

Abstract
Large proteins have multiple domains that are potentially capable of binding many kinds of partners. It is conceivable, therefore, that such proteins could function as an intricate framework of assembly protein complexes. To comprehensively study protein–protein interactions between large KIAA proteins, we have constructed a library composed of 1087 KIAA cDNA clones based on prior functional classifications done in silico. We were guided by two principles that raise the success rate for detecting interactions per tested combination: we avoided testing low-probability combinations, and reduced the number of potential false negatives that arise from the fact that large proteins cannot reliably be expressed in yeast. The latter was addressed by constructing a cDNA library comprised of random fragments encoding large proteins. Cytoplasmic domains of KIAA transmembrane proteins (>1000 amino acids) were used as bait for yeast two-hybrid screening. Our analyses reveal that several KIAA proteins bearing a transmembrane region have the capability of binding to other KIAA proteins containing domains (e.g., PDZ, SH3, rhoGEF, and spectrin) known to be localized to highly specialized submembranous sites, indicating that they participate in cellular junction formation, receptor or channel clustering, and intracellular signaling events. Our representative library should be a very useful resource for detecting previously unidentified interactions because it complements conventional expression libraries, which seldom contain large cDNAs.
[Interaction data accession numbers are BIND ID 12487–12570. Supplemental material is available online at http://www.genome.org.]
It is often technically challenging to analyze extraordinarily large proteins because of their size. In particular, it is extremely difficult to clone the full-length cDNAs encoding such huge proteins and equally difficult to purify intact, large mRNAs and their proteins. For these reasons, the analysis of huge proteins proceeds relatively slowly. However, because such large proteins often are known to play central roles in nervous system development, brain function, and neurological disorders (Lieber et al. 1992; D'Arcangelo et al. 1995), we believe that extraordinarily large proteins are important players in biology, warranting in-depth study. Moreover, their exceptionally large molecular size most likely contributes to a unique biological function. We are therefore interested in the comprehensive characterization of these large proteins in the brain; and over the course of the past six years, we have conducted a cDNA-sequencing project focused on human brain cDNA clones that encode large proteins.
In order to identify unknown human genes overlooked using conventional methods, we have used the following two approaches. The first approach is based on random sampling of long cDNA clones that produce proteins larger than 50 kD in an in vitro transcription/translation system (Nagase et al. 2000). Thus far, we have determined >1900 cDNA sequences (named KIAA) that have been subsequently deposited into public databases. The data obtained to date indicate that: (1) the predicted functions of large proteins are mainly classified into cell communication/signaling (e.g., receptor, adhesion molecule, and GTPase-activating protein), cell structure/motility (e.g., cytoskeletal proteins, membrane skeletal proteins, and motor proteins), and nucleic acid management (e.g., DNA-binding protein; Nagase et al. 1997); (2) although difficult to identify using conventional methods, homologous genes are efficiently isolated by this approach; and (3) several large unknown proteins with unknown functions are predicted from our cDNA sequences. The second approach is to systematically identify genes encoding proteins with a particular domain of interest. The epidermal growth factor (EGF)-like domain is thought to play an important role in extracellular events, including cell adhesion and receptor–ligand interactions. Because the amino acid sequences of EGF-like domains in various known proteins have little in common, except for the conserved location of six cysteine residues, genes coding for EGF-like domain-containing proteins could not be systematically identified either by colony hybridization under relaxed conditions or by the PCR method using degenerate primers. We have therefore developed a computer-assisted method, called motif-trap screening, for this purpose and have succeeded in identifying many large proteins with multiple EGF-like motifs and/or cadherin domains (Nakayama et al. 1998; Nagase et al. 2000; Nakajima et al. 2001).
One of the main research themes of our group is the comprehensive understanding of large proteins in the brain. Our ultimate goal is to deeply understand interactions between transmembrane proteins (Nakayama et al. 1998; Nagase et al. 2000; Nakajima et al. 2001; Mitsui et al. 2002) and proteins localized in submembrane regions (Ohara et al. 1998, 1999, 2000; Yamakawa et al. 1999). This work is prompted by the idea that interactions between transmembrane proteins and proteins localized in submembrane regions are necessary for developmental processes, synaptic plasticity, and neuronal events dependent on electrical activity (e.g., Wyszynski et al. 1997; Lisman and McIntyre 2001). One particular protein complex in neurons capturing the spotlight is the postsynaptic density (PSD), which appears as an electron-dense complex upon electron microscopic examination. PSDs are comprised of many components, including calmodulin kinase, glutamate receptors, and PSD95 (Kennedy 2000). As of yet, however, little information exists regarding the nature of the high-molecular-weight proteins present in PSDs. Large proteins have multiple domains potentially capable of binding many kinds of partners. It is conceivable, therefore, that such large proteins could function as an intricate framework of assembly protein complexes.
Therefore, as part of an ongoing study of large-molecular-weight proteins, we have characterized interactions between large proteins. Two types of approaches were used to analyze genome-wide protein–protein interactions using the yeast two-hybrid system, namely, matrix compilation and library screening (Legrain and Selig 2000).
Many technical problems are encountered, however, when using standard methods to analyze large proteins with yeasts. Even when using either one-by-one or high-throughput matrix approaches (Uetz et al. 2000; Ito et al. 2001), stable activator domain-fused, full-length proteins must be produced reliably in yeast. It is rarely possible, however, to stably produce large proteins derived from other species in yeasts. Analysis of large proteins using library screening is also technically challenging. If conventional methods were to be used, most higher eukaryotic cDNA libraries would contain mainly highly redundant cDNAs and fewer long cDNAs (Ohara et al. 1997). Because commercially available expression libraries for yeast two-hybrid systems simply represent conversions of cDNA libraries produced using conventional methods, one cannot expect to find large amounts of long cDNAs in these commercially available libraries. Even if an expression library is prepared using random primers, it is doubtful that such libraries would contain cDNAs encoding the N-terminal regions of large proteins and contain enough cDNA fragments covering full-length, long cDNAs. For these reasons, we have constructed a normalized representative library composed of long cDNAs. This novel library is comprised of KIAA cDNAs identified in our laboratory. Using the yeast two-hybrid system and this unique expression library, we have identified many protein–protein interactions using the cytoplasmic domains of large transmembrane proteins as bait.
RESULTS
Construction of a Functionally Classified Library Composed of Long cDNAs
Figure 1 illustrates our strategy for efficiently examining interactions between a large number of protein–protein pairs. When combinations of unclassified protein pairs are tested (Fig. 1A), interactions are detected with relatively low frequency. However, if protein pairs are matched according to similar function or cellular location derived from experimental results or bioinformatics, the probability of detecting interactions is much greater. Thus, in this study, we focused on interactions between proteins localized in the submembrane region and the cytoplasmic domain of transmembrane proteins. Because putative transcription factors in the nuclear and extracellular domains of the transmembrane protein are not located within the cytoplasm, to avoid false-positive interactions, these domains were removed from the prey library, as described in Methods. Proteins of unknown function and location were not removed from the prey library.
Figure 1.

Diagram of our strategy for constructing a functionally classified library composed of long cDNAs. (A) Illustration of different combinations according to functionally classified order. Because there are ∼30,000 human proteins, our ultimate aim would be to prepare a complete protein–protein interaction map by assessing interactions between ∼30,000 × ∼30,000 protein combinations. One dot represents the interaction between one vertical-ordered protein (i.e., left, ordinate) and one horizontal-ordered protein (i.e., left, abscissa). The set of possible interactions to be assayed is extremely large. If proteins are, instead, first categorized according to their location and function, as shown in the right panel, interaction dots are clustered to smaller regions, and thus the set of possible interactions to be investigated is reduced considerably. (B) Schematic diagram of library construction. Black and white bars represent open reading and untranslated regions, respectively. Triangle indicates the first methionine codon. After functional classification in silico, randomly fragmented cDNAs were introduced into vectors for yeast two-hybrid screening using the Gateway system.
To date, we have documented 1987 long cDNAs (named KIAA) that encode large proteins. The average length of the protein predicted from KIAA cDNAs is 872 amino acids. Of these KIAA proteins, 175 are >1500 amino acids in length. Because it is difficult to stably express large ORFs in yeast, we chose a strategy that enables the expression of short fragments of proteins containing each domain at equal ratios. Of the 1642 KIAA genes present during the time of library construction, 1087 KIAA genes for proteins localized in the submembrane region that can potentially interact with the cytoplasmic domain of transmembrane proteins were selected (Supplemental Table 1 online contains a list of these KIAA genes). To produce random fragments, these cDNAs were pooled at almost equal ratios and sonicated. cDNA fragments encoding 333–666 amino acids were isolated and introduced into entry vectors (Fig. 1B). These lengths were chosen because we reasoned from our experience that they could be expressed in yeast stably. Because distinction of their three reading frame and orientation cannot be achieved using this method, only one-sixth of the cDNA fragments produced AD fusion proteins with the correct reading frames for the prey proteins.
The total numbers of entry clones, expression clones in Escherichia coli, and expression clones in yeast implemented in this study were 3.5 × 106, 7.3 × 106, and 16 × 106, respectively. The average sizes of the cDNA inserts, the ORFs, and the vectors were approximately 5 kb, 2.6 kb, and 3 kb, respectively. Therefore, 167 different parts of any particular protein per ORF were produced in an in-frame orientation. In fact, as shown in Figure 2, after two-hybrid screening, many independent clones containing various protein regions with core-binding domains were isolated. This indicates that this library is composed of many kinds of expressible cDNA fragments, probably covering all ORF regions.
Figure 2.

Yeast two-hybrid screening and subsequent determination of interacting domains. The domain organization for bait (left) and prey (right) KIAA proteins are shown. Blue bars indicate independent clones (one bar represents one prey). The length of the blue bar indicates the sequence of the KIAA protein contained within an isolated prey. The interacting domains located in the bait and preys are indicated by pink (bait) and green (prey) brackets. In the case of KIAA1634, we estimated that KIAA1132 can bind two different domains: the first and second PDZ domain, as shown.
Selection of Cytoplasmic Domains of Extra-Large KIAA Transmembrane Proteins as Baits
For baits, we selected 71 KIAA cDNAs on the basis of three features: (1) Expression in the brain. Only KIAA cDNAs derived from human brains were used. The initial KIAA library contained both brain-derived and human KG-1 myeloblast-derived cDNAs. (2) Protein size. KIAA cDNAs encoding peptides of >1000 amino acids in length were used. (3) Protein location and type (i.e., transmembrane proteins). The hydrophobicity of each KIAA protein was predicted using the SOSUI program, and only proteins with more than one transmembrane region (not including the N-terminal signal sequence) were used.
To reduce self-activation-associated noise measured with the two-hybrid screening, an assay for self-activation was performed before screening. As shown in Table 1, 19% of the baits assayed displayed self-activation. This percentage is consistent with previous results obtained from comprehensive research of yeast proteomes (Uetz et al. 2000). Although it is known that acidic amphipathic domains are often responsible for unwanted transcriptional activation or noise (Ruden et al. 1991), to the best of our knowledge, bait self-activation has never been assessed in silico. Thus, in the present study, we provide a comprehensive list of bait amino acid sequences displaying self-activation activity. These data supply important basic information that can be used to create computer programs for predicting potential bait self-activation sequences (see Supplemental Table 2 online). Such a program would be very useful in the field of higher eukaryotic proteome research for raising the efficiency of screening by eliminating 19% of the noise caused by bait self-activation.
Yeast Two-Hybrid Screening Using Bait and Prey Vectors With the Gateway System
The Gateway system is a useful tool to easily transfer one or more cDNA fragments into various vectors, all the while maintaining orientation and reading frame in parallel reactions contained within one tube. We introduced this system into our vector for yeast two-hybrid screening because we plan to convert our representative cDNA library and the screened prey cDNA into other types of expression vectors (for an example, see Fig. 3 and Discussion). The ProQuest Gateway recombination system was made commercially available after we constructed the library described above using the standard Gateway recombination and yeast two-hybrid screening methods. The backbone of our yeast two-hybrid screening, however, is the MATCHMAKER GAL4-based two-hybrid system supplied by Clontech. Unlike the ProQuest system, both the yeast DNA-BD and AD vectors contain multicopy plasmids. Yeast AH109 containing the bait plasmid and its mating partner, Y187, containing the prey plasmid were mated together, and the resulting diploid yeasts were selected according to nutritional requirements for Ade, His, Leu, and Trp. Candidates for a two-hybrid interaction were retested with the same nutritional selective plates containing X-α-Gal for the colorimetric detection of the MEL1 reporter gene product, α-galactosidase. These three reporters are driven by different promoters. This triple selection method using 3-amino-1,2,3-triazole (3-AT) to suppress leaky His3 expression is thought to be one of the most effective selection systems for yeast two-hybrid screening at the present time.
Figure 3.
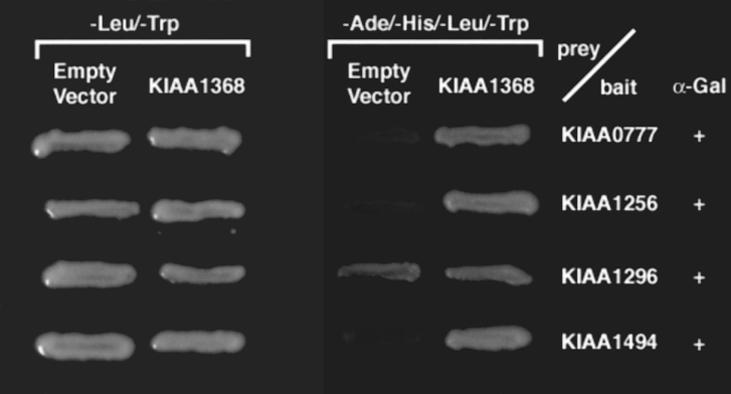
Reassessment of interactions after exchanging cDNAs in baits and preys via the Gateway system. Image of yeast two hybrids grown on selection media. After exchanging the plasmids contained in the original bait and prey using the Gateway system, new bait plasmids (KIAA0777, KIAA1256, KIAA1296, or KIAA1496), new prey plasmid (KIAA1368), and an empty vector (pACTGW-attR) were transformed into yeast AH109. The viability of the transformants was assessed on SD/−Leu/−-Trp and SD/−Ade/−His/−Leu/−Trp plates. The + symbol located in the right-hand column indicates that the MEL1 gene product, α-galactosidase, is actively expressed. Blue-colored growth on nutritional selection plates in the presence of X-α-Gal indicates active expression of α-galactosidase.
Yeast Two-Hybrid Screening and Confirmation of Protein Interactions
In the present study, 2 × 108 CFU of prey library per bait was used; therefore, if the mating efficiency is >1.75% (normally it is >3%), we estimate that all possible protein–protein combinations (3.5 × 106 independent clones) will be tested. Because a large number of clones were screened, several independent clones containing the same binding domain were isolated in many cases (Table 1; Fig. 2). When two or more clones containing the same binding domain were detected, at least in yeast, the interaction between two proteins is real and reproducible. An example of KIAA1368 is shown in Figure 2. Using its cytoplasmic domain as bait, 14 prey colonies were isolated, and their inserts were sequenced. Every sixth prey was either KIAA1296 or KIAA1494. Every other prey was KIAA0777 or KIAA1256. Although prey clones originating from KIAA1296 are independent clones with different sizes of cDNA fragments, all preys contained an identical region consisting of KIAA1296 protein. Thus, as shown in Figure 2, one advantage of screening randomly generated fragments is that functional interacting domains can be directly mapped alongside the screening experiment that identified the interacting protein. With regard to the interacting domains of the four preys described above, namely, the binding partners of KIAA1368/semaphorin 6A (Fig. 2), all of the binding regions of these four prey KIAA proteins contained the SH3 (Src homology 3) domain. Moreover, the amino acid sequence of the binding region of KIAA1296 is similar to that of KIAA0777 and KIAA1494 (59% and 38% sequence identity, respectively).
Table 1.
Summary of Experimental Results
| Description | Total |
|---|---|
| Number of baits assessed | 71 |
| Number PCR products obtained | 65 |
| Number of baits cloned | 64 |
| Self-activationa | 12 |
| Number of baits that underwent two-hybrid screening | 52 |
| Number of baits identified to have interactions | 32 |
| Total number of protein pairs exhibiting interactions | 84 |
| Interaction identified multiple times | 42 |
Self-activation was assessed on SD/−Ade/−His/−Trp plates.
To confirm the yeast two-hybrid screening results, the cDNAs in the baits and preys used in the initial screening were exchanged, and the resulting plasmid DNAs were transformed into haploid yeast, AH109, to assess for protein–protein interactions. Using the Gateway system, bait and prey inserts can easily be exchanged. Figure 3 shows the result of retesting protein interactions after this exchange. Transformants bearing baits containing KIAA0777, KIAA1256, or KIAA1494 and empty prey vectors resulted in no growth on the selection plates. The transformants bearing bait containing KIAA1296 and empty prey vectors, however, displayed self-activation. On the other hand, transformants bearing baits containing KIAA0777, KIAA1256, or KIAA1494 and prey containing KIAA1368/semaphorin 6A exhibited growth on selection plates containing X-α-Gal. These colonies were blue, indicating that the MEL1 reporter, α-galactosidase, was activated. Retests involving KIAA0777, KIAA1256, and KIAA1494 clones confirmed the interaction with KIAA1368/semaphorin 6A.
The bait–prey exchange experiments confirmed the results of the initial two-hybrid screening data and demonstrated that these initial results represented true interactions, even in cases in which only one clone (e.g., KIAA0077 and KIAA1256) was obtained for DNA sequencing. Moreover, bait–prey exchange experiments that examined interactions between KIAA1206 and KIAA0076, KIAA0380, KIAA0708, or KIAA1634 (data not shown) were also confirmed.
Remarkable Interaction Pairs Following Their Annotation and Predicted Functional Domain
As shown in Table 2, we have identified 84 protein–protein interactions (more detailed accounts of these interactions are available at http://www.kazusa.or.jp/huge/ppi/). Since our initial publications (Ohara et al. 1997; Nagase et al. 1999), some KIAA proteins have been identified and described by other groups using different criteria, and rodent KIAA homologs have been analyzed extensively. The function of most KIAA proteins, however, remains unknown. Our data presented here shed light on the potential functions of some of these KIAA proteins because specific binding sites on KIAA partners were identified. Furthermore, a number of notable interaction pairs were identified in our comprehensive analysis (Fig. 2). KIAA1368/semaphorin 6A, a transmembrane semaphorin that can act as a repulsive axon guidance cue, bound KIAA proteins containing multiple SH3 domains. Multiple SH3 domains are suggested to act as an adapter that mediates signal transduction (Tong et al. 2000; Li et al. 2001). KIAA1206/Plexin-B3, a neuronal receptor for semaphorin, bound proteins containing a PDZ domain and proteins that regulate small G proteins. Its binding partner, KIAA0380, contains both PDZ and RhoGEF domains encoding the guanine nucleotide exchange factor for Rho/Rac/Cdc42-like GTPases. It is worth noting that Vikis et al. (2000) reported that human plexin-B1 specifically interacts with active Rac in a ligand-dependent manner. Hu et al. (2001) also reported that, in Drosophila, plexin B mediated axon guidance by simultaneously inhibiting Rac activity and enhancing RhoA signaling. Moreover, one of its other binding partners, KIAA0076, contains a RhoGAP domain for GTPase activator proteins, although its scores for sequence family classification using Pfam search is low (E-value, 9.3). As shown in Table 2, KIAA0708, a homolog of KIAA0076, can also bind KIAA1206/Plexin-B3. Comparing the interacting regions of KIAA0076 to that of KIAA0708, we determined that these regions bore the same amino acid sequences, which may represent a new binding domain responsible for organizing protein complexes in the submembrane region. KIAA1132, an uncharacterized adhesion molecule, also binds proteins containing PDZ domains and KIAA1092/phospholipase C, which is thought to be involved in the production of second messengers.
Table 2.
Protein-Protein Interaction Pairs Between Large Proteins
| Bait | Description | Prey | Description |
|---|---|---|---|
| KIAA0319 | — | KIAA0396(2)a | FEM-1-like death receptor-binding protein |
| KIAA1299(1) | SH2-B β-signaling protein | ||
| KIAA0343 | — | KIAA0417(2) | — |
| KIAA1251(1) | Trabeculin-α | ||
| KIAA1634(6) | Membrane-associated guanylate kinease-related MAGI-3 | ||
| KIAA0348 | Synaptojanin 2 | KIAA1256(3) | Intersectin 2 (SH3D1B) |
| KIAA0358 | MAP kinase-activating death domain protein | KIAA0453(1) | — |
| KIAA0364 | Immunoglobulin-like domain-containing 1 protein | KIAA1131(1) | — |
| KIAA1464(1) | RanBPM homolog (66%)b | ||
| KIAA0387 | Protein-tyrosine phosphatase X precursor | KIAA0097(2) | CH-TOG PROTEIN |
| KIAA0463 | Plexin 2 precursor | KIAA0144(1) | — |
| KIAA1014(1) | Formin-binding protein 30 | ||
| KIAA1199(1) | — | ||
| KIAA1374(1) | — | ||
| KIAA0543 | — | KIAA1043(1) | — |
| — | KIAA1218(1) | — | |
| KIAA0578 | Neurexin I-α | KIAA0300(1) | PDZ domain-containing protein AIPC |
| KIAA0440(1) | High-risk human papilloma viruses E6 oncoproteins targeted protein E6TP1 α | ||
| KIAA0865(1) | — | ||
| KIAA1251(2) | Trabeculin-α | ||
| KIAA0603c | — | KIAA0332(2) | — |
| KIAA0765(2) | RNA binding motif protein 12 | ||
| KIAA1498(2) | p400 SWI2/SNF2-related protein | ||
| KIAA1546(2) | — | ||
| KIAA0620 | — | KIAA1251(1) | Trabeculin-α |
| KIAA0690 | — | KIAA0109(1) | Clathrin coat assembly protein AP50 |
| KIAA0812c | MEGF2/Flamingo | KIAA0640(9) | SWAP-70 homolog |
| KIAA0728(3) | Bullous pemphigoid antigen 1-a (Bpag1) | ||
| KIAA0816 | MEGF7 | KIAA1526(2) | — |
| KIAA0868 | Contactin associated protein-like 2 (Caspr2) | KIAA0155(10) | TPR-containing/SH2-binding phosphoprotein |
| KIAA1224(1) | — | ||
| KIAA1251(2) | Trabeculin-α | ||
| KIAA0911 | Calsyntenin-1 | KIAA0045(2) | Thyroid hormone receptor interactor 12 |
| KIAA0067(1) | ERG-associated protein ESET | ||
| KIAA0204(3) | CTCL tumor antigen se20-9 | ||
| KIAA0572(2) | 80-kDa MCM3-associated protein | ||
| KIAA1447(3) | — | ||
| KIAA0921 | Neurexin 2-α | KIAA1251(1) | Trabeculin-α |
| KIAA0960 | — | KIAA0400(3) | — |
| KIAA0987 | Band 4.1-like protein 3 | KIAA0912(1) | — |
| KIAA1032(2) | Munc13–1 | ||
| KIAA1062 | ATP-binding cassette, sub-family A | KIAA1633(5) | — |
| KIAA1132 | — | KIAA0705(8) | Synaptic scaffolding protein S-SCAM |
| KIAA1092(2) | PLC-L2 mRNA for phospholipase C-L2 | ||
| KIAA1634(14) | Membrane-associated guanylate kinase-related MAGI-3 | ||
| KIAA1206 | Plexin-B3 (PLXNB3) | KIAA0076(3) | — |
| KIAA0275(1) | Testican-2 | ||
| KIAA0380(5) | RhoGEF glutamate transport modulator GTRAP48 | ||
| KIAA0705(1) | Synaptic scaffolding protein S-SCAM | ||
| KIAA0708(1) | KIAA0076 homolog (62%) | ||
| KIAA0952(1) | — | ||
| KIAA1634(3) | Membrane-associated guanylate kinase-related MAGI-3 | ||
| KIAA1216 | Vitamin D3 receptor interacting protein | KIAA0235(1) | Pumilio 2 |
| KIAA0593(1) | Thyroid hormone receptor-associated protein complex component TRAP240 | ||
| KIAA1037(1) | — | ||
| KIAA1041(1) | — | ||
| KIAA1278 | FUSED serine/threonine kinase | KIAA0316(2) | — |
| KIAA0807(3) | Microtubule-associated serine/threonine protein kinase | ||
| KIAA1633(1) | — | ||
| KIAA1368 | Semaphorin 6A | KIAA0777(1) | SH3-containing protein p4015 |
| KIAA1256(1) | Intersectin 2 (SH3D1B) | ||
| KIAA1296(6) | R85FL | ||
| KIAA1494(6) | SH3 domains-containing protein POSH | ||
| KIAA1394 | — | KIAA0045(4) | Thyroid hormone receptor interactor 12 |
| KIAA1549 | — | KIAA0728(1) | Bullous pemphigoid antigen 1-a |
| KIAA1119(1) | MYOSIN VB | ||
| KIAA1121(1) | Calcium-dependent actin-binding protein | ||
| KIAA1256(1) | Intersectin 2 (SH3D1B) | ||
| KIAA1461(1) | — | ||
| KIAA1573 | — | KIAA0240(1) | — |
| KIAA1177(2) | HCNP protein; XPA-binding protein 2 | ||
| KIAA1224(2) | — | ||
| KIAA1464(1) | RanBPM homolog (66%) | ||
| KIAA1679 | — | KIAA1464(5) | RanBPM homolog (66%) |
| KIAA1746 | — | KIAA0465(1) | Actin binding protein ABP620 |
| KIAA1763 | CASPR4 | KIAA0561(1) | Microtubule- and syntrophin-associated serine–threonine protein kinase homolog (63%) |
| KIAA0875(1) | F-box only protein 21 | ||
| KIAA1251(1) | Trabeculin-α | ||
| KIAA1464(8) | RanBPM homolog (66%) | ||
| KIAA1780c | MEGF10 | KIAA0109(5) | Clathrin coat assembly protein AP50 |
| KIAA0207(2) | Growth factor receptor-bound protein 10 | ||
| KIAA0577(2) | Putative pre-mRNA splicing factor RNA helicase | ||
| KIAA0788(2) | U5 small nuclear ribonucleoprotein 200 kDa helicase | ||
| KIAA1464(2) | RanBPM homolog (66%) |
The number in parentheses represents the number of independent, isolated, positive clones.
Homologous genes whose sequences are more than 80% identified are detailed in the description column. Concerning annotation worth being described but lower than 80% identity, the percent identity is given in parentheses.
When 10 or more preys were identified, only preys prepared from two or more independent clones were listed because the bait may contain a sticky region.
All interaction data in this report are available in the public database BIND (BIND ID 12487–12570; http://www.bind.ca).
We found that 16 KIAA proteins interact with the PDZ domain and target the C-terminal tail, as shown in Table 3A. KIAA0343, KIAA0816, KIAA1132, and KIAA1206 were designated as class I, and KIAA0578 and KIAA1763 were categorized as class II, according to the classification scheme of Tochio et al. (1999). Although the C terminus of KIAA1206, TDL, does not exactly match the class I consensus sequence (S/T-X-I/V), the properties of Leu are very similar to that of Ile; thus, KIAA1206 was designated as possessing a variant class I PDZ domain and was grouped into class I, according to Harris and Lim (2001). Because the C terminus of KIAA1216, HSM, was never described before (Harris and Lim 2001; Vaccaro et al. 2001), PDZ domains of KIAA0316 and KIAA0807 might be assigned to a new specificity class, which we temporarily designated as class new. Interactions between the PDZ domain and the target C terminus occur in vivo in many cases (Harris and Lim 2001). Although the interactions shown in Table 2 were not reported previously, our data strongly support that true interactions were detected in our screening. In addition, as shown in Table 3B, several interactions involving the SH3 domain were also identified.
Table 3.
Well-Characterized Interaction of Protein Pairs and the Submembrane Localization Predicted by Domains
| A. Interaction of Proteins Containing PDZ Domain and C-Terminal Tailsa | ||||
| Bait | C-Terminus | Consensus | Class of PDZ domain | Prey |
|---|---|---|---|---|
| KIAA0343 | −SFV | S/T-X-I/V | Class I | KIAA1634(6) |
| KIAA0578 | −YYV | F/Y-X-A/F/V | Class II | KIAA0300(1), KIAA0440(1) |
| KIAA0816 | −SQV | S/T-X-I/V | Class I | KIAA1526(2) |
| KIAA1132 | −TLV | S/T-X-I/V | Class I | KIAA0705(8), KIAA1634(14) |
| KIAA1206 | −TDL | S/T-X-I/V | Class Ib | KIAA0380(5), KIAA0705(1), KIAA1634(3) |
| KIAA1278 | −HSM | — | Class new | KIAA0316(2), KIAA0807(3) |
| KIAA1763 | −YFF | F/Y-X-A/F/V | Class II | KIAA0561(1) |
| B. Interaction of Proteins Containing SH3 and Cytoplasmic Domainsc | ||
| Bait | Alias | Prey |
|---|---|---|
| KIAA0348 | Synaptojanin 2 | KIAA1256(3) |
| KIAA0960 | — | KIAA0400(2) |
| KIAA1368 | Semaphorin 6A | KIAA0777(1), KIAA1256(1), KIAA1296(6), KIAA1494(6) |
| KIAA1549 | — | KIAA1256(1) |
| C. Interaction Pairs in Which the Prey Contains a Domain Associated with the Membrane and/or Located in Structural Complexes Near the Submembrane Regiond | |||
| Bait | Prey | Domain | Accession number |
|---|---|---|---|
| KIAA0319 | KIAA0396 | Ankyrin repeat | PF0023 |
| KIAA1299 | Pleckstrin homology (PH) domain | PF00169 | |
| KIAA0343 | KIAA1251 | Spectrin repeat | PF00435 |
| KIAA0348 | KIAA1256 | Dbl domain (dbl/cdc24 rhoGEF family) | PF00621 |
| Pleckstrin homolog (PH) domain | PF00169 | ||
| KIAA0463 | KIAA1374 | G-protein β WD-40 repeats | PS50294 |
| KIAA0812 | KIAA0640 | Pleckstrin homolog (PH) domain | PF00169 |
| KIAA0728 | Spectrin repeat | PF00435 | |
| KIAA0868 | KIAA1251 | Spectrin repeat | PF00435 |
| KIAA0921 | KIAA1251 | Spectrin repeat | PF00435 |
| KIAA0960 | KIAA0400 | Ankyrin repeat | PF00023 |
| Pleckstrin homolog (PH) domain | PF00169 | ||
| KIAA1132 | KIAA0705 | Guanylate kinase | PF00625 |
| KIAA1092 | Pleckstrin homology (PH) domain | PF00169 | |
| Phosphatidylinositol-specific phospholipase C | PF00388 | ||
| KIAA1206 | KIAA0380 | Dbl domain (dbl/cdc24 rhoGEF family) | PF00621 |
| KIAA1216 | KIAA1037 | G-protein β WD-40 repeats | PS50294 |
| KIAA1368 | KIAA1256 | Dbl domain (dbl/cdc24 rhoGEF family) | PF00621 |
| Pleckstrin homolog (PH) domain | PF00169 | ||
| KIAA1549 | KIAA0728 | Spectrin repeat | PF00435 |
| KIAA1746 | KIAA0465 | Spectrin repeat | PF00435 |
| KIAA1763 | KIAA1251 | Spectrin repeat | PF00435 |
| KIAA1780 | KIAA0207 | Pleckstrin homology (PH) domain | PF00169 |
Only proteins containing the PDZ are listed. Classification criteria of Tochio et al. (1999) for the PDZ domain target sequence was used.
Did not exactly match class I consensus sequence, but was categorized as class I according to Harris and Lim (2001).
Only proteins containing the SH3 domain are described.
This list also contains domains aside from the domains in the interaction region. PDZ and SH3 domains that are described in Parts A and B are not included.
As shown in Table 3, KIAA proteins that interact with the cytoplasmic domains of transmembrane proteins often contain distinctive domains (e.g., PDZ domain, SH3 domain, spectrin domain, or Pleckstrin homology domain) known to be localized within the submembrane region. These observations are also consistent with the target at which this study aims, and support our idea that the interactions observed in our two-hybrid screening were, indeed, true interactions. Taken together, many of the interactions we identified in this study are most likely to be involved in extra-large machinery composed of multiple protein components, acting as scaffolds that organize signaling complexes.
DISCUSSION
Strategies for Efficient Testing of Protein–Protein Interactions
In the present study, we sought to efficiently examine interactions between large-molecular-weight proteins. Inspection of the draft DNA sequence of the human genome indicates that there are an estimated 30,000–40,000 genes that encode proteins (Lander et al. 2001). The ultimate aim would be to test all possible interaction combinations between proteins encoded by the predicted human ORFs (i.e., 0.9–1.6 × 109 combinations). The vastness of such an undertaking can be better appreciated when comparing the total number of these combinations with the size of the human genome (i.e., 3 × 109 bp). To obtain a higher ratio of interaction per tested combination, we attempted to avoid testing combinations with very low probabilities of interaction based on disjunctive locations and functions of proteins. Figure 1 summarizes this strategy. When combinations of unclassified protein pairs are tested (Fig. 1A), interactions are rarely detected. However, if protein pairs are matched according to similar function or location, the probability of detecting interactions is much greater. For example, one would expect that a protein localized in the nucleus binds other proteins localized in the nucleus. Similarly, a protein localized in the cytoplasm would likely interact with other proteins in the cytoplasm. One can be confident of a small probability of false positives if reporter genes of a yeast two-hybrid screening system are activated because of an interaction between a nuclear protein and an extracellular protein. A priori classification of potential protein–protein pairs based on known function or location will decrease the number of false positives by avoiding combinations that are unlikely to colocalize in vivo. There are exceptions, however, in which some proteins may move to different parts of the cell, where they may interact with other proteins. Our guiding principal, therefore, for this study was that only protein–protein combinations with a higher probability of interaction would be tested first.
Another problem encountered in examining interactions between large-molecular-weight proteins using the yeast two-hybrid system is that it is difficult to stably express large ORFs in yeast. To address this problem, we chose a strategy that enables the expression of short fragments of proteins containing each domain at equal ratios. cDNAs were pooled at almost equal ratios and sonicated to produce random fragments. Short cDNA fragments (333–666 amino acids) that could be stably expressed in yeast were isolated and introduced into entry vectors (Fig. 1B). Although one report describes a yeast expression vector (bearing in-frame cloned ORFs) that can be selected in enrichment media (Holz et al. 2001), selection with a yield of >60% for ORF clones with correct reading frames is still difficult to achieve. Moreover, bias arising from such preselection enrichment techniques cannot be avoided. Because prey clones containing cDNAs fused out-frame with the AD produce mostly short peptides that do not have binding activity, we chose normalization without selection even if effective clones are decreased to one-sixth of all clones. Instead of excluding out-of-frame clones, special attention was paid to maintaining an adequately large number of independent clones. By preparing the clones in this manner, we were assured that a maximum number of protein pairs could be assessed using two-hybrid screening.
The Success Rate for Detecting Interactions per Tested Combination
Previously, Uetz et al. (2000) obtained the success rate of one interaction per 30,000 combinations by systematic analysis of the yeast two-hybrid system for yeast proteins. Using a mammalian two-hybrid screening method to assess interactions between mouse proteins, Suzuki et al. (2001) detected approximately one interaction for every 80,000 combinations tested. In the present study, we detected 84 interactions per 52 baits tested in a library composed of 1087 KIAA cDNAs. Although we cannot compare our interaction values directly with those of Suzuki et al. (2001), because the two screening systems implemented were different, our ratio of interactions per combination tested was relatively higher. For one combination (i.e., one bait for every one KIAA cDNA in the library), our success rate was one interaction for every 673 combinations tested. There are several reasons for this higher ratio. First, because each protein was produced from a short piece of cDNA containing a binding domain, rather than a longer cDNA containing the full ORF, a larger amount of bait protein may have been expressed stably in the yeast. This decreases the number of false negatives inherent to the use of full ORFs in two-hybrid screening using the matrix approach. Second, in many cases, the cytoplasmic domain of transmembrane proteins binds two or more proteins (Kennedy 2000; Angst et al. 2001; Harris and Lim 2001). For this reason, the cytoplasmic domain is thought to be a good target for detecting interactions efficiently. Thus, our baits, which contain such cytoplasmic domains, tend to interact with many more proteins compared with the average interaction per protein. As a result, we detected many interactions at a high rate. Third, when the screening is performed with a functionally classified library, the likelihood of detecting true interactions is maximized by first removing the region encoding the nuclear proteins and extracellular proteins that extend into the extracellular matrix. Fourth, false positives cannot be excluded when using the yeast two-hybrid system to assess protein–protein interactions. However, because two or more clones containing overlapping cDNAs were isolated, our interaction data are consistent with previously described criteria from comprehensive protein–protein analyses (Uetz et al. 2000; Ito et al. 2001). Interaction analyses using other methodologies (e.g., BIAcore analysis, GST pull down, coimmunoprecipitation, and immunohistochemistry for localization) should be performed to confirm our yeast two-hybrid data. The Gateway system, the method we used to introduce the cDNA fragment into our vectors for two-hybrid screening, is also helpful to transfer the screened candidate cDNA fragment into other expression vectors for other methodologies in order to confirm and analyze extensively our yeast two-hybrid data.
The Unique Position Our Screening Occupies in the Field of Comprehensive Protein–Protein Interaction Research
Data resulting from our comprehensive screening of KIAA proteins comprise one step in the ongoing analyses of protein–protein interactions. Ultimately, our data, when taken together with protein-interaction data from other groups, will contribute to the complete mapping of all possible protein–protein interactions. Of course, our library contains only a limited number of long cDNAs; thus, interaction information about proteins encoded by genes not presently found in our library is unknown. Nevertheless, our library is inherently useful, and, unlike with other conventional libraries, we have a clear grasp of all the cDNAs of our library. When more cDNAs are made available as a result of further cDNA analysis, we will be able to construct new libraries containing cDNAs not contained in our present library. Because our libraries will never contain overlapping sequences, duplicate testing of protein–protein pairs derived from combinations from all of these libraries (i.e., the mixed matrix approach) will be avoided.
As an alternative approach to analyze large proteins, we have an ongoing project to construct another type of library specifically intended to analyze long cDNAs. We previously compiled a specialized cDNA library for the isolation of unidentified long cDNAs that encode large proteins (Ohara et al. 1997). The work was prompted by the fact that typical cDNA libraries generated using conventional procedures based on growth rate tend to bias the selection of plasmids containing short cDNAs as opposed to long cDNAs. To avoid this growth-rate bias, long cDNAs are now being selected following separation via agarose gel electrophoresis; this procedure enables the enrichment of long cDNAs of approximately the same size. Using this series of strictly size-fractionated cDNAs as starting material, we are able to construct another library using the same procedure as reported in this study, namely, random fragmentation and introduction to two-hybrid vectors. Although this library should contain a large variety of long cDNAs, one problem encountered is redundancy at the level of expression. Because of the distinctive features of each of these libraries, we suggest that one of these two libraries should be chosen to match the user's specific needs.
Why We Focus on Protein–Protein Interactions Between Large Proteins: The Putative Role of Large Proteins in Biological Multiprotein Complexes
One good example of a large protein that participates in biological multiprotein complexes is RNA polymerase II. Because its components and subunit assembly are well characterized (Woychik and Hampsey 2002), it serves as a good model of how large proteins function in multiprotein complexes. Eukaryotic RNA polymerase II, which catalyzes DNA-dependent synthesis of mRNA, is composed of 12 subunits. Its overall configuration is “jaw-shaped.” Two of its largest subunits (1733 and 1224 amino acids in Saccharomyces cerevisiae) contribute to this shape (Allison et al. 1985; Goffeau et al. 1996) in which the largest subunit (Rpb1) forms most of the lower jaw, and the second largest subunit (Rpb2) forms most of the upper jaw. These two large subunits contain both catalytic domains and binding sites to the other subunits, and play a central role in RNA polymerase function, structure, and subunit assembly.
Another example of a large protein involved in multiprotein complexes is the polycomb group in Drosophila. These proteins form large multiprotein complexes composed of 30 proteins (Saurin et al. 2001). Polycomb repressive complex 1, which was identified via biochemical purification techniques, contains at least two large proteins, posterior sex combs (PSC, 1603 amino acids; Brunk et al. 1991) and polyhomeotic-proximal chromatin protein (PH, 1589 amino acids; DeCamillis et al. 1992). Genetic experimental results show that PSC and PH are essential in maintaining a repressed transcriptional state for homeotic genes; to date, however, their roles in complex assembly have not been described in detail.
Recently, Gavin et al. (2002) described 232 distinct multiprotein complexes in S. cerevisiae using tandem-affinity purification (TAP) and mass spectrometry techniques. To compare these complexes with the human equivalents, using TAP, they purified human Ccr4/Not and Bet3 complexes from transfected cell cultures generated by retrovirus-mediated gene transfer. It is worthy to note that these two complexes contained large proteins (i.e., KIAA1007, KIAA1194, and KIAA1012) previously identified by our laboratory.
Taken together, these examples clearly indicate that most biological multiprotein complexes contain one or more large proteins that are believed to be essential components, both functionally and structurally. These examples are fortuitous cases, however, because it is generally difficult to purify large proteins from higher eukaryotic cells.
Because large proteins may bind both large and small proteins, interactions between large and small proteins would need to be tested with the two-hybrid screening method in our extensive research. As an initial goal, we focused on especially large protein complexes that are detectable at the electron microscopic level with the aim of examining how these complexes assemble. Because large proteins possess multiple, potential binding domains, we speculate that large proteins bind other large proteins at these binding sites to create a framework of proteins that could be characterized as a superstructure containing many scaffolds (i.e., a scaffold of scaffolds).
Concluding Remarks
Soon after the introduction of the draft sequence of the human genome, many groups initiated comprehensive functional analyses of the genome; however, if special attention is not focused on collecting data about large proteins, most of these studies will most likely accumulate information primarily about small proteins and short cDNAs. Data from our study is one step to bridge this important and technically difficult gap by providing protein–protein interaction information about large proteins.
METHODS
Construction of a Prey Library Composed of Long cDNAs (KIAA Clones)
In this study, we focused on binding of the cytoplasmic domain of a large, transmembrane protein with proteins localized in the submembrane region. To minimize false-positive identification of interactions with proteins believed not to be colocalized in vivo, the nuclear and extracellular proteins located in domains outside of the cytoplasm (i.e., putative transcription factors and extracellular domains of the transmembrane protein) were removed from the prey library. This was achieved by analyzing the amino acid sequence of KIAA genes using Pfam searches and the SOSUI program. KIAA genes containing motifs for transcription factors (e.g., Pfam ID, PF00010, PF00096, PF00170, PF00249, PF00628, PF00642, PF00643, and PF01530) and the predicted transmembrane region were omitted from the library used for this study. Plasmids totaling 1087 KIAA clones (Supplemental Table 1 online) were pooled at almost equal ratios and sonicated, and blunt ends were produced using mung bean nuclease and T4 DNA polymerase (TAKARA).
DNA fragments ranging from 1 kb to 2 kb were isolated electrophoretically in agarose gels (low melting point temperature), then cloned between the DraI and EcoRV sites of the pENTR 1A vector (Invitrogen). A pool of cDNA plasmids was recovered from 3.5 × 106 primary transformants (ElectroMax DH10B cells; Invitrogen) and grown on agar plates containing 30 μg/mL kanamycin. To construct the destination plasmid pACTGW-attR, plasmid attR-pSP73 (Ohara and Temple 2001) was double-digested with HindIII/SacI, and an attR cassette containing the ccdB and chloramphenicol-resistance genes was isolated and ligated into the SmaI–XhoI site of pACT2 (Clontech) with blunt ends. A pool of cDNA plasmids was converted to expression clones via the Gateway LR reaction (Invitrogen) using pACTGW-attR. The reaction products were transformed into Escherichia coli cells. Plasmids were recovered from transformants (7.3 × 106 ) on ampicillin-containing agar plates after growth at 30°C for 14 h. The cDNA plasmids were transformed into yeast, Y187 (MATα). Transformants (1.6 × 107 ) were grown on synthetic medium SD/−Leu plates and pooled. Aliquots from the pooled yeast transformant were used for mating in two-hybrid screening.
Preparation of Bait and Mating-Mediated Yeast Two-Hybrid Screening
To construct the destination plasmid pASGW-attR, plasmid attR-pSP73 was double-digested with HindIII/SacI, and an attR cassette containing the ccdB and chloramphenicol-resistance genes was isolated and ligated into the EcoRI–SalI site of pAS2-1 (Clontech) with blunt ends. Among the KIAA proteins, KIAA genes encoding transmembrane regions were selected using predictions from the SOSUI program. Using KIAA cDNA as a template, each cDNA fragment containing a cytoplasmic domain was amplified by PCR and oligomers GWF and GWR: 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTT G(X)24-3′ and 5′-GGGGACCACTTTGTACAAGAAAGCTGGG TTA(X)24-3′, respectively, where (X)24 is the gene-specific sequence. (Supplemental Table 2 online describes these cDNAs) Following the BP reaction, the attB-PCR product was introduced into expression clones via the Gateway LR reaction using pASGW-attR and a donor plasmid (Invitrogen). These clones were introduced into E. coli DH5α, and the inserts of all bait plasmids were confirmed by DNA sequencing. The bait plasmid was transformed into yeast AH109 (MATa), and transformants were grown on SD/−Trp plates. Self-activation assays were performed on SD/−Ade/−His/−Trp plates. Two transformants per each bait exhibiting no background growth on −Ade/−His were pooled, cultured, and mated with an aliquot of yeast prey containing the transformant library. Diploid cells were spread onto 9 cm × 13 cm 20–25 plates containing SD/−Ade/−His/−Leu/−Trp/15 mM 3-AT (3-amino-1,2,4-triazole). After 7–10 d of growth at 30°C, typically 30 colonies were replated onto SD/−Ade/−His/−Leu/−Trp/15 mM 3-AT plates containing X-α-gal. Blue colonies were selected and cultured in SD/−Leu medium. Each plasmid was purified from the yeast and used as a template for PCR with oligomers OADN (5′-TCG ATG ATG AAG ATA CCC CAC-3′) and OADC (5′-AAG AAA TTG AGA TGG TGC ACG-3′). DNA sequences of prey clones were determined to identify the interacting region of the KIAA protein. The resulting sequences were compared with the HUGE database (http://www.kazusa.or.jp/huge/) using BLAST2.
Exchanging of Bait and Prey Sequences and Confirmation of Binding Activity by Retest
To confirm the binding activity detected by the library screening of yeast two-hybrids, bait and prey sequences were exchanged using the Gateway reaction, and both were transfected into haploid AH109 yeasts. Baits were prepared by introducing the PCR product (from OADN and OADC oligomers) into expression clones via the Gateway BP and LR reaction and pASGW-attR and donor plasmids. Preys were similarly prepared by introducing the PCR product (from GWF and GWR oligomers) into expression clones using the Gateway BP and LR reaction and pACTGW-attR and donor plasmids. Gateway reaction products were transformed into E. coli DH5α. After purification of the plasmids, the new bait and prey plasmids or the empty vector were transformed into yeast AH109; viability was checked on SD/−Leu/−Trp and SD/−Ade/−His/−Leu/−Trp plates.
WEB SITE REFERENCES
http://www.kazusa.or.jp/huge/; HUGE database.
http://www.kazusa.or.jp/huge/ppi/; detailed accounts of 84 protein–protein interactions.
Acknowledgments
We are grateful to S. Minorikawa, E. Suzuki, S. Tsuda, and N. Kashima for their excellent technical assistance. We also thank Dr. Takahiro Nagase and all our colleagues from the Kazusa cDNA project team for their cooperation. This study was supported by grants from the Kazusa Research Institute and in part by Special Coordination Funds of the Ministry of Education, Culture, Sports, Science and Technology, the Japanese Government.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL nmanabu@kazusa.or.jp; FAX 81 438-52-3914.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.406902. Article published online before print in October 2002.
REFERENCES
- Allison LA, Moyle M, Shales M, Ingles CJ. Extensive homology among the largest subunits of eukaryotic and prokaryotic RNA polymerases. Cell. 1985;42:599–610. doi: 10.1016/0092-8674(85)90117-5. [DOI] [PubMed] [Google Scholar]
- Angst BD, Marcozzi C, Magee AI. The cadherin superfamily. J Cell Sci. 2001;114:625–626. doi: 10.1242/jcs.114.4.625. [DOI] [PubMed] [Google Scholar]
- Brunk BP, Martin EC, Adler PN. Drosophila genes Posterior Sex Combs and Suppressor two of zeste encode proteins with homology to the murine bmi-1 oncogene. Nature. 1991;353:351–353. doi: 10.1038/353351a0. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- DeCamillis M, Cheng NS, Pierre D, Brock HW. The polyhomeotic gene of Drosophila encodes a chromatin protein that shares polytene chromosome-binding sites with Polycomb. Genes & Dev. 1992;6:223–232. doi: 10.1101/gad.6.2.223. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- Holz C, Lueking A, Bovekamp L, Gutjahr C, Bolotina N, Lehrach H, Cahill DJ. A human cDNA expression library in yeast enriched for open reading frames. Genome Res. 2001;11:1730–1735. doi: 10.1101/gr.181501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Marton TF, Goodman CS. Plexin B mediates axon guidance in Drosophila by simultaneously inhibiting active Rac and enhancing RhoA signaling. Neuron. 2001;32:39–51. doi: 10.1016/s0896-6273(01)00453-6. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc Natl Acad Sci. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Legrain P, Selig L. Genome-wide protein interaction maps using two-hybrid systems. FEBS Lett. 2000;480:32–36. doi: 10.1016/s0014-5793(00)01774-9. [DOI] [PubMed] [Google Scholar]
- Li W, Fan J, Woodley DT. Nck/Dock: An adapter between cell surface receptors and the actin cytoskeleton. Oncogene. 2001;20:6403–6417. doi: 10.1038/sj.onc.1204782. [DOI] [PubMed] [Google Scholar]
- Lieber T, Wesley CS, Alcamo E, Hassel B, Krane JF, Campos-Ortega JA, Young MW. Single amino acid substitutions in EGF-like elements of Notch and Delta modify Drosophila development and affect cell adhesion in vitro. Neuron. 1992;9:847–859. doi: 10.1016/0896-6273(92)90238-9. [DOI] [PubMed] [Google Scholar]
- Lisman JE, McIntyre CC. Synaptic plasticity: A molecular memory switch. Curr Biol. 2001;11:R788–R791. doi: 10.1016/s0960-9822(01)00472-9. [DOI] [PubMed] [Google Scholar]
- Mitsui K, Nakajima D, Ohara O, Nakayama M. Mammalian fat3: A large protein that contains multiple cadherin and EGF-like motifs. Biochem Biophys Res Commun. 2002;290:1260–1266. doi: 10.1006/bbrc.2002.6338. [DOI] [PubMed] [Google Scholar]
- Nagase T, Ishikawa K, Nakajima D, Ohira M, Seki N, Miyajima N, Tanaka A, Kotani H, Nomura N, Ohara O. Prediction of the coding sequences of unidentified human genes. VII. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1997;4:141–150. doi: 10.1093/dnares/4.2.141. [DOI] [PubMed] [Google Scholar]
- Nagase T, Ishikawa K, Kikuno R, Hirosawa M, Nomura N, Ohara O. Prediction of the coding sequences of unidentified human genes. XV. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 1999;6:337–345. doi: 10.1093/dnares/6.5.337. [DOI] [PubMed] [Google Scholar]
- Nagase T, Kikuno R, Nakayama M, Hirosawa M, Ohara O. Prediction of the coding sequences of unidentified human genes. XVIII. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2000;7:273–281. doi: 10.1093/dnares/7.4.271. [DOI] [PubMed] [Google Scholar]
- Nakajima D, Nakayama M, Kikuno R, Hirosawa M, Nagase T, Ohara O. Identification of three novel non-classical cadherin genes through comprehensive analysis of large cDNAs. Mol Brain Res. 2001;94:85–95. doi: 10.1016/s0169-328x(01)00218-2. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Nakajima D, Nagase T, Nomura N, Seki N, Ohara O. Identification of high-molecular-weight proteins with multiple EGF-like motifs by motif-trap screening. Genomics. 1998;51:27–34. doi: 10.1006/geno.1998.5341. [DOI] [PubMed] [Google Scholar]
- Ohara O, Temple G. Directional cDNA library construction assisted by the in vitro recombination reaction. Nucleic Acids Res. 2001;29:E22. doi: 10.1093/nar/29.4.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara O, Nagase T, Ishikawa K, Nakajima D, Ohira M, Seki N, Nomura N. Construction and characterization of human brain cDNA libraries suitable for analysis of cDNA clones encoding relatively large proteins. DNA Res. 1997;4:53–59. doi: 10.1093/dnares/4.1.53. [DOI] [PubMed] [Google Scholar]
- Ohara O, Ohara R, Yamakawa H, Nakajima D, Nakayama M. Characterization of a new β-spectrin gene which is predominantly expressed in brain. Mol Brain Res. 1998;57:181–192. doi: 10.1016/s0169-328x(98)00068-0. [DOI] [PubMed] [Google Scholar]
- Ohara R, Yamakawa H, Nakayama M, Yuasa S, Ohara O. Cellular and subcellular localization of a newly identified member of the protein 4.1 family, brain 4.1, in the cerebellum of adult and postnatally developing rats. Dev Brain Res. 1999;117:127–138. doi: 10.1016/s0165-3806(99)00110-8. [DOI] [PubMed] [Google Scholar]
- Ohara R, Yamakawa H, Nakayama M, Ohara O. Type II brain 4.1 (4.1B/KIAA0987), a member of the protein 4.1 family, is localized to neuronal paranodes. Mol Brain Res. 2000;85:41–52. doi: 10.1016/s0169-328x(00)00233-3. [DOI] [PubMed] [Google Scholar]
- Ruden DM, Ma J, Li Y, Wood K, Ptashne M. Generating yeast transcriptional activators containing no yeast protein sequences. Nature. 1991;350:250–252. doi: 10.1038/350250a0. [DOI] [PubMed] [Google Scholar]
- Saurin AJ, Shao Z, Erdjument-Bromage H, Tempst P, Kingston RE. A Drosophila polycomb group complex includes Zeste and dTAFII proteins. Nature. 2001;412:655–660. doi: 10.1038/35088096. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Fukunishi Y, Kagawa I, Saito R, Oda H, Endo T, Kondo S, Bono H, Okazaki Y, Hayashizaki Y. Protein–protein interaction panel using mouse full-length cDNAs. Genome Res. 2001;11:1758–1765. doi: 10.1101/gr.180101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochio H, Zhang Q, Mandal P, Li M, Zhang M. Solution structure of the extended neuronal nitric oxide synthase PDZ domain complexed with an associated peptide. Nat Struct Biol. 1999;6:417–421. doi: 10.1038/8216. [DOI] [PubMed] [Google Scholar]
- Tong XK, Hussain NK, de Heuvel E, Kurakin A, Abi-Jaoude E, Quinn CC, Olson MF, Marais R, Baranes D, Kay BK, et al. The endocytic protein intersectin is a major binding partner for the Ras exchange factor mSos1 in rat brain. EMBO J. 2000;19:1263–1271. doi: 10.1093/emboj/19.6.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Vaccaro P, Brannetti B, Montecchi-Palazzi L, Philipp S, Citterich MH, Cesareni G, Dente L. Distinct binding specificity of the multiple PDZ domains of INADL, a human protein with homology to INAD from Drosophila melanogaster. J Biol Chem. 2001;276:42122–42130. doi: 10.1074/jbc.M104208200. [DOI] [PubMed] [Google Scholar]
- Vikis HG, Li W, He Z, Guan KL. The semaphorin receptor plexin-B1 specifically interacts with active Rac in a ligand-dependent manner. Proc Natl Acad Sci. 2000;97:12457–12462. doi: 10.1073/pnas.220421797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woychik NA, Hampsey M. The RNA polymerase II machinery: Structure illuminates function. Cell. 2002;108:453–463. doi: 10.1016/s0092-8674(02)00646-3. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Yamakawa H, Ohara R, Nakajima D, Nakayama M, Ohara O. Molecular characterization of a new member of the protein 4.1 family (brain 4.1) in rat brain. Mol Brain Res. 1999;70:197–209. doi: 10.1016/s0169-328x(99)00139-4. [DOI] [PubMed] [Google Scholar]