antisense primer 895–912. Total RNA was isolated with the RNaesy kit (Qiagen) according to the supplier's protocol and dTsc1, dTsc2, and dPTEN mRNA levels were analyzed by Quantitative Real Time PCR using the following probes: sense-primer 1640–1664; Taq 1667–1694; antisense-primer 1696–1716 of AF144232, sense-primer 1831–1850; Taq 1857–1874; antisense-primer 1878–1896 of AF172995, and sense-primer 1120–1138; Taq 1144–1166; antisense-primer 1179–1197 of AF173560.
In vitro kinase activity assays
Protein extracts of cultured cells or of larvae were prepared as reported (Oldham et al. 2000). Kinase activity of dS6K and dPKB was measured essentially as described (Radimerski et al. 2002). Drosophila atypical PKC was immunoprecipitated with the nPKCζ (C-20) antibody (Santa Cruz Biotechnology) and activity assayed as for dS6K except for use of myelin basic protein (0.5 μg) as substrate.
RAD feeding
RAD was prepared freshly from a 20-mM stock in ethanol by diluting to 30 μM in water, and 100 μL were distributed over the surface of standard Drosophila cornmeal–molasses medium (10 mL), onto which flies had previously deposited roughly 150 eggs in short-term collections.
Acknowledgments
We thank I. Hariharan, C. Wilson, S. Oldham, and E. Hafen for the gift of flies, B. Hemmings for providing the dPKB antibody, N. Tapon and N. Ito for providing cDNAs for dTsc1 and dTsc2, and E. Hafen and R. Lamb for critical reading of the manuscript. This work was supported by the Novartis Research Foundation and by grants provided by the Roche Research Foundation to T.R. and G.T. and the Swiss Cancer League to J.M. and G.T.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gthomas@fmi.ch; FAX 41-61-697-3976.
Supplemental material is available at http://www.genesdev.org.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.239102.
References
- Canal I, Acebes A, Ferrus A. Single neuron mosaics of the Drosophila gigas mutant project beyond normal targets and modify behavior. J Neurosci. 1998;18:999–1008. doi: 10.1523/JNEUROSCI.18-03-00999.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens JC, Worby CA, Simonson-Leff N, Muda M, Maehama T, Hemmings BA, Dixon JE. Use of double-stranded RNA interference in Drosophila cell lines to dissect signal transduction pathways. Proc Natl Acad Sci. 2000;97:6499–6503. doi: 10.1073/pnas.110149597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Pan D. TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes & Dev. 2001;15:1383–1392. doi: 10.1101/gad.901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Neufeld TP, Pan D. Drosophila PTEN regulates cell growth and proliferation through PI3K-dependent and -independent pathways. Dev Biol. 2000;221:404–418. doi: 10.1006/dbio.2000.9680. [DOI] [PubMed] [Google Scholar]
- Goberdhan DC, Paricio N, Goodman EC, Mlodzik M, Wilson C. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes & Dev. 1999;13:3244–3258. doi: 10.1101/gad.13.24.3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19:6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]
- Ito N, Rubin GM. gigas, a Drosophila homolog of tuberous sclerosis gene product-2, regulates the cell cycle. Cell. 1999;96:529–539. doi: 10.1016/s0092-8674(00)80657-1. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang ZH, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, Onda H. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- Macleod K. Tumor suppressor genes. Curr Opin Genet Dev. 2000;10:81–93. doi: 10.1016/s0959-437x(99)00041-6. [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G. Drosophila S6 kinase: A regulator of cell size. Science. 1999;285:2126–2129. doi: 10.1126/science.285.5436.2126. [DOI] [PubMed] [Google Scholar]
- Montagne J, Radimerski T, Thomas G. Insulin signaling: Lessons from the Drosophila tuberous sclerosis complex, a tumor suppressor. Sci STKE. 2001;2001:E36. doi: 10.1126/stke.2001.105.pe36. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes & Dev. 2000;14:2689–2694. doi: 10.1101/gad.845700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter CJ, Huang H, Xu T. Drosophila tsc1 functions with tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell. 2001;105:357–368. doi: 10.1016/s0092-8674(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Radimerski T, Montagne J, Rintelen F, Stocker H, van Der Kaay J, Downes CP, Hafen E, Thomas G. dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol. 2002;4:251–255. doi: 10.1038/ncb763. [DOI] [PubMed] [Google Scholar]
- Staveley BE, Ruel L, Jin J, Stambolic V, Mastronardi FG, Heitzler P, Woodgett JR, Manoukian AS. Genetic analysis of protein kinase B (AKT) in Drosophila. Curr Biol. 1998;8:599–602. doi: 10.1016/s0960-9822(98)70231-3. [DOI] [PubMed] [Google Scholar]
- Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. Living with lethal PIP3 levels: Viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science. 2002;295:2088–2091. doi: 10.1126/science.1068094. [DOI] [PubMed] [Google Scholar]
- Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell. 2001;105:345–355. doi: 10.1016/s0092-8674(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Young J, Povey S. The genetic basis of tuberous sclerosis. Mol Med Today. 1998;4:313–319. doi: 10.1016/s1357-4310(98)01245-3. [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes & Dev. 2000;14:2712–2724. doi: 10.1101/gad.835000. [DOI] [PMC free article] [PubMed] [Google Scholar]