

Abstract
We investigated substitution patterns and neighboring-nucleotide effects for 2,576,903 single nucleotide polymorphisms (SNPs) publicly available through the National Center for Biotechnology Information (NCBI). The proportions of substitutions were A/G, 32.77%; C/T, 32.81%; A/C, 8.98%; G/T, 9.06%; A/T, 7.46%; and C/G, 8.92%. The two nucleotides immediately neighboring the variable site showed major deviation from genome-wide and chromosome-specific expectations, although lesser biases extended as far as 200 bp. On the 5′ side, the biases for A, C, G, and T were 1.43%, 4.91%, −1.70%, and −4.62%, respectively. These biases were −4.44%, −1.59%, 5.05%, and 0.99%, respectively, on the 3′ side. The neighboring-nucleotide patterns for transitions were dominated by the hypermutability effects of CpG dinucleotides. Transitions were more common than transversions, and the probability of a transversion increased with increasing A + T content at the two adjacent sites. Neighboring-nucleotide biases were not consistent among chromosomes, with Chromosomes 19 and 22 standing out as different from the others. These data provide genome-wide information about the effects of neighboring nucleotides on mutational and evolutionary processes giving rise to contemporary patterns of nucleotide occurrence surrounding SNPs.
Substitution patterns at polymorphic sites and bias patterns in nucleotides neighboring polymorphic sites are important for understanding molecular mechanisms of mutation and genome evolution. Single nucleotide polymorphism (SNP) data and information about surrounding sequence motifs are suitable for studying mutational processes in human and other genomes (Zavolan and Kepler 2001). However, in humans previous analyses of SNP variation and neighboring-nucleotide effects have largely been limited to pseudogenes or a limited number of genes with known effects, many of which are disease-causing (e.g., Gojobori et al. 1982; Li et al. 1984; Cooper and Krawczak 1990; Krawczak et al. 1998). In plants, effects of neighboring nucleotides have been extensively studied in chloroplasts (e.g., Morton 1995; Morton et al. 1997).
There is considerable recent interest in SNPs within every gene in the genome or regularly spaced across the genome as tools for association-mapping of disease-susceptibility genes (Risch and Merikangas 1996) or identifying polymorphic sites within a known gene that are associated with a trait of interest and may be functional (Huang et al. 2001). There are more than two and one-half million SNPs available in the public domain. At present, most of the SNPs are deposited by The SNP Consortium (TSC), the Sanger Genome Center, and Washington University (Marth et al. 2001). This large data set provides us with an opportunity to investigate substitution patterns as well as neighboring-nucleotide effects representative of the whole genome, including genic and intergenic regions. The data set is also large enough to investigate patterns for each substitution type and for each chromosome. We investigated substitution patterns and neighboring-nucleotide effects for 2,576,903 SNPs publicly available through the National Center for Biotechnology Information (NCBI). To uncover the actual extent of the nucleotide bias, we normalized with respect to the averaged nucleotide proportion in the human genome and the relevant chromosome. Finally, a large number of transversions were studied to reveal the patterns of mutation avoiding obvious CpG effects.
RESULTS
Substitution Patterns and Frequency Bias
There were 2,576,903 substitutions used in this analysis. Substitution of A to G or G to A is denoted A/G, because the direction of the nucleotide change is unknown. The other nucleotide substitutions follow similarly. There were 844,427 A/G substitutions, 845,441 C/T substitutions, 231,506 A/C substitutions, 233,387 G/T substitutions, 192,285 A/T substitutions, and 229,857 C/G substitutions. The number of A/G substitutions was close to that of C/T substitutions, and the number of A/C substitutions was close to that of G/T substitutions, reflecting complementary strand symmetry. A/T substitutions were the least frequent among the six types, a pattern observed in pseudogenes or noncoding regions and indicating a lower mutation rate (Li et al. 1984; Zhao et al. 2000). Transitions accounted for 65.6% of the total substitutions in this genome-wide collection of SNP data.
To examine the nucleotide bias at polymorphic sites, the overall nucleotide composition in the human genome was estimated using the genomic sequences downloaded from NCBI (ftp://ftp.ncbi.nih.gov/genomes/H_sapiens; September 6, 2001, release). Considering a total of 2.86 × 109 bases, the proportions of the four nucleotides were 29.55% A, 20.44% C, 20.46% G, and 29.54% T. The GC content was 40.90%. At polymorphic sites, the nucleotide composition was 24.61% A, 25.36% C, 25.37% G, and 24.66% T. As a simple difference, the bias was −4.94%, 4.92%, 4.91%, and −4.88%, respectively, relative to the whole genome.
Neighboring-Nucleotide Effects
The proportion of each nucleotide neighboring the polymorphic site is shown in Table 1. The proportions at the two nearest positions on each side showed a large bias relative to the average in the human genome. For example, the nucleotide C (25.35%) on the immediate 5′-adjacent site occurred more frequently than the genome average of 20.44%, and the nucleotide G (25.51%) on the immediate 3′-adjacent site occurred more frequently than the genome average of 20.46%. The frequency of each nucleotide at the flanking sites was next normalized by subtracting the corresponding average value in the human genome. The pattern of bias after normalization is plotted in Figure 1. On the 3′ side of the substitution, the frequency of G was 5.05% higher than the genome average, which is close to the proportion at the substitution site. At the +2 site, the proportion of G was 2.51% higher than the genome average. The nucleotide T, which occurred 4.88% less frequently than the genome average at the substitution site, occurred 0.99% more frequently at the +1 site. However, its frequency was 1.08% lower than the average at the +2 site. The nucleotides A and C occurred 4.44% and 1.59% lower than the average at the +1 position, respectively. In general, the nucleotide patterns on the 5′ side of the substitution complemented that observed on the 3′ side, although the extent of bias was less (Fig. 1). Away from the immediate site, there was a trend toward C and G having a higher proportion than the genome average, whereas A and T had a lower proportion. This trend extended as far as 200 bp to each side.
Table 1.
Proportion of Neighboring Nucleotides
| Position (bp)a | 5′ side | 3′ side | Both sides | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (%) | C (%) | G (%) | T (%) | A (%) | C (%) | G (%) | T (%) | A (%) | C (%) | G (%) | T (%) | |
| 1 | 30.98 | 25.35 | 18.76 | 24.92 | 25.11 | 18.85 | 25.51 | 30.53 | 28.04 | 22.10 | 22.13 | 27.72 |
| 2 | 28.51 | 23.05 | 20.29 | 28.15 | 28.33 | 20.24 | 22.97 | 28.46 | 28.42 | 21.65 | 21.63 | 28.30 |
| 3 | 29.39 | 21.14 | 19.99 | 29.48 | 29.62 | 20.03 | 21.04 | 29.30 | 29.51 | 20.58 | 20.52 | 29.39 |
| 4 | 29.94 | 21.22 | 20.39 | 28.45 | 28.64 | 20.39 | 21.20 | 29.77 | 29.29 | 20.81 | 20.79 | 29.11 |
| 5 | 29.64 | 20.91 | 20.71 | 28.75 | 28.92 | 20.70 | 20.80 | 29.57 | 29.28 | 20.80 | 20.76 | 29.16 |
| 6 | 29.33 | 20.53 | 21.44 | 28.70 | 28.87 | 21.40 | 20.56 | 29.17 | 29.10 | 20.96 | 21.00 | 28.94 |
| 7 | 29.66 | 20.14 | 20.92 | 29.28 | 29.38 | 20.98 | 20.16 | 29.48 | 29.52 | 20.56 | 20.54 | 29.38 |
| 8 | 29.77 | 20.43 | 21.07 | 28.73 | 28.97 | 21.03 | 20.41 | 29.59 | 29.37 | 20.73 | 20.74 | 29.16 |
| 9 | 29.16 | 20.82 | 20.79 | 29.23 | 29.36 | 20.81 | 20.79 | 29.04 | 29.26 | 20.82 | 20.79 | 29.14 |
| 10 | 29.06 | 20.73 | 20.95 | 29.26 | 29.41 | 20.94 | 20.69 | 28.95 | 29.24 | 20.84 | 20.82 | 29.11 |
| 11–20 | 29.36 | 20.70 | 20.84 | 29.10 | 29.31 | 20.83 | 20.66 | 29.20 | 29.34 | 20.77 | 20.75 | 29.15 |
| 21–50 | 29.20 | 20.92 | 20.82 | 29.06 | 29.25 | 20.83 | 20.88 | 29.05 | 29.21 | 20.89 | 20.86 | 29.04 |
| 51–100 | 29.32 | 20.69 | 20.73 | 29.26 | 29.39 | 20.78 | 20.66 | 29.17 | 29.32 | 20.76 | 20.72 | 29.19 |
| 101–200 | 29.41 | 20.65 | 20.65 | 29.30 | 29.41 | 20.71 | 20.64 | 29.24 | 29.32 | 20.76 | 20.73 | 29.18 |
| 201–300 | 29.61 | 20.37 | 20.38 | 29.63 | 29.68 | 20.52 | 20.44 | 29.36 | 29.48 | 20.63 | 20.60 | 29.29 |
| Genomeb | 29.55 | 20.44 | 20.46 | 29.54 | 29.55 | 20.44 | 20.46 | 29.54 | 29.55 | 20.44 | 20.46 | 29.54 |
Position relative to the polymorphic site. For positions farther than 10 bp, the average proportion of each nucleotide in the range is shown.
The average proportion of four nucleotides in the human genome based on NCBI Human RefSeq.
Figure 1.

Neighboring-nucleotide biases. A minus sign indicates the 5′ side, a positive sign the 3′ side, and a ± sign is two-sided.
Neighboring Effects on the Six Categories of Substitution
The neighboring-nucleotide effects were next examined separately for each of the six categories of observable substitutions: C/T, A/G, G/T, A/C, C/G, and A/T. The nature of the observed biases varied greatly among the six substitution categories, especially at the immediate adjacent sites. The frequency of G at +1 was 33.62% for C/T substitutions, 2.4 times that observed for A/C substitutions (i.e., 14.27%). On the other hand, the frequency of A at +1 was 34.59% for A/C substitutions, 1.6 times that observed for G/T substitutions (i.e., 21.63%).
Figure 2 shows the normalized bias of the neighboring nucleotides for each substitution category. Figure 2, A and B, is for the transitions, whereas Figure 2C–F is for the transversions. The pattern observed for C/T (Fig. 2A) and A/G (Fig. 2B) was different from that observed for all of the other categories (Fig. 2C–F). For the transitions, the pattern was dominated by the effect of CpG dinucleotides. For the C/T category, there was a large excess of G at +1 (13.16%), whereas in the A/G category, there was a large excess of C at −1 (13.02%). More subtly for the C/T category, the proportion of A was 5.16% higher than expected at −1, but decreased to 4.79% lower than expected at −2. The proportion of T shows the opposite pattern; it was 6.61% lower than expected at −1, and 3.07% higher than expected at −2. These data indicate that A at −1 has a positive influence on the substitution rate of C/T, whereas T at −1 has a negative influence.
Figure 2.


Neighboring-nucleotide effects on the six categories of substitution. The nucleotide bias is normalized to the average genome values. A minus sign indicates the 5′ side, a positive sign is the 3′ side, and a ± sign is two-sided. The nucleotides are labeled green (A), blue (C), pink (G), and red (T).
The neighboring effects on transversions are complex. First, the two nucleotides involved in the substitution usually occurred more often than expected in adjacent positions, and this observed bias extended as far as 200 bases to each side. Second, the patterns of neighboring-nucleotide proportions for G/T and A/C were the same, because G is paired to C and T is paired to A (Fig. 2C,D). For example, in the G/T category, nucleotide G at the +1 site occurred 7.78% more frequently than the average and A occurred 7.92% less frequently than the average (Fig. 2C). Third, the categories C/G and A/T are complementary to themselves. For the C/G substitution category, the frequency of G on the 3′ side was above average at all the sites except for the immediate adjacent positions (i.e., +1 site; Fig. 2E). The G was 3.68% below the genome average at the +1 site, but was 6.06% above the average at the +2 site. This sharp difference at the nearest two positions was unique for C/G substitutions. For A/T substitutions, the proportion of T was above average at the remaining sites (Fig. 2F). Although these biases grew progressively smaller, the nucleotide proportions did not reach their genome average until nearly 200–300 bases away from the substitution site. The mechanism for the bias at the immediate adjacent nucleotides may rely on the high mutation rate of CpG dinucleotides and the transition of C to T in these mutations. The mechanism for the extended bias at the remaining sites is unknown to us.
We next examined the number of transitions and transversions and the proportion of transversions in 16 categories grouped by A + T content at the two immediate adjacent sites. The proportion of transversions was largest (45.9%) when TNA occurred, more than twice that when CNG occurred (20.1%), where N denotes any substitution. The proportion of transversions was higher for those sites flanked with an A + T context equal to 2 (38.8%), moderate for an A + T context equal to 1 (33.1%), and lower for an A + T context equal to 0 (30.3%).
Neighboring Effects at the Chromosome Level
The nucleotide content varied greatly among chromosomes, ranging from 48.33% GC content on Chromosome 19 to 38.26% GC content on Chromosome 4 (Table 2). Therefore, it is important to examine the nucleotide bias at adjacent sites in the context of the chromosome containing the substitution. Even after controlling for the GC content of each chromosome, there was a marked excess of C at position −1 and G at position +1. There was also some notable variation among the chromosomes. In particular, Chromosomes 19 and 22 stood out as being different from the other chromosomes with respect to patterns of neighboring-nucleotide variation. At position −1, they both had a decrease in A, whereas all of the other chromosomes had an increase. Likewise, the proportion of C at −1 was markedly higher than on the other chromosomes. At position +1, these two chromosomes had a decrease in T, whereas all of the other chromosomes had an increase. Similarly, the proportion of G at +1 was higher than on the other chromosomes. At the substitution site itself, the nucleotide bias of Chromosomes 19 and 22 was less than that of the other chromosomes.
Table 2.
Nucleotide Biasa at the Substitution Site and Immediate Adjacent Sites for Each Chromosome
| Chromosome | GC (%)b | −1 | 0 | +1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A (%) | C (%) | G (%) | T (%) | A (%) | C (%) | G (%) | T (%) | A (%) | C (%) | G (%) | T (%) | ||
| 1 | 41.72 | 1.01 | 5.37 | −1.59 | −4.79 | −4.61 | 4.50 | 4.53 | −4.42 | −5.03 | −1.39 | 5.48 | 0.94 |
| 2 | 40.14 | 2.07 | 4.55 | −1.67 | −4.95 | −5.39 | 5.33 | 5.27 | −5.22 | −4.55 | −1.61 | 4.65 | 1.51 |
| 3 | 39.86 | 2.35 | 3.90 | −1.74 | −4.51 | −5.50 | 5.43 | 5.34 | −5.27 | −4.26 | −1.51 | 4.09 | 1.68 |
| 4 | 38.26 | 2.49 | 3.91 | −1.49 | −4.90 | −5.94 | 6.00 | 5.76 | −5.82 | −4.33 | −1.35 | 3.86 | 1.81 |
| 5 | 39.72 | 0.92 | 5.02 | −1.39 | −4.55 | −5.47 | 5.25 | 5.41 | −5.19 | −4.58 | −1.55 | 5.29 | 0.82 |
| 6 | 39.62 | 1.19 | 4.88 | −1.35 | −4.73 | −5.43 | 5.38 | 5.41 | −5.36 | −5.20 | −0.98 | 5.22 | 0.97 |
| 7 | 40.65 | 1.47 | 4.99 | −1.78 | −4.67 | −5.38 | 5.30 | 4.95 | −4.86 | −4.55 | −1.94 | 5.26 | 1.24 |
| 8 | 40.07 | 2.58 | 3.40 | −1.79 | −4.20 | −5.37 | 5.56 | 5.52 | −5.71 | −3.91 | −1.85 | 3.40 | 2.35 |
| 9 | 41.38 | 2.24 | 4.48 | −2.27 | −4.45 | −5.11 | 5.22 | 4.73 | −4.84 | −4.16 | −1.98 | 4.54 | 1.61 |
| 10 | 41.56 | 1.22 | 5.51 | −1.74 | −5.00 | −4.75 | 4.72 | 4.42 | −4.39 | −4.86 | −1.57 | 5.66 | 0.77 |
| 11 | 41.65 | 0.81 | 5.62 | −2.55 | −3.88 | −4.45 | 4.54 | 4.50 | −4.59 | −3.68 | −2.11 | 5.44 | 0.35 |
| 12 | 40.77 | 1.39 | 5.37 | −1.84 | −4.92 | −4.94 | 4.91 | 4.98 | −4.95 | −4.68 | −1.42 | 4.89 | 1.20 |
| 13 | 38.63 | 1.17 | 4.71 | −1.32 | −4.56 | −5.64 | 5.63 | 5.57 | −5.57 | −4.78 | −1.31 | 4.87 | 1.22 |
| 14 | 40.76 | 1.30 | 4.46 | −1.88 | −3.88 | −4.77 | 4.74 | 4.84 | −4.81 | −3.38 | −2.12 | 4.67 | 0.83 |
| 15 | 42.12 | 1.26 | 4.94 | −1.88 | −4.33 | −4.82 | 4.66 | 4.62 | −4.46 | −4.25 | −1.80 | 4.88 | 1.16 |
| 16 | 44.83 | 0.99 | 5.34 | −1.84 | −4.49 | −4.09 | 3.79 | 3.87 | −3.57 | −4.61 | −1.76 | 5.40 | 0.97 |
| 17 | 45.02 | 0.90 | 6.37 | −2.43 | −4.83 | −3.61 | 3.62 | 3.00 | −3.00 | −4.59 | −2.19 | 6.59 | 0.20 |
| 18 | 39.84 | 1.49 | 4.78 | −2.16 | −4.11 | −5.20 | 5.12 | 5.39 | −5.30 | −4.06 | −1.77 | 5.44 | 0.38 |
| 19 | 48.33 | −0.42 | 7.51 | −2.54 | −4.57 | −1.98 | 1.82 | 1.99 | −1.84 | −4.73 | −2.63 | 7.79 | −0.45 |
| 20 | 44.11 | 0.92 | 6.34 | −2.02 | −5.25 | −3.31 | 3.54 | 3.62 | −3.85 | −4.91 | −1.51 | 5.86 | 0.54 |
| 21 | 40.89 | 2.94 | 3.87 | −2.27 | −4.55 | −5.13 | 5.04 | 4.52 | −4.44 | −4.48 | −2.46 | 4.67 | 2.27 |
| 22 | 47.64 | −0.40 | 7.74 | −2.28 | −5.06 | −2.48 | 2.37 | 2.26 | −2.15 | −5.18 | −2.09 | 7.87 | −0.60 |
| X | 39.39 | 1.33 | 3.95 | −0.88 | −4.39 | −5.41 | 5.24 | 5.83 | −5.66 | −4.85 | −0.91 | 3.98 | 1.79 |
| Y | 39.11 | 3.18 | 3.02 | −1.84 | −4.37 | −5.05 | 4.90 | 6.40 | −6.25 | −3.38 | −0.87 | 3.85 | 0.40 |
| Genome | 40.90 | 1.43 | 4.91 | −1.70 | −4.62 | −4.94 | 4.92 | 4.91 | −4.88 | −4.44 | −1.59 | 5.05 | 0.99 |
Bias is expressed as a deviation from the chromosome-specific average.
The average GC content on chromosome.
Figure 3 shows the relationship between GC content difference on each chromosome from the overall genome average (40.90%) and the corresponding bias of nucleotide C at the −1 site (−1 C) and nucleotide G at the +1 site (+1 G). The GC content was below the genome average on 13 chromosomes, above the average on 9 chromosomes, and close to the average on the other two. In general, higher GC content in a chromosome was associated with higher proportional bias for −1 C and +1 G. Using a single linear regression model, we have
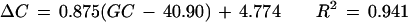 |
for −1 C and
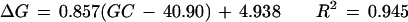 |
for +1 G, where ΔC is the bias for −1 C, ΔG is the bias for +1 G, and GC is the GC content for the chromosome.
Figure 3.

Linear correlation between the GC content difference from the genome average and the proportion of bias at −1 C and +1 G observed on each chromosome.
Ranks of Nucleotide Proportion at Adjacent Sites
Table 3 shows the ranking of the nucleotide bias at the immediately adjacent sites, where ≫ denotes a >5% difference between two nucleotide proportions, > denotes a 1%–5% difference, and ≈ denotes a <1% difference. An upper-case letter denotes a greater observed proportion than the genome average, and a lower-case letter denotes a lower observed proportion than the genome average. Overall, the ranks were C > A > g > t at the −1 site and G > T > c > a at the +1 site. A T at the −1 site and an A at the +1 site occurred more than 4% less frequently than the genome average, indicating a strong bias at these adjacent sites. For transitions, the order was essentially the same as the one observed for all substitutions (Table 3). The results were different for transversions. For transversions, the proportion of nucleotides could be ranked as A > G > c ≈ t at the −1 site and T ≈ C ≈ g ≈ a at the +1 site. Therefore, it appears as if purines had a positive influence on transversions at the −1 site, but pyrimidines had a negative influence. The ranking at the two immediate adjacent sites was opposite as a result of DNA strand complementation.
Table 3.
Ranks of Adjacent Nucleotide Proportions
| Type | Observed proportion | Proportion bias | ||
|---|---|---|---|---|
| −1 | +1 | −1 | +1 | |
| Substitution | A ≫ C ≈ T ≫ Ga | T ≫ G ≈ A ≫ C | C > A > g > tb | G > T > c > a |
| Transition | A > C ≫ T ≫ G | T > G ≫ A ≫ C | C ≫ A > g > t | G ≫ T > c > a |
| Transversion | A > T ≫ G > C | T > A ≫ C ≈ G | A > G > c ≈ t | T ≈ C ≈ g ≈ a |
| A/G | C ≫ A > T ≫ G | T ≫ A ≈ G > C | C ≫ a > g > t | T > G > c ≫ a |
| C/T | A ≫ C ≈ T > G | G ≫ T > A ≫ C | A > C > g ≫ t | G ≫ t > c > a |
| A/C | A > C ≫ T > G | A ≫ T ≈ C ≫ G | C > A > g ≫ t | A ≈ C ≫ t > g |
| G/T | T ≫ A > G ≫ C | T > G ≫ A > C | G ≈ T ≫ a > c | G ≫ T > c ≫ a |
| A/T | A > T ≫ C > G | T > A ≫ G > C | A > C > t > g | T > G > a > c |
| C/G | A > T ≫ G > C | T > A ≫ C > G | A > G > t > c | T > C ≈ a > g |
≫ denotes greater than 5% difference between two nucleotide proportions, > 1%–5% difference, and ≈ less than 1% difference.
A lower-case letter denotes a negative bias of the observed nucleotide proportion compared with the genome average.
DISCUSSION
In this study, we have examined the patterns of nucleotide occurrence neighboring ∼2.6 million SNPs across the human genome. Because this study was not limited to particular genes or motifs (e.g., pseudogenes) or isolated regions of the genome, the results presented here are more representative of the human genome than previous studies. The numbers of A/G and C/T substitutions were similar to one another, and the numbers of A/C and G/T substitutions were similar to one another as a result of complementary strand symmetry. The proportion of nucleotides at the positions neighboring an SNP showed a large bias relative to the average in the human genome. For example, on the 3′ side of the substitution, the frequency of G was 5.05% higher than the genome average. There was a trend, extending some 200 bases, that C and G had higher proportions than their genome averages, whereas A and T had lower proportions. When SNPs were examined by category, neighboring-nucleotide patterns for transitions were dominated by the mutation effect of CpG dinucleotides. Surprisingly, the neighboring-nucleotide patterns varied among chromosomes, with Chromosomes 19 and 22 standing out as being different from the others. At position −1, they both had a decrease in A relative to the chromosome-specific average, whereas all of the other chromosomes had an increase. The nucleotide bias at the immediate adjacent sites were C > A > g > t at the −1 site and G > T > c > a at the +1 site. These data provide a comprehensive view of the effects of neighboring nucleotides on mutations and subsequent evolutionary processes giving rise to the patterns observed today.
We have made use of existing data from dbSNP to describe the patterns of neighboring nucleotides surrounding SNPs. A validation study of 1200 SNPs from the SNP consortium and Washington University, and deposited in dbSNP, revealed that >80% of the SNPs were polymorphic in a multiethnic study sample (Marth et al. 2001). Many of the other SNPs used in this analysis have not been systematically validated. It is our opinion, however, that lack of validation of some SNPs would reduce the overall number being considered, but would not greatly influence the pattern of surrounding nucleotide variation. Another limitation of this study includes the inability to determine the direction of the mutation (e.g., C → A or A → C), and, therefore, the strand that was mutated. Finally, the very large number of SNPs used in this analysis means that even small differences are statistically significant. Therefore, the present treatment of the data is explanatory, permitting the reader to make their own judgments as to the significance of an observed difference.
On average, the results of this study were dominated by the effects of transitions and the high mutation rate of CpG dinucleotides. Transitions accounted for 65.6% of the total substitutions in this genome-wide collection of SNP data. The excess of transitions is largely believed to be attributable to the abundant hypermutable methylated dinucleotide 5′-CpG-3′ (Cooper and Krawczak 1990). Bird (1986) estimated that 60%–90% of CpG dinucleotides may be methylated in vertebrate genomes. Deamination of 5′-methylcytosine in CpG leads to TpG, and CpA in the complement strand (Krawczak et al. 1998). As a result, the frequency of G at the 5′-adjacent site and C at the 3′-adjacent site was strongly positively biased. Furthermore, the proportion of doublet CGs in the genome was 0.99%, 3.19% below the expected value of 4.18% estimated from the genome reference sequences. The proportions of doublet TGs and CAs were 2.44% higher than expected, reflecting the substitution of CG → TG and CG → CA. In contrast to transitions, the adjacent nucleotide bias was small for transversions. The neighboring-nucleotide effects on transversions were complex and varied by the specific category of transversion.
Across the genome-wide collection of SNPs, the rank order of nucleotide proportions was C > A > g > t at the −1 site and G > T > c > a at the +1 site. This order is different from that calculated from 3243 substitutions in gene and pseudogene sequences published by Blake et al. (1992), which had C ≈ A ≫ t > g at the −1 site and G ≫ A > t ≫ c at the +1 site after being normalized by the average nucleotide proportion in the gene sequences. The GC content of their gene and pseudogene sequences was 57%, compared with 41% for the genome average. Their order was also different from that observed for Chromosomes 19 and 22, which had a high GC content (∼48%).
Although the frequency of transitions was more common in these data, the probability of a transversion increased with the A + T content of adjacent nucleotides, a result that is consistent with that observed in plant chloroplasts (Morton et al. 1997). However, the influence of A + T context on transversion in the human genome was smaller than that reported for chloroplasts. In contrast to what had been observed in the chloroplast genome (Morton et al. 1997), we observed that the probability of a transversion increased when the number of purines increased (i.e., 0 → 1 → 2) at the immediate adjacent sites. This difference may be owing to the different nucleotide composition of the human genome compared with that of the plant chloroplast genome. This difference may also be caused by the different mutational mechanisms in plant versus mammalian genomes.
One other difference between the results reported here and those reported by other investigators is the distance that the neighboring-nucleotide bias extends away from the SNP location. Although this study supports the observation of Krawczak et al. (1998), who studied 7271 substitutions in the coding regions of 547 genes, that neighboring-nucleotide bias exists, it disagrees that the bias is confined to only 2 bp from the substitution site. Considering the data from the 2.6 million SNPs studied here, the extent of nucleotide bias extended as far away as 200 bp to each side. The mechanism for this long-distance bias is unknown to us, but likely reflects the overall nucleotide bias of the region (e.g., GC-rich) and not the effects of individual positions.
To reduce the possible bias introduced by very AT-rich or GC-rich regions, we excluded those SNPs occurring in regions (up to 300 bp to each side) in which the A + T content was >70% or the G + C content was >60%. This removed 7.3% of the SNPs by the A + T exclusion and 3.4% by the G + C exclusion. The analyses were then repeated on the restricted data. The general conclusions reported here were the same in the restricted data set and in unrestricted genome-wide collection of SNPs. For example, the biases for A, C, G, and T at the +1 site were −4.63%, −1.45%, 5.21%, and 0.89%, respectively.
METHODS
SNP and Sequence Data
SNPs were downloaded from ftp://ftp.ncbi.nih.gov/snp/human on December 26, 2001 (Build 101, December 13, 2001, release). A total of 2,584,300 reference SNPs were analyzed. We selected the dbSNP database because of the availability of the SNP flanking sequences and the ability to download and manipulate the entire collection. Human genomic DNA sequences were downloaded from ftp://ftp.ncbi.nih.gov/genomes/H_sapiens (September 6, 2001, release) on January 9, 2002.
Data Analysis
We chose only SNPs that have two different nucleotides at the polymorphic site, thus, excluding 7397 SNPs (0.29% of the total). We scored the number for each category of substitution A/G, C/T, A/C, G/T, A/T, and C/G, respectively. The number of transitions was scored from the substitutions A/G and C/T, and the number of transversions was scored from A/C, G/T, A/T, and C/G. We labeled the position at the 5′ side as a negative number, at the 3′ side as a positive number, and for the two sides combined as a ±. For example, −1 stands for the 5′-immediate adjacent nucleotide of the polymorphic site and ±1 for the average of the two immediate adjacent sites. The proportion of neighboring-nucleotides was computed as far as 300 bp to both sides. In the first 10 bp to each side, the proportion of each nucleotide was calculated by
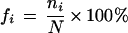 |
where ni is the score of nucleotide A, C, G, or T; and N is the total score of four nucleotides at the site. In the ranges of 11–20 bp, 21–50 bp, 51–100 bp, 101–200 bp, and 201–300 bp to each side, the proportions of nucleotides were averaged. The number of each substitution type on each chromosome was computed, and the patterns among them were compared.
We developed software to analyze the nucleotide composition in the human genome sequence as well as the neighboring-nucleotide composition around SNPs. These programs were written in Perl and C, and are available upon request.
WEB SITE REFERENCES
ftp://ftp.ncbi.nih.gov/snp/human; National Center for Biotechnology Information (NCBI) dbSNP FTP site.
ftp://ftp.ncbi.nih.gov/genomes/H_sapiens; National Center for Biotechnology Information (NCBI) RefSeq FTP site.
Acknowledgments
We thank Yixi Zhong and David Hewett-Emmett for their assistance. This work was supported by grants from the National Heart Lung and Blood Institute and the National Institute of General Medical Sciences. Z.Z. is supported by a training fellowship from the W.M. Keck Foundation to the Gulf Coast Consortia through the Keck Center for Computational and Structural Biology.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL eric.boerwinkle@uth.tmc.edu; FAX (713) 500-0900.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.287302.
REFERENCES
- Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- Blake RD, Hess ST, Nicholson-Tuell J. The influence of nearest neighbors on the rate and pattern of spontaneous point mutations. J Mol Evol. 1992;34:189–200. doi: 10.1007/BF00162968. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M. The mutational spectrum of single base-pair substitutions causing human genetic disease: Patterns and predictions. Hum Genet. 1990;85:55–74. doi: 10.1007/BF00276326. [DOI] [PubMed] [Google Scholar]
- Gojobori T, Li W-H, Graur D. Patterns of nucleotide substitution in pseudogenes and functional genes. J Mol Evol. 1982;18:360–369. doi: 10.1007/BF01733904. [DOI] [PubMed] [Google Scholar]
- Huang Q, Morrison AC, Boerwinkle E. Linkage disequilibrium structure and its impact on the localization of a candidate functional mutation. Genet Epidemiol. 2001;21:S620–S625. doi: 10.1002/gepi.2001.21.s1.s620. [DOI] [PubMed] [Google Scholar]
- Krawczak M, Ball EV, Cooper DN. Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet. 1998;63:474–488. doi: 10.1086/301965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H, Wu C-I, Luo C-C. Nonrandomness of point mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J Mol Evol. 1984;21:58–71. doi: 10.1007/BF02100628. [DOI] [PubMed] [Google Scholar]
- Marth G, Yeh R, Minton M, Donaldson R, Li Q, Duan S, Davenport R, Miller RD, Kwok P-Y. Single-nucleotide polymorphisms in the public domain: How useful are they? Nat Genet. 2001;27:371–372. doi: 10.1038/86864. [DOI] [PubMed] [Google Scholar]
- Morton BR. Neighboring base composition and transversion/transition bias in a comparison of rice and maize chloroplast noncoding regions. Proc Natl Acad Sci. 1995;92:9717–9721. doi: 10.1073/pnas.92.21.9717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton BR, Oberholzer VM, Clegg MT. The influence of specific neighboring bases on substitution bias in noncoding regions of the plant chloroplast genome. J Mol Evol. 1997;45:227–231. doi: 10.1007/pl00006224. [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- Zavolan M, Kepler TB. Statistical inference of sequence-dependent mutation rates. Curr Opin Genet Dev. 2001;11:612–615. doi: 10.1016/s0959-437x(00)00242-2. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Li J, Fu Y-X, Ramsay M, Jenkins T, Leskinen E, Pamilo P, Trexler M, Patthy L, Jorde LB, et al. Worldwide DNA sequence variation in a 10-kilobase noncoding region on human Chromosome 22. Proc Natl Acad Sci. 2000;97:11354–11358. doi: 10.1073/pnas.200348197. [DOI] [PMC free article] [PubMed] [Google Scholar]