

Abstract
The design and synthesis of a small library of 8-amidoflavone, 8-sulfonamidoflavone, 8-amido-7-hydroxyflavone, and heterocyclic analogues of flavopiridol is reported. The potential activity of these compounds as kinase inhibitors was evaluated by cytotoxicity studies in MCF-7 and ID-8 cancer cell lines and inhibition of CDK2-Cyclin A enzyme activity in vitro. The antiproliferative and CDK2-Cyclin A inhibitory activity of these analogues was significantly lower than the activity of flavopiridol. Molecular docking simulations were carried out and these studies suggested a different binding orientation inside the CDK2 binding pocket for these analogues compared to flavopiridol.
1. Introduction
The cyclin-dependent kinases (CDK) represent a class of enzymes that play a central role in cell-cycle progression and cellular proliferation. 1 CDKs are multi-subunit enzymes composed of at least a catalytic subunit (CDK) and a regulatory subunit (cyclin). They exert their effect via activation of host proteins through phosphorylation of key serine or threonine residues by ATP. The phosphorylated proteins modulate the activity of a variety of cellular proteins. It is widely accepted that inhibition of CDKs could provide control of the inappropriate cellular proliferation characteristic of certain cancers. A number of reviews have recently appeared which detail the possible use of CDKs as novel therapeutic targets for cancer chemotherapy.2 Inhibitors developed to date can be divided into three general classes: i) ATP-competitive inhibitors that contain key structural hydrogen-bonding motifs to bind to the ATP pocket; ii) noncompetitive inhibitors that bind to the region of natural peptide inhibitors, typically these are small synthetic peptides (~20 residues); and iii) dual inhibitors, representing molecules incorporating both of these attributes.3
Three common classes have emerged among the kinase inhibitors found to date: bis-indoles, purine-containing analogues, and flavones (Figure 1). Staurosporine, an ATP-competitive PKC inhibitor, 4 and UCN-1 are representative compounds in the bis-indole class. A number of purine-containing compounds that bind to the ATP pocket have also shown promise, including purvalanols A and B,5 and olomucine.4c,5 Representative compounds in the flavone class of CDK inhibitors include flavopiridol(1), 6 2-thioflavopiridol (2a), 7 2-oxoflavopiridol (2b),7 and quercetin (3),8 and related analogues such as 4.7 Flavopiridol is a novel semi synthetic derivative of the plant alkaloid rohitukine (5), itself isolated from an Indian plant, Dysoxylum sp.9
Figure 1.
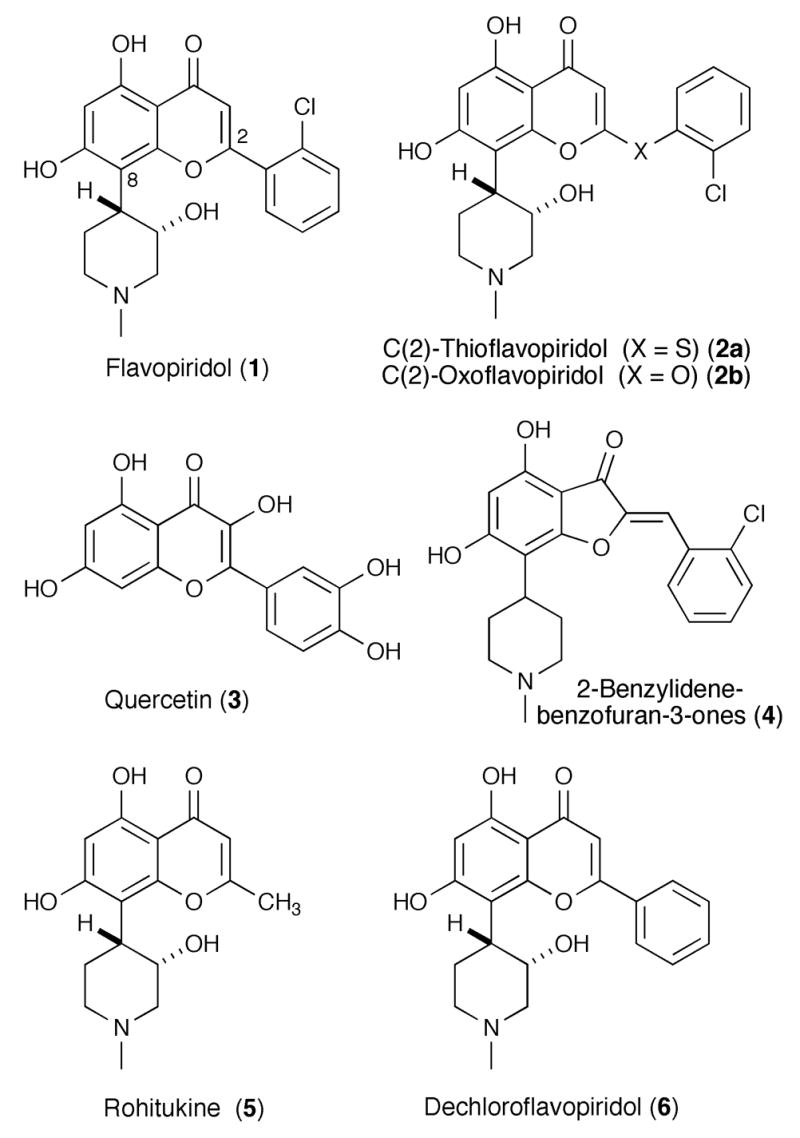
Structures of flavopiridol and representative flavopiridol analogues.
Flavopiridol is the first CDK inhibitor to undergo clinical trials against a variety of cancers. 10 Flavopiridol was shown to inhibit the proliferation of mammalian cell lines at nanomolar concentrations. Flavopiridol is non-selective, showing in vitro activity against CDK1, CDK2, CDK4 and protein-tyrosine kinase, with some activity for the EGF-receptor tyrosine kinase.6e
Due to the overall success of flavopiridol, and because of the availability of the X-ray structure of dechloroflavopiridol (6), co-crystallized with CDK2, revealing key hydrogen bonds (Figure 2A),6g–i we decided to pursue the development of prototypical libraries based on the flavone scaffold. In spite of flavopiridol’s potent activity, two major challenges remain; the development of analogues with improved kinase inhibitory selectivity and higher binding affinity.3,11
Figure 2.
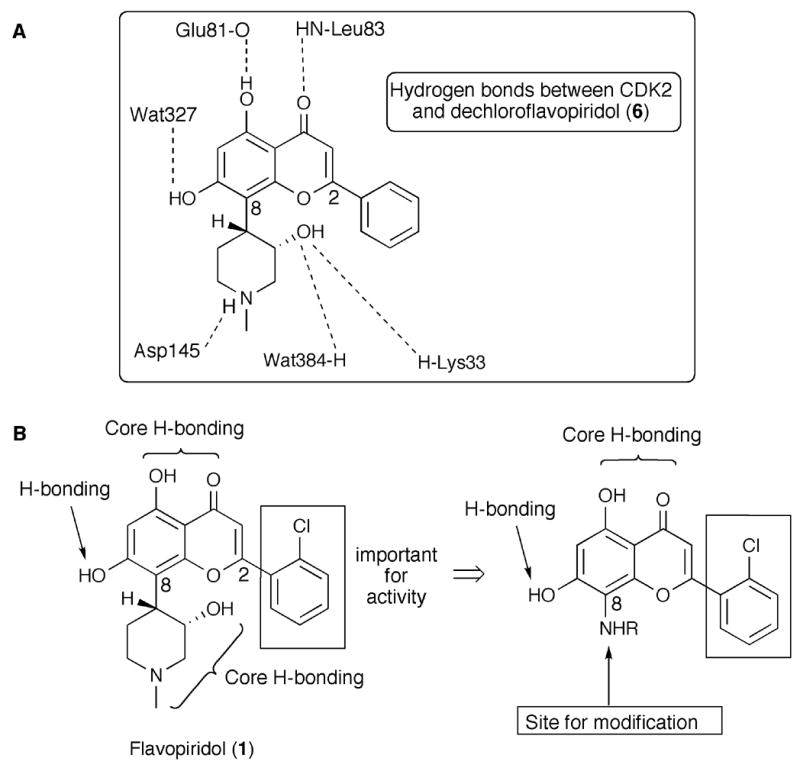
Key H-bonding interactions between CDK2 and dechloroflavopiridol (Fig. 2A). SAR relationship for flavopiridol and targeted compounds (Fig. 2B).
Recent work by Aronov and Murcko on kinase inhibitors suggests a distinct structural pattern for "frequent-hitters" emphasizing a five-point-of-attachment pharmacophore for the ATP binding site of kinases.12 Flavopiridol and the analogues to be described herein, depart from this structural pattern, and therefore can be reasonably supposed to show selectivity. SAR studies demonstrated that the flavone class of CDK inhibitory compounds is amenable to structural modifications at the C2 and C8 positions of the flavone core.3a
Another important consideration is that high activity and selectivity require the formation of at least two key hydrogen bonds between the substrate and the ATP binding pocket.3 To this date, none of the flavone inhibitors have shown picomolar potencies and therefore it has been hypothesized that an additional binding interaction will be required to achieve both better potency and selectivity.3a
Accordingly, we initially designed a key 8-aminoflavone intermediate (Figure 2B), which was designed to retain the hydrogen bonding interactions with Glu81, Leu83, possibly also Wat327 (Figure 2A), and the interactions of the 2-(2-chlorophenyl) group of 1 with the protein, avoiding the "frequent-hitters" pharmacophore.12 The 8-amino group provides a new site for introduction of various hydrogen bond donor/acceptor motifs aimed at providing additional interactions with the ATP binding pocket and surrounding areas so as to potentially impart potency and selectivity. Guided by the aforementioned SAR studies,3 we initiated the synthesis of four classes of 8-amino-modified flavones related to flavopiridol (Figure 3). The synthesis of the key 8-aminoflavone intermediates 10 and 16 are outlined in Scheme 1 and Scheme 2, respectively.
Figure 3.
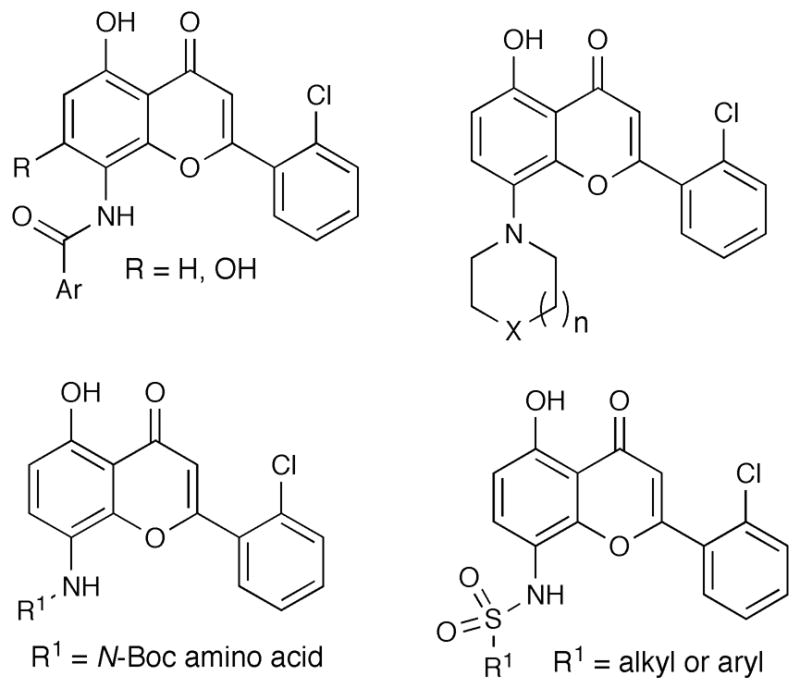
Targeted classes of flavopiridol analogues.
Scheme 1.
Scheme 2.
2. Chemistry
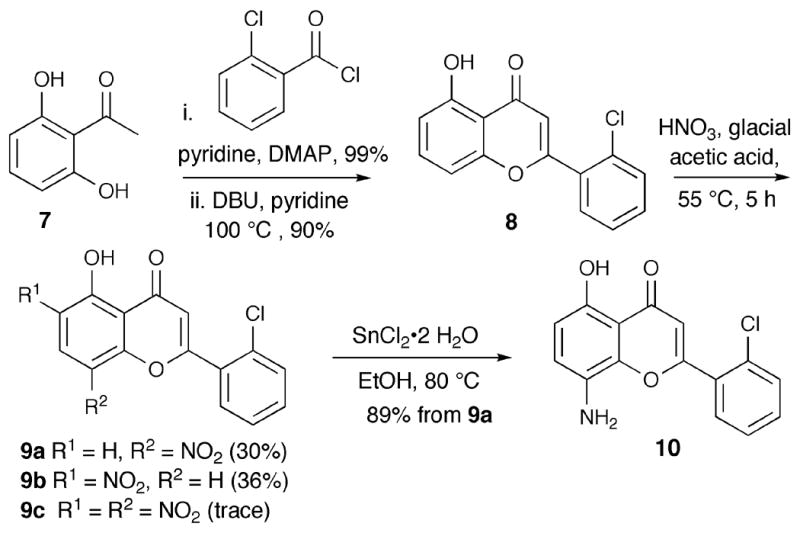
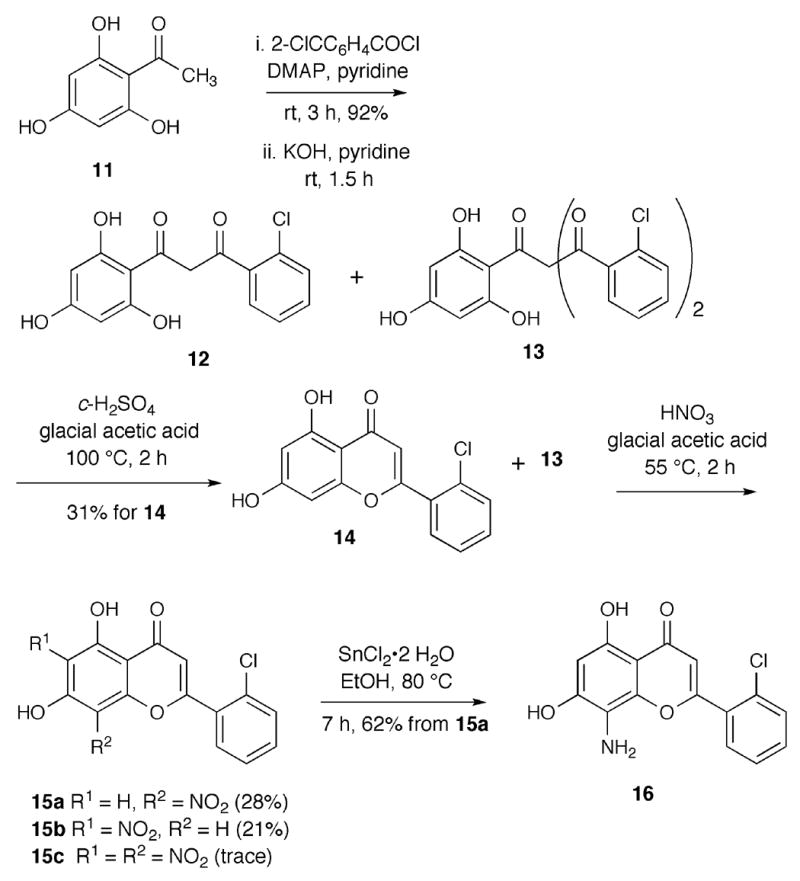
The 8-aminoflavone 10 was synthesized from 2′,6′-dihydroxyacetophenone (7) in four steps (Scheme 1). Reaction of 7 with two equivalents of 2-chlorobenzoyl chloride and a catalytic amount of dimethylaminopyridine (DMAP) in pyridine provided 2′,6′-di(2-chlorobenzoyl)acetophenone. Subsequent Baker-Venkataraman rearrangement 13 using DBU produced flavone 8 in excellent yield over two steps. Nitration with nitric acid and glacial acetic acid at 55 °C generated a 1:1 mixture of 8-nitro- and 6-nitroflavones 9a and 9b, along with traces of the 6,8-dinitroflavone. The nitroflavones 9a and 9b were separated using silica gel column chromatography. The corresponding 8-aminoflavone 10 was obtained by reduction of 9a in the presence of tin chloride dihydrate. The structures of the two nitroflavones 9a and 9b (9a is less polar than 9b) were assigned after X-ray crystallography of 10.
The 8-amino-7-hydroxyflavone 16 was synthesized from 2′,4′,6′-trihydroxyacetophenone (11) in five steps (Scheme 2). Acetophenone 11, was treated with three equivalents of 2-chlorobenzoyl chloride and a catalytic amount of DMAP in pyridine to form 2′,4′,6′-tris(2-chlorobenzoyl)acetophenone, which was treated with KOH (5.0 equiv.) at room temperature for 1.5 h to furnish intermediates 12 and 13 (3:1 ratio for 12:13).14 The mixture of 12 and 13 was subjected to H2SO4 and glacial acetic acid conditions at 100 °C to provide 14 in 31% yield. Nitration of 14 gave a mixture of regioisomers 15a and 15b (1:1 ratio) and a trace amount of 15c. The nitro compound 15a was obtained after column chromatography and 15b was separated from 15c by reverse-phase HPLC on a C-18 column. Subsequent reduction of 15a using tin chloride dihydrate afforded the targeted 8-amino-2-(2-chlorophenyl)-5,7-dihydroxychro men-4-one (16) in good yield.
Parallel synthesis was employed to react various benzoyl chlorides with 8-aminoflavone 10 using Hünig's base to generate 8-amidoflavones 17a–e (Scheme 3).
Scheme 3.
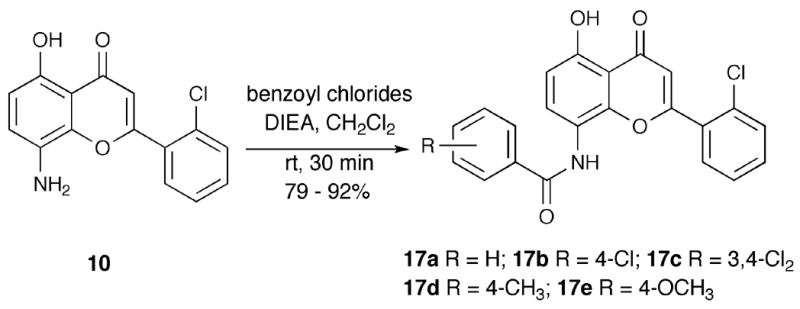
Acylation of 16 furnished N,O-bis-acylated products 18, which were hydrolyzed with KOH to obtain N-acylated products 19 (Scheme 4). The structure of 18b (R = Cl), as well as the assignment of regioisomers 15a and 15b, were confirmed by X-ray crystallography of 18b (R = Cl).
Scheme 4.
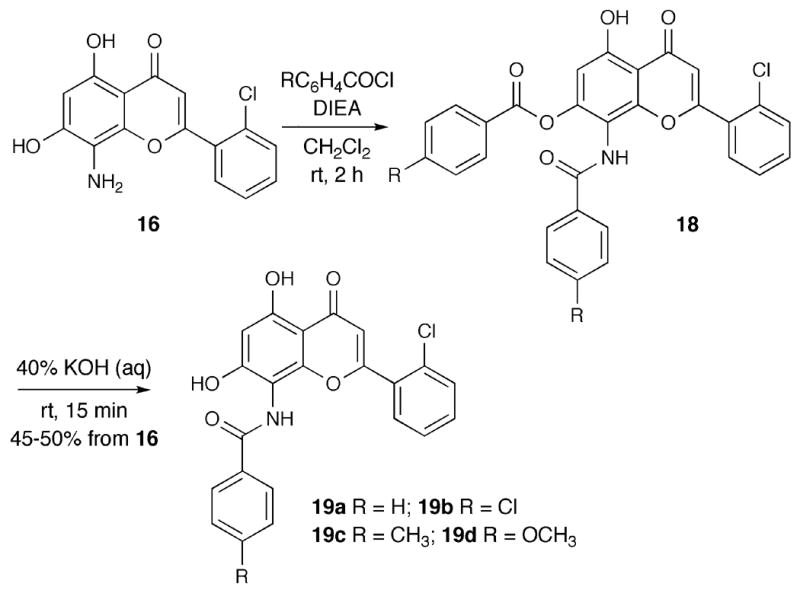
Heterocyclic analogues at C8 were prepared using various alkyl dibromides, which were reacted with 8-aminoflavone 10 under basic conditions to afford the corresponding C8-pyrrolidinyl flavopiridol analogue 20a, C8-piperidinylflavopiridol analogue 20b, and C8-morpholinylflavopiridol analogue 20c (Scheme 5).
Scheme 5.
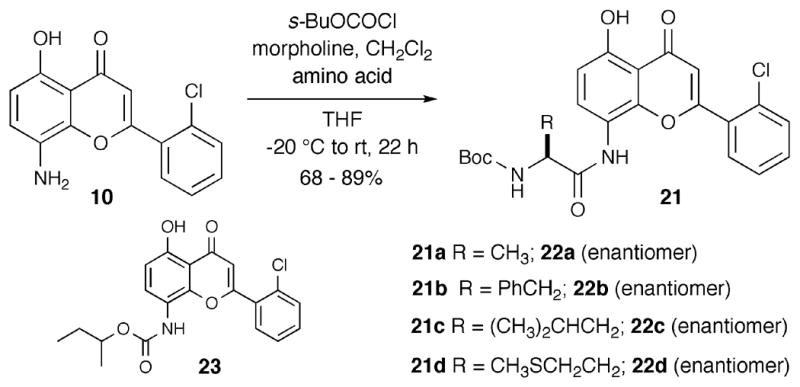
Reactions between N-Boc-protected amino acids and 8-aminoflavone 10 were carried out using 4-methyl morpholine (NMM) as base in THF with sec-butyl chloroformate activation (formation of the mixed anhydride) to yield amides 21a–d and 22a–d (Scheme 6). 8-Aminoflavone 10 also reacted with sec-butyl chloroformate itself to give by-product 23 in less than 10% yield.
Scheme 6.
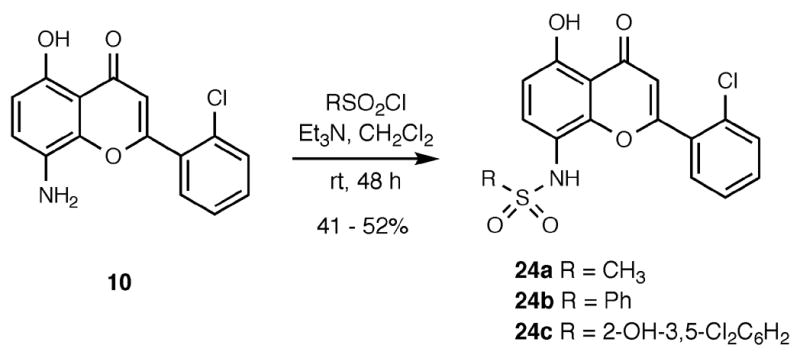
The sulfonamido flavones 24a–c were prepared by treating 8-aminoflavone 10 with a variety of sulfonyl chlorides under basic conditions to yield products 24 in moderate yields (Scheme 7).
Scheme 7.
3. Biological Evaluation and Computational Study
Flavopiridol (1), and compounds 17–24 were tested for their anti-proliferative properties against ovarian cancer ID-8 and breast cancer MCF-7 cell lines and their activities are shown in Table 1. The data reveal that all compounds in Table 1 were less active than flavopiridol. The effect on growth of MCF-7 and ID-8 cells measures the biological efficacy of the flavone derivatives and is a cumulative effect of the inhibition of the target enzyme, the ability of the compounds to reach their intracellular target, stability and solubility of the compounds. A measure of the inhibitory activity of compounds to its intended enzyme target is an important parameter for effective drug design. Therefore, we determined the effect of the flavone derivatives on CDK2-Cyclin A enzymatic activity in vitro. The enzyme activity was measured using a peptide with a recognition sequence specific for CDK2-Cyclin A which contains a chelation enhanced fluorophore, sox (8-hydroxy-5-(N,N-dimethylsulfonamido)-2-methylquinoline).
Table 1.
Antiproliferative and CDK2-Cyclin A inhibitory activities of 8-aminoflavopiridol analogues.
| Compounds | ID-8a IC50 (μM) | MCF-7a IC50 (μM) | CDK2-Cyclin A (μM) |
|---|---|---|---|
| Flavopiridol (1) | 0.0070 | 0.026 | 1.5 |
| 17a | 13 | 7.1 | 417 |
| 17b | 5.5 | 4.6 | 417 |
| 17c | 13 | 15 | 217 |
| 17d | 16 | 10 | N.D. |
| 17e | 5.0 | 3.5 | 383 |
| 19a | N.D. | 8.5 | 91 |
| 19b | N.D. | 13 | 339 |
| 19c | N.D. | 9.7 | 90 |
| 19d | N.D. | 13 | 54 |
| 20a | 9.3 | 20 | 417 |
| 20b | 5.7 | 17 | 417 |
| 20c | 7.9 | 20 | 417 |
| 21a | 104 | 5.5 | N.D. |
| 21b | 12 | 4.5 | 417 |
| 21c | 17 | 2.5 | 417 |
| 21d | 13 | 2.8 | N.D. |
| 22a | 9.8 | 5.5 | 417 |
| 22b | 5.3 | 7.1 | 417 |
| 22c | 5.1 | 4.0 | 417 |
| 22d | 6.2 | 20 | N.D. |
| 23 | 18 | 30 | 417 |
| 24a | 16 | 25 | 219 |
| 24b | 9.5 | 16 | 178 |
| 24c | 24 | 17 | 94 |
Proliferation experiments were performed as previously described.16
CDK2-Cyclin A enzymatic assay is described in the Experimental procedures section. N.D. = not determined.
Phosphorylation of the peptide by the enzyme leads to chelation of Mg2+ and formation of a bridge between the sox moiety and the phosphate resulting in an increase in fluorescence signal. 15 Table 1 shows the IC50 for the compounds. Flavopiridol inhibits CDK2-Cyclin A activity with an IC50 of 1.5 μM. None of the derivatives are good inhibitors with 19d being the best with an IC50 of 54 μM. The results are very similar to the cytotoxicity of these compounds to MCF-7 and ID-8 cells.
In an attempt to understand the decrease of potency of the new analogues compared to flavopiridol, molecular docking simulations of flavone derivatives binding to the CDK2 ATP receptor were carried out using the FlexX 4 program. 17 The results of these studies, revealed a different binding orientation inside the CDK2 binding pocket for these analogues compared to flavopiridol (Figure 4). The receptor model was constructed from the bare uninhibited CDK2 crystal structure including all residues within 7.0 Å of the documented binding pocket.18 Thirty docking poses were requested for each ligand, and structural predictions were based on the conformation of that pose achieving the top FlexX score. The difference in orientation likely arises from the presence of a second carbonyl in molecule 19b (Fig 4b). This second carbonyl, located on the semi-flexible C8-amide chain, can sample a range of torsional orientations and thus is able to adapt to a more optimal electrostatic interaction with Lys33 than is possible for the rigid carbonyl on the chromenone ring.
Figure 4.
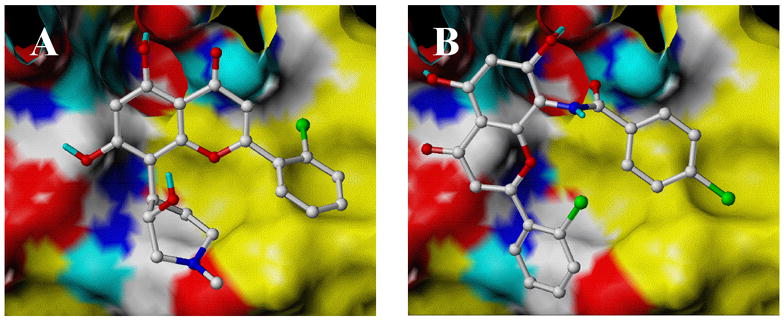
Docked conformations in the CDK2 receptor for flavopiridol (1, Fig. 4A) and flavopiridol analogue 19b (Fig. 4B).
These computational studies have led us to pursue rational design-guided efforts in the preparation of analogues where the amino group has been altered to a bioisosteric equivalent. In this series, a nanomolar inhibitor was discovered. This work will be detailed in a future publication.
In conclusion, we have designed and synthesized a small chemical library of 8-amidoflavone, 8-sulfonylamidoflavone, 8-amido-7-hydroxyflavone, and heterocyclic analogues of flavopiridol. The cytotoxicity of these compounds was evaluated in MCF-7 and ID-8 cell lines, and several were shown to inhibit ID-8 and MCF-7 cancer cell proliferation in the single digit micromolar range. None of the compounds were effective inhibitors of CDK2-Cyclin A. Computational studies have directed us toward novel C8 bioisosteric analogues with promising bioactivity.
4. Experimental Procedures
Column chromatography was carried out employing silica gel (230–400 mesh). Analytical thin-layer chromatography (TLC) was performed on a silica gel 60F254 plate. THF and CH2Cl2 were freshly distilled or purified over an aluminum column before use. Other anhydrous solvents were purchased.
1H NMR and 13C NMR spectra were recorded on a 300 MHz spectrometer (300 and 75.6 MHz, respectively), a 400 MHz spectrometer (400 and 100 MHz, respectively), or a 500 MHz spectrometer (500 and 125.5 MHz, respectively). NMR spectra were recorded in CDCl3 unless otherwise indicated. Abbreviations are as follows: s, singlet; d, doublet; t, triplet; bs, broad singlet, bd, broad doublet. High-resolution mass spectrometry (HRMS) spectra were obtained on a double-focusing mass spectrometer. X-ray crystallography data were collected using an area detector mounted on a D8 platform goniometer using graphite-monochromated Mo Kα radiation (λ= 0.71073Å). The X-ray crystallographic data for compounds 10, and 18b have been deposited with the Cambridge Crystallographic Data Center and have been allocated the deposition numbers CCDC 293761 (10) and CCDC 293762 (18b). Optical rotations were obtained at room temperature. Melting points are uncorrected.
2-(2-Chlorophenyl)-5-hydroxychromen-4-one (8)
To a solution of 2′,6′-dihydroxyacetophenone (2.5 g, 16 mmol) in pyridine (20 mL) was added 2-chlorobenzoyl chloride (5 mL, 38 mmol) and a catalytic amount of dimethylaminopyridine (DMAP). The reaction mixture was stirred at room temperature for 2 h and then poured into ice water (150 mL). The solution was extracted with ethyl acetate (EtOAc). The organic layer was washed with 3N-HCl, NaHCO3 (sat’d aq) and brine. The organic layer was dried (Na2SO4) and the solvent was removed under reduced pressure. The residue was purified by flash silica gel column chromatography (1:9 EtOAc/hexane) to afford 7.0 g (99%) of 2′,6′-di(2-chlorobenzoyl)acetophenone as a white solid: mp 67–68 °C; Rf 0.43 (2:1 hexane/EtOAc); 1H NMR (300 MHz): δ 8.02 (d, J = 8.4 Hz, 2H), 7.3–7.6 (m, 7H), 7.27 (d, J = 8.4 Hz, 2H), 2.51 (s, 3H); 13C NMR (75.6 MHz): δ 198.3, 168.3, 147.8, 134.7, 133.7, 132.1, 131.5, 131.0, 128.5, 128.4, 127.0, 120.8, 31.5; HRMS (FAB) m/z calcd for C22H15O5Cl2 [M + H]+ 429.0297, found 429.0278. To a solution of 2′,6′-di(2-chlorobenzoyl)acetophenone (7.0 g, 16 mmol) in pyridine (40 mL) was added DBU (5.8 mL, 38 mmol). The reaction mixture was heated at 100 °C for 11 h and then poured into ice water (150 mL). The solution was extracted with EtOAc. The organic layer was washed with 3N-HCl, NaHCO3 (sat’d aq) and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by flash silica gel column chromatography (1:9 EtOAc/hexane) to afford 3.9 g (90%) of 8 as a white solid: mp 168-168.5 °C; Rf 0.59 (2:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.5 (s, 1H), 7.66 (dd, J = 1.8, 7.5 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.56 (d, J = 8.3 Hz, 1H), 7.49 (dt, J = 1.6, 7.8 Hz, 1H), 7.44 (dt, J = 1.2, 7.5 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 6.63 (s, 1H); 13C NMR (100 MHz): δ 183.8, 164.1, 161.2, 157.2, 136.0, 133.3, 132.5, 131.8, 131.3, 131.0, 127.6, 112.0, 111.9, 111.2, 107.6; HRMS (FAB) m/z calcd for C15H10O3Cl [M + H]+ 273.0318, found 273.0323.
2-(2-Chlorophenyl)-5-hydroxy-8-nitrochromen-4-one (9a)
To a solution of 8 (360 mg, 1.32 mmol) in glacial acetic acid (10 mL) was added nitric acid (1.3 mL). The reaction mixture was heated at 55 °C (external temperature) for 5 h and then poured into ice water (100 mL). The solution was extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (1:9 EtOAc/hexane) to afford 150 mg (30%) of 9a as a white solid, 180 mg (36%) of 9b as a pale yellow solid and a trace of 9c. Compound 9a: mp 194–195 °C; Rf 0.57 (1:1 hexane/EtOAc); 1H NMR (300 MHz): δ 13.7 (s, 1H), 8.45 (d, J = 9.3 Hz, 1H), 7.98 (m, 1H), 7.65-7.45 (m, 3H), 7.01 (s, 1H), 6.90 (d, J = 9.3 Hz, 1H); 13C NMR (75.6 MHz): δ 182.4, 166.7, 163.4, 133.4, 133.1, 132.9, 131.4, 131.2, 129.7, 127.7, 112.6, 111.5, 110.3; HRMS (FAB) m/z calcd for C15H9O5NCl [M + H]+ 318.0169, found 318.0166.
9b: mp 200-200.5 °C; Rf 0.45 (1:1 hexane/EtOAc); 1H NMR (300 MHz): δ 14.5 (s, 1H), 8.41 (d, J = 9.3 Hz, 1H), 7.66 (dd, J = 1.5, 7.5 Hz, 1H), 7.40–7.62 (m, 3H), 7.04 (d, J = 9.3 Hz, 1H), 6.77 (s, 1H); 13C NMR (75.6 MHz): δ 183.3, 164.6, 159.4, 157.7, 133.1, 132.9, 132.4, 131.2, 130.7, 130.3, 127.5, 112.2, 111.7, 107.5; HRMS (FAB) m/z calcd for C15H9O5NCl [M + H]+ 318.0169, found 318.0169.
8-Amino-2-(2-chlorophenyl)-5-hydroxychromen-4-one (10)
To a solution of 9a (541 mg, 1.7 mmol) in ethanol (200 mL) was added tin chloride hydrate (2.3 g, 10.2 mmol). The reaction mixture was heated at 80 °C for 5 h and then the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (500 mL) and the solution was washed with water. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to obtain 435 mg (89%) of 10 as a reddish powder: mp 197–198 °C; Rf 0.52 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 11.65 (s, 1H), 7.64 (dd, J = 1.7, 7.5 Hz, 1H), 7.57 (dd, J = 1.3, 8.0 Hz, 1H), 7.51 (dt, J = 1.7, 7.9 Hz, 1H), 7.45 (dt, J = 1.4, 7.4 Hz 1H), 7.06 (d, J = 8.7 Hz, 1H), 6.73 (d, J = 8.7 Hz, 1H), 6.57 (s, 1H), 3.77 (br s, 2H); 13C NMR (100 MHz): δ 183.4, 163.2, 152.1, 144.2, 132.8, 132.1, 131.4, 130.9, 130.7, 127.2, 126.8, 111.3, 111.0, 110.9; HRMS (FAB) m/z calcd for C15H11O3NCl [M + H]+ 288.0427, found 288.0415.
2-(2-Chlorophenyl)-5,7-dihydroxychromen-4-one (14)
To a solution of 11 (2.0 g, 0.011 mol) in pyridine (20 mL) was added 2-chlorobenzoyl chloride (5 mL, 0.022 mmol) and a catalytic amount of DMAP. The reaction mixture was stirred at room temperature for 3 h and then was poured into ice water (150 mL). The solution was extracted with EtOAc. The organic layer was washed with 3N-HCl, NaHCO3 (sat’d aq) and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was recrystallized with EtOAc/hexane to afford 5.8 g (92%) of 2′,4′,6′-tri(2-chlorobenzoyloxy)acetophenone as a white solid: mp 141–142 °C; Rf 0.78 (1:1 hexane/EtOAc); 1H NMR (300 MHz)): δ 8.05 (m, 3H), 7.3–7.6 (m, 9H), 2.52 (s, 3H); 13C NMR (75.6 MHz): δ 197.5, 162.8, 162.7, 151.9, 148.4, 134.9, 133.8, 133.7, 133.5, 132.5, 132.2, 131.6(2), 131.5, 128.5, 128.1, 127.0, 126.9, 126.7, 114.6, 31.5; HRMS (FAB) m/z calcd for C29H18O7Cl3 [M + H]+ 583.0118, found 583.0120. To a solution of 2′,4′,6′-tri(2-chlorobenzoyloxy)acetophenone (5.0 g, 8.6 mmol) in pyridine (40 mL) was added KOH (2.0 g, 5 equiv). The reaction mixture was stirred at room temperature for 1.5 h and then poured into ice-cold 10% AcOH (150 mL). The solution was extracted with EtOAc. The organic layer was washed with 3N-HCl, NaHCO3 (sat’d aq) and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to obtain a mixture of 12 and 13 (3:1 ratio by 1H-NMR). The mixture was then treated with c-H2SO4 (3 mL) in glacial acetic acid (50 mL). The solution was heated at 100 °C for 2 h and then poured into ice water (100 mL). The solution was extracted with EtOAc. The organic layer was washed with brine and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to obtain a mixture of unreacted 13 and 14. The mixture was purified by silica gel column chromatography (1:9 EtOAc/CH2Cl2) to obtain a mixture of 13 (0.36 g, 10%) as a pale yellow solid and 14 (0.76 g, 31%) as a white solid. 13: mp 253–254 °C; Rf 0.84 (1:1 acetone/CH2Cl2); 1H NMR (acetone-d6, 300 MHz): δ 12.3 (s, 1H), 10.0 (br s, 1H) 7.66 (dd, J = 1.7, 7.7 Hz, 1H), 7.60 (m, 1H), 7.35–7.55 (m, 5H), 7.34 (dt, J = 1.2, 7.4 Hz, 1H), 6.50 (s, 1H), 6.36 (s, 1H); 13C NMR (acetone-d6, 75.6 MHz): δ 189.8, 180.5, 165.9, 163.7, 158.7, 136.4, 133.8, 133.7, 133.2, 132.8, 132.0, 131.8, 131.4, 130.7, 128.0, 127.8, 124.5, 105.2, 100.6, 95.2; HRMS (FAB) m/z calcd for C22H13O5Cl2 [M + H]+ 427.0140, found 427.0140. 14: mp 258 °C dec.; Rf 0.82 (1:1 acetone/CH2Cl2); 1H NMR (DMSO-d6, 400 MHz): δ 12.7 (br s, 1H), 11.0 (br s, 1H) 7.78 (d, J = 7.8 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.61 (t, J = 7.4 Hz, 1H), 7.54 (t, J = 7.4 Hz, 1H), 6.57 (s, 1H), 6.41 (s,1H), 6.25 (s,1H); 13C NMR (DMSO-d6, 100 MHz): δ 181.5, 164.7, 162.8, 161.5, 157.8, 131.7, 131.6, 131.4, 130.9, 130.5, 127.8, 110.6, 103.9, 99.2, 94.1; HRMS (FAB) m/z calcd for C15H10O4Cl [M + H]+ 289.0268, found 289.0263.
2-(2-Chlorophenyl)-5,7-dihydroxy-8-nitrochromen-4-one (15a)
To a solution of 14 (0.5 g, 1.73 mmol) in glacial acetic acid (10 mL) was added nitric acid (1.5 mL). The reaction mixture was heated at 55 °C for 2 h and then poured into ice water (100 mL). The solution was extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to give a mixture of products 15. The residue was purified by silica gel column chromatography (1:4 EtOAc/CH2Cl2) to afford 160 mg (28%) of 15a a pale yellow solid. Elution with methanol provided a mixture of 15b and 15c. This mixture was purified by prep-HPLC (C-18 column: 4.6 x 150 mm, water/acetonitrile) to yield 121 mg (21%) of 15b as a yellow solid. Compound 15a: mp 223–225 °C; Rf 0.52 (2:1 hexane/EtOAc); 1H NMR (400 MHz): δ 14.0 (s, 1H), 12.0 (d, J = 9.3 Hz, 1H), 7.97 (m, 1H), 7.40–7.70 (m, 3H), 7.06 (s, 1H), 6.51 (s, 1H); 13C NMR (100 MHz): δ 181.6, 167.5, 163.1, 163.0, 153.8, 133.2, 133.0, 131.6, 131.2, 129.7, 127.8, 118.3, 113.3, 105.8, 101.0; HRMS (FAB) m/z calcd for C15H9O6NCl [M + H]+ 334.0118, found 334.0115. 15b: 1H NMR (DMSO-d6, 500 MHz): δ 13.0 (s, 1H), 7.77 (d, J = 7.5 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.57 (t, J = 7.5 Hz, 1H), 6.84 (s, 1H), 6.42 (s, 1H); 13C NMR (75.6 MHz): δ 180.8, 162.4, 162.2, 157.7, 149.8, 133.1, 132.7, 131.3, 130.7, 130.2, 129.9, 121.8, 111,6, 102.8, 99.1; HRMS (FAB) m/z calcd for C15H9O6NCl [M + H]+ 334.0118, found 334.0129.
8-Amino-2-(2-chlorophenyl)-5,7-dihydroxychromen-4-one (16)
To a solution of 15a (130 mg, 0.37 mmol) in ethanol (70 mL) were added tin chloride dihydrate (530 mg, 2.35 mmol). The reaction mixture was heated at 80 °C for 7 h and then the solvent was removed under reduced pressure. The residue was dissolved in EtOAc (300 mL) and the solution was washed with water. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure to afford 73 mg (62%) of 16 as a reddish solid: mp 227 °C dec.; Rf 0.31 (1:1 hexane/EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ 11.9 (s, 1H), 7.88 (dd, J = 1.5, 7.6 Hz, 1H), 7.68 (dd, J = 1.0, 7.1 Hz, 1H), 7.61 (dt, J = 1.5, 7.5 Hz, 1H), 7.54 (dt, J = 1.2, 7.5 Hz, 1H), 6.52 (s, 1H), 6.32 (s, 1H); 13C NMR (DMSO-d6, 100 MHz): δ 182.3, 172.1, 162.7, 151.9, 144.1, 133.0, 132.1, 132.0, 131.5, 131.0, 128.2, 116.8, 110.4, 104.0, 99.0; HRMS (FAB) m/z calcd for C15H11O4NCl [M + H]+ 304.0377, found 304.0370.
General Procedure for Reactions of 10 with Benzoyl Chlorides
The procedure for the synthesis of 17a was also used for the preparation of 17b – 17e.
N-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]benzamide (17a)
To a solution of 10 (14.0 mg, 0.0435 mmol) in CH2Cl2 (2 mL) were added DIEA (17 μL, 0.087 mmol) and benzoyl chloride (11 μL, 0.087 mmol). The reaction mixture was stirred at room temperature for 30 min and then quenched with ice water (20 mL). The solution was extracted with EtOAc and then the organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography (1:4 EtOAc/hexane) to provide 16 mg (85%) of 17a as a yellow solid: mp 224–225 °C; Rf 0.76 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.60 (d, J = 9.0 Hz, 1H), 8.27 (br s, 1H), 7.91 (d, J = 7.3 Hz, 2H), 7.65 (dd, J = 1.3, 7.3 Hz, 1H), 7.6-7.4 (m, 4H), 7.50 (d, J = 7.2 Hz, 2H), 6.92 (d, J = 9.0 Hz, 1H), 6.62 (s, 1H); 13C NMR (100 MHz): δ 182.8, 163.2, 156.5, 146.3, 134.2, 132.4 (2), 131.9, 131.1, 130.9, 130.7, 128.8, 128.7, 128.5, 127.4, 126.9, 117.9, 111.4, 111.1, 110.2; HRMS (FAB) m/z calcd for C22H15O4NCl [M + H]+ 392.0690, found 392.0714.
4-Chloro-N-[2-(2-chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]benzamide (17b)
15.1 mg (82%); mp 246–247.5 °C; Rf 0.73 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.56 (d, J = 9.0 Hz, 1H), 8.22 (br s, 1H), 7.85 (d, J = 8.5 Hz, 2H), 7.64 (dd, J = 1.7, 7.7 Hz, 1H), 7.61 (dd, J = 1.2, 7.0 Hz 1H), 7.56 (dt, J = 1.6, 7.4 Hz, 1H), 7.49 (d, J = 8.5 Hz, 2H), 7.46 (dt, J = 1.2, 7.4 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 6.62 (s, 1H); 13C NMR (100 MHz): δ 183.0, 164.4, 163.4, 156.9, 146.8, 138.5, 132.7, 132.5, 132.0, 131.4, 131.1, 130.8, 129.2, 128.7, 128.5, 127.7, 117.9, 111.6, 111.3, 110.4; HRMS (FAB) m/z calcd for C22H14O4NCl2 [M + H]+ 426.0300, found 426.0277.
3,4-Dichloro-N-[2-(2-chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]benzamide (17c)
15.8 mg (79%); mp 222–223 °C; Rf 0.91 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.0 (s, 1H), 8.54 (d, J = 9.0 Hz, 1H), 8.21 (br s, 1H), 8.02 (d, J = 2.0 Hz, 1H), 7.74 (dd, J = 2.1, 8.4 Hz, 1H), 7.64 (m, 3H), 7.56 (dt, J = 1.6, 7.5 Hz, 1H), 7.48 (dt, J = 1.2, 7.6 Hz, 1H), 6.92 (d, J = 9.0 Hz, 1H), 6.63 (s, 1H); 13C NMR (100 MHz): δ 182.7, 163.2, 162.9, 156.8, 148.3, 136.4, 133.9, 133.3, 132.6, 132.2, 131.2, 130.9, 130.8, 130.5, 129.1, 128.1, 127.6, 126.0, 117.4, 111.3, 111.1, 110.2; HRMS (FAB) m/z calcd for C22H13O4NCl3 [M + H]+ 459.9910, found 459.9903.
N-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]-4-methylbenzamide (17d)
16.9 mg (92%); mp 252.5–254 °C; Rf 0.72 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.61 (d, J = 8.9 Hz, 1H), 8.26 (brs, 1H), 7.81 (d, J = 7.9 Hz, 2H), 7.64 (d, J = 7.5 Hz, 1H), 7.60 (d, J = 7.9 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.31 (d, J = 7.9 Hz, 2H), 6.91 (d, J = 9.0 Hz, 1H), 6.62 (s, 1H), 2.44 (s, 3H); 13C NMR (100 MHz): δ 183.1, 165.5, 163.4, 156.7, 146.5, 142.8, 132.7, 131.6, 131.4, 131.2, 131.0, 129.6, 128.7, 127.7, 127.2, 118.4, 111.6, 111.4, 110.5, 21.6; HRMS (FAB) m/z calcd for C23H17O4NCl [M + H]+ 426.0846, found 426.0842.
N-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]-4-methoxybenzamide (17e)
15.8 mg (86%); mp 210–210.5 °C; Rf 0.76 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.0 (s, 1H), 8.56 (d, J = 9.0 Hz, 1H), 8.20 (br s, 1H), 7.87 (d, J = 8.7 Hz, 2H), 7.64 (dd, J = 1.3, 7.7 Hz, 2H), 7.59 (d, J = 7.8 Hz, 1H), 7.52 (dt, J = 1.3, 7.9 Hz, 1H), 7.45 (t, J = 7.6 Hz, 3H), 6.98 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 9.0 Hz, 1H), 6.60 (s, 1H); 13C NMR (100 MHz): δ 183.0, 165.0, 163.3, 162.6, 156.5, 146.4, 132.6, 131.2, 131.1, 130.9, 128.9, 128.7, 127.6, 126.5, 118.4, 114.1, 111.5, 111.2, 110.4, 55.5; HRMS (FAB) m/z calcd for C23H17O5NCl [M + H]+ 422.0795, found 422.0781.
2-(2-Chlorophenyl)-8-(4-chlorophenylamido)-5-hydroxy-4-oxo-4H-chromen-7-yl 4-Chlorobenzoate (18b)
To a solution of 16 (30.0 mg, 0.099 mmol) in CH2Cl2 (3 mL) were added DIEA (34.5 μL, 0.20 mmol) and benzoyl chloride (25.2 μL, 0.20 mmol). The reaction mixture was stirred at room temperature for 40 min and then quenched with ice water (20 mL). The solution was extracted with EtOAc and the organic layer was dried over anhydrous Na2SO4 and the removed under reduced pressure. The residue was purified by silica gel column chromatography (1:5 EtOAc/hexane) to yield 20 mg (50%) of 18b as a yellow solid: mp 241°C; Rf 0.57 (1:1 hexane/EtOAc); 1H NMR (DMSO-d6, 400 MHz): δ 12.8 (s, 1H), 10.3 (s, 1H), 7.83 (d, J = 1.2Hz, 1H), 7.66 (m, 5H), 7.55(d, J = 1.3, 2H), 7.40 (m, 4H), 7.09 (s, 1H), 6.87(s, 1H); 13C NMR (DMSO-d6, 100MHz): δ 183.3, 171.4, 163.5, 161.5, 157.2, 156.3, 147.2, 133.1, 132.8, 131.6, 131.5, 131.3, 131.2, 129.5, 128.5, 127.4, 111.0, 104.9, 104.5, 99.8; MS (FAB) m/z calcd for C29H17Cl3NO6 [M + H]+ 581.8, Found 582.1.
General Procedure for Reactions with Benzoyl Chlorides
The procedure for the synthesis of 19a was also used for the preparation of 19b – 19d.
N-[2-(2-Chlorophenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl]benzamide (19a)
To a solution of 16 (30.0 mg, 0.099 mmol) in CH2Cl2 (2 mL) were added DIEA (19 μL, 0.11mmol) and benzoyl chloride (13 μL, 0.11 mmol). The reaction mixture was stirred at room temperature for 2 h and then the quenched with ice water (20 mL). The solution was extracted with EtOAc and then the solvent was removed until the volume of the solution was around 6 mL. To the solution was added 40% KOH (4 mL). The solution was stirred at room temperature for 15 min. and then the organic layer was washed with water, 3N-HCl and brine. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (1:5 EtOAc/hexane) to yield 20 mg (50%) of 19a as a yellow solid: mp 205–206 °C; Rf 0.44 (1:1 hexane/EtOAc); 1H NMR (300 MHz): δ 12.2 (s, 1H), 11.2 (bs, 1H), 8.50 (br s, 1H), 7.93 (dd, J = 1.3, 7.2 Hz, 2H), 7.62 (dt, J = 1.6, 7.6 Hz, 2H), 7.4–7.6 (m, 5H), 6.54 (s, 1H), 6.53 (s, 1H); 13C NMR (100 MHz): δ 181.7, 174.3, 164.8, 167.4, 162.7, 159.7, 157.1, 148.8, 133.0, 132.7, 131.2 (2), 131.1, 129.1, 127.8, 127.6, 111.6, 106.8, 104.8, 102.7; HRMS (FAB) m/z calcd for C22H15O5NCl [M + H]+ 408.0639, found 408.0638.
4-Chloro-N-[2-(2-chlorophenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl]benzamide (19b)
20 mg (45%); mp 234 °C; Rf 0.38 (1:1 hexane/EtOAc); 1H NMR (acetoned6, 500 MHz): δ 12.6 (s, 1H), 9.21 (bs, 1H), 8.04 (d, J = 8.4 Hz, 2H), 7.84 (dd, J = 1.6, 7.7 Hz, 1H), 7.54 (dt, J = 1.3, 8.0 Hz, 1H), 7.51 (dd, J = 1.6, 7.9 Hz, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.44 (dt, J = 1.3, 8.0 Hz, 1H), 6.61 (s, 1H), 6.39 (s, 1H); 13C NMR (acetone-d6, 125.5 MHz): δ 182.6, 166.8, 163.0, 161.3, 153.6, 138.3, 133.4, 133.1, 132.9, 131.9, 131.8, 131.6, 130.3, 129.3, 128.3, 111.8, 105.8, 105.3, 100.6; HRMS (FAB) m/z calcd for C22H14O5NCl2 [M + H]+ 442.0249, found 442.0250.
N-[2-(2-Chlorophenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl]-4-methylbenzamide (19c)
21 mg (49%); mp 210–212 °C; Rf 0.50 (1:1 hexane/EtOAc); 1H NMR (DMSO-d6, 300 MHz): δ 12.7 (s, 1H), 11.1 (br s, 1H), 9.52 (s, 1H), 7.86 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 7.5 Hz, 1H), 7.4–7.7 (m, 3H), 7.29 (d, J = 7.8 Hz, 2H), 6.69 (s, 1H), 6.42 (s, 1H), 2.36 (s, 3H); 13C NMR (DMSO-d6, 100 MHz): δ 181.5, 166.0, 162.1, 161.2, 159.3, 153.4, 141.3, 132.7, 131.3, 131.1, 130.7, 130.4, 128.8, 127.7, 127.6, 110.5, 105.0, 103.7, 98.9, 20.9; HRMS (FAB) m/z calcd for C23H17O5NCl [M + H]+ 422.0795, found 422.0786.
N-[2-(2-Chlorophenyl)-5,7-dihydroxy-4-oxo-4H-chromen-8-yl]-4-methoxybenzamide (19d)
24 mg (55%); mp 208 °C; Rf 0.53 (1:1 hexane/EtOAc); 1H NMR (300 MHz): δ 12.2 (s, 1H), 11.2 (br s, 1H), 8.38 (br s, 1H), 7.88 (d, J = 8.8 Hz, 2H), 7.62 (m, 2H), 7.54 (dt, J = 1.6, 7.5 Hz, 1H), 7.46 (dt, J = 1.0, 7.5 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 6.52 (s, 1H), 6.51 (s, 1H), 3.88 (s, 3H); 13C NMR (100 MHz): δ 181.7, 166.9, 163.4, 162.6, 159.4, 157.1, 148.6, 132.6, 132.4, 131.2, 131.1 129.5, 127.7, 124.3, 114.3, 111.6, 106.9, 104.7, 102.7, 55.6, 29.7; HRMS (FAB) m/z calcd for C23H17O6NCl [M + H]+ 438.0744, found 438.0729.
General Procedure for the Formation of heterocyclic analogues
The procedure for the synthesis of 20a was also used for the preparation of 20b and 20c.
2-(2-Chlorophenyl)-5-hydroxy-8-pyrrolidin-1-yl-chromen-4-one (20a)
To a solution of 10 (20 mg, 0.070 mmol) in DMF (1 mL) were added DIEA (100 μL) and 1,4-dibromobutane (40 μL, 0.35 mmol). The reaction mixture was heated at 65 °C for 18 h and then the solution was diluted with EtOAc (50 mL). The solution was washed with Na2CO3 (50 mL) and then the organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography (1:5 EtOAc/hexane) to afford 17 mg (71%) of 20a as a yellow solid; mp 144–145 °C; Rf 0.68 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.0 (s, 1H), 7.64 (dd, J = 1.8, 7.4 Hz, 1H), 7.55 (dd, J = 1.2, 7.9 Hz, 1H), 7.45 (m, 2H), 7.08 (d, J = 8.9 Hz, 1H), 6.78 (d, J = 8.9 Hz, 1H), 6.59 (s, 1H), 3.33 (t, J = 6.4 Hz, 4H), 1.94 (apparent quintet, J = 3.4, 6.5 Hz, 4H); 13C NMR (100 MHz): δ 183.8, 163.0, 152.5, 147.2, 132.8, 132.0, 131.8, 131.3, 130.8, 130.7, 127.2, 121.5, 111.7, 111.4, 110.9, 51.0, 24.9; HRMS (FAB) m/z calcd for C19H17O3NCl [M + H]+ 342.0897, found 342.0882.
2-(2-Chlorophenyl)-5-hydroxy-8-piperidin-1-yl-chromen-4-one (20b)
23 mg (91%); mp 120–121 °C; Rf 0.87 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.2 (s, 1H), 7.75 (dd, J = 2.0, 7.3 Hz, 1H), 7.56 (dd, J = 1.7, 7.6 Hz, 1H), 7.45 (m, 2H), 7.27 (d, J = 8.8 Hz, 1H), 6.78 (d, J = 8.8 Hz, 1H), 6.75 (s, 1H), 3.00 (t, J = 5.1 Hz, 4H), 1.73 (quintet, J = 5.9, 11.4 Hz, 4H), 1.58 (quintet, J = 6.1, 11.6 Hz, 2H); 13C NMR (100 MHz): δ 183.8, 162.8, 155.2, 149.9, 134.1, 132.8, 132.0, 131.4, 131.1, 130.8, 127.2, 125.7, 111.8, 111.4, 110.7, 53.4, 26.4, 24.2; HRMS (FAB) m/z calcd for C20H19O3NCl [M + H]+ 356.1053, found 356.1030.
2-(2-Chlorophenyl)-5-hydroxy-8-morpholin-4-yl-chromen-4-one (20c)
14 mg (55%); mp 137–139 °C; Rf 0.81 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.2 (s, 1H), 7.69 (dd, J = 1.7, 7.7 Hz, 1H), 7.57 (dd, J = 1.2, 7.9 Hz, 1H), 7.47 (m, 2H), 7.28 (d, J = 8.8 Hz, 1H), 6.80 (d, J = 8.8 Hz, 1H), 6.71 (s, 1H), 3.85 (t, J = 4.5 Hz, 4H), 3.08 (t, J = 4.6 Hz, 4H); 13C NMR (100 MHz): δ 183.6, 163.1, 155.9, 150.0, 132.7, 132.5, 132.2, 131.4, 131.1, 130.8 127.3, 125.8, 111.9, 111.4, 110.9, 67.2, 52.1; HRMS (FAB) m/z calcd for C19H17O4NCl [M + H]+ 358.0846, found 358.0828.
General Procedure for the Reactions with Amino Acids
The procedure for the synthesis of 21a was also used for the preparation of 21b – 20d.
(R)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]ethyl}carbamic Acid tert-Butyl Ester (21a)
To a solution of D-N-Boc-alanine (19 mg, 0.1 mmol) in THF (2 mL) were added 4-methylmorpoline (10 μL, 0.1 mmol) and iso-butylchloroformate (10 μL, 0.08 mmol) at −20 °C. 8-Amino-2-(2-chlorophenyl)-5-hydroxychromen-4-one (10, 24 mg, 0.084 mmol) was dissolved in THF (1 mL) and then the solution was added into the reaction mixture at −20 °C and stirred at the same temperature for 30 min. The reaction mixture was stirred at room temperature for 22 h and then the solution was diluted with EtOAc (30 mL). The organic layer was washed with brine and then dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography (1:5 EtOAc/hexane) to afford a mixture of 32 mg (84%) of 21a as a yellow solid and 3 mg (9%) of 23 as a yellow solid. 21a: mp 174–175 °C; Rf 0.65 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.60 (br s, 1H), 8.38 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 6.8 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.51 (dt, J = 1.4, 7.3 Hz, 1H), 7.45 (t, J = 7.3 Hz, 1H), 6.81 (d, J = 9.0 Hz, 1H), 6.63 (s, 1H), 5.05 (br s, 1H), 4.35 (br s, 1H), 1.44 (d, J = 9.6 Hz, 3H), 1.30 (s, 9H); 13C NMR (100 MHz): δ 182.9, 171.0, 163.1, 156.7,155.8, 146.6, 132.7, 132.4, 131.1, 130.8, 128.5, 127.5, 117.8, 111.8, 111.0, 110.3, 80.4, 50.8, 28.1, 17.7; HRMS (FAB) m/z calcd for C23H24O6N2Cl [M + H]+ 459.1323, found 459.1315; [α]20D +6.1 (c 0.85, CHCl3). 23: mp 136.5–137 °C; Rf 0.87 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.0 (s, 1H), 8.21 (br s, 1H), 7.64 (dd, J = 1.7, 7.6 Hz, 1H), 7.59 (dd, J = 1.1, 7.9 Hz, 1H), 7.53 (dt, J = 1.8, 7.5 Hz, 1H), 7.46 (dt, J = 1.3, 7.6 Hz, 1H), 6.87 (br s, 1H), 6.86 (d, J = 9.0 Hz, 1H), 6.59 (s, 1H), 3.97 (d, J = 6.6 Hz, 2H), 1.99 (m, 1H), 0.96 (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz): δ 183.0, 163.3, 132.8, 132.4, 131.2, 130.9, 130.8, 127.4, 111.5, 111.2, 110.4, 71.7, 27.9, 19.0; HRMS (FAB) m/z calcd for C20H19O5NCl [M + H]+ 388.0952, found 388.0944.
(R)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-2-phenylethyl}carbamic Acid tert-Butyl Ester (21b)
31 mg (68%); mp 217–218 °C; Rf 0.75 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.0 (s, 1H), 8.43 (d, J = 9.0 Hz, 1H), 8.05 (br s, 1H), 7.61 (dd, J = 1.3, 7.7 Hz, 1H), 7.56 (dd, J = 1.3, 7.9 Hz, 1H), 7.52 (dt, J = 1.7, 7.9 Hz, 1H), 7.48 (dt, J = 1.6, 7.5 Hz, 1H), 7.1–7.25 (m, 5H), 6.83 (d, J = 9.0 Hz, 1H), 6.58 (s, 1H), 5.08 (br s, 1H), 4.49 (br s, 1H), 3.16 (d, J = 6.3 Hz, 2H), 1.32 (s, 9H); 13C NMR (100 MHz): δ 182.9, 169.6, 163.1, 156.7, 155.5, 146.2, 136.2, 132.7, 132.4, 131.2, 131.0, 130.8, 129.2, 128.7, 128.1, 127.5, 127.0, 117.5, 111.6, 111.1, 110.3, 80.5, 56.7, 38.3, 28.1; HRMS (FAB) m/z calcd for C29H28O6N2Cl [M + H]+ 535.1636, found 535.1641; [α]20D +38 (c 0.22, CHCl3).
(R)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-3-methylbutyl}carbamic Acid tert-Butyl Ester (21c)
35 mg (84%) was prepared in a similar procedure as 21a; mp 169 °C; Rf 0.80 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.43 (br s, 1H), 8.35 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 6.7 Hz, 1H), 7.58 (dd, J = 1.2, 7.8 Hz, 1H), 7.51 (dt, J = 1.7, 7.4 Hz, 1H), 7.46 (dt, J = 1.4, 7.5 Hz, 1H), 6.82 (d, J = 9.0 Hz, 1H), 6.63 (s, 1H), 4.96 (br s, 1H), 4.27 (br s, 1H), 1.77 (m, 2H), 1.55 (m, 1H), 1.33 (s, 9H), 0.95 (d, J = 3.0 Hz, 3H), 0.94 (d, J = 2.9 Hz, 3H); 13C NMR (100 MHz): δ 182.9, 171.0, 163.1, 156.8, 155.7, 146.8, 132.7, 132.4, 131.1, 130.8, 128.8, 127.5, 117.6, 111.8, 111.1, 110.4, 80.3, 53.8, 40.9, 28.2, 24.8, 22.9, 22.0; HRMS (FAB) m/z calcd for C26H30O6N2Cl [M + H]+ 501.1792, found 501.1787; [α]20D +19 (c 0.58, CHCl3).
(R)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-3-methylsulfanylpropyl}carbamic Acid tert-Butyl Ester (21d)
36 mg (82%); mp 162–163 °C; Rf 0.65 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.1 (s, 1H), 8.51 (br s, 1H), 8.39 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 7.0 Hz, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.52 (dt, J = 1.7, 7.5 Hz, 1H), 7.47 (dt, J = 1.2, 7.5 Hz, 1H), 6.84 (d, J = 9.0 Hz, 1H), 6.61 (s, 1H), 5.24 (d, J = 5.4 Hz, 1H), 4.45 (m, 1H), 2.59 (m, 2H), 2.21 (m, 1H), 2.06 (s, 3H), 2.00 (m, 1H), 1.35 (s, 9H); 13C NMR (100 MHz): δ 183.1, 170.1, 163.3, 157.1, 146.8, 133.0, 132.6, 131.3, 131.2, 131.0, 128.7, 127.6, 117.7, 112.0, 111.3, 110.5, 80.7, 54.3, 31.2, 30.4, 28.3, 15.4; HRMS (FAB) m/z calcd for C25H28O6N2ClS [M+ H]+ 519.1357, found 519.1350; [α]20D +11 (c 0.15, CHCl3).
(S)-(1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]ethyl)carbamic Acid tert-Butyl Ester (22a)
34 mg (89%); mp 176 °C; HRMS (FAB) m/z calcd for C23H24O6N2Cl [M + H]+ 459.1323, found 459.1311; [α]20D −5.9 (c 0.80, CHCl3).
(S)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-2-phenylethyl}carbamic Acid tert-Butyl Ester (22b)
32 mg (72%); mp 217–218 °C; HRMS (FAB) m/z calcd for C29H27O6N2Cl M+ 534.1558, found 534.1549; [α]20D −35 (c 0.46, CHCl3).
(S)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-3-methylbutyl}carbamic acid tert-Butyl Ester (22c)
31 mg (74%) was prepared in a similar procedure as 21a; mp 170–171 °C; HRMS (FAB) m/z calcd for C26H30O6N2Cl [M + H]+ 501.1792, found 501.1788; [α]20D −18 (c 0.24, CHCl3).
(S)-{1-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-ylcarbamoyl]-3-methylsulfanyl-propyl}carbamic Acid tert-Butyl Ester (22d)
31 mg (71%); mp 159–160 °C; HRMS (FAB) m/z calcd for C25H28O6N2ClS [M + H]+ 519.1357, found 519.1335; [α]20D −12 (c 0.49, CHCl3).
General Procedure for Reactions with Sulfonyl Chlorides
The procedure for the synthesis of 24a was also used for the preparation of 24b and 24c.
N-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]methanesulfonamide (24a)
To a solution of 10 (30 mg, 0.104 mmol) in CH2Cl2 (3 mL) were added Et3N (22 μL, 0.16 mmol) and methylsulfonyl chloride (10 μL, 0.13 mmol). The reaction mixture was stirred at room temperature for 48 h and then the solution was diluted with EtOAc (30 mL). The organic layer was washed with brine and then dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (1:5 EtOAc/hexane) to afford 15 mg (52%) of 24a as a yellow solid: mp 162–164 °C; Rf 0.59 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.3 (s, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.63 (dd, J = 1.6, 7.6 Hz, 1H), 7.59 (dd, J = 1.4, 8.0 Hz, 1H), 7.54 (dt, J = 1.4, 8.0 Hz, 1H), 6.47 (dt, J = 1.7, 7.5 Hz, 1H), 6.88 (d, J = 8.9 Hz, 1H), 6.61 (s, 1H), 6.54 (br s, 1H), 2.99 (s, 3H); 13C NMR (100 MHz): δ 183.0, 163.8, 159.2, 149.2, 132.9, 132.8, 132.1, 131.3, 131.0, 130.9, 127.8, 115.7, 112.2, 112.1, 110.9, 40.2; HRMS (FAB) m/z calcd for C16H13O5NSCl [M + H]+ 366.0203, found 366.0193.
N-[2-(2-Chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]benzenesulfonamide (24b)
18 mg (41%); mp 176–177 °C; Rf 0.64 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.3 (s, 1H), 7.78 (d, J = 8.9 Hz, 1H), 7.59 (dd, J = 1.3, 8.3 Hz, 2H), 7.55 (m, 2H), 7.40 (m, 2H), 7.32 (dd, J = 1.2, 7.7 Hz, 1H), 7.18 (t, J = 7.6 Hz, 2H), 6.85 (d, J = 8.9 Hz, 1H), 6.68 (br s, 1H), 6.48 (s, 1H); 13C NMR (100 MHz): δ 182.8, 163.0, 159.5, 149.7, 139.0, 133.9, 133.3, 132.8, 132.7, 131.3, 130.9, 130.5, 128.9, 127.5, 127.2, 115.2, 112.0, 111.8, 110.5; HRMS (FAB) m/z calcd for C21H15O5NSCl [M + H]+ 428.0359, found 428.0352.
3,5-Dichloro-N-[2-(2-chlorophenyl)-5-hydroxy-4-oxo-4H-chromen-8-yl]-2-hydroxybenzenesulfonamide (24c)
23 mg (44%); mp 192–194 °C; Rf 0.32 (1:1 hexane/EtOAc); 1H NMR (400 MHz): δ 12.4 (br s, 1H), 8.17 (br s, 1H), 7.72 (d, J = 8.9 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.54 (dt, J = 2.4, 8.0 Hz, 1H), 7.46 (m, 2H), 7.36 (d, J = 2.4 Hz, 1H), 7.28 (d, J = 2.4 Hz, 1H), 6.87 (d, J = 8.9 Hz, 1H), 6.86 (br s, 1H), 6.55 (s, 1H); 13C NMR (100 MHz): δ 182.7, 163.3, 160.7, 135.0, 134.9, 133.0, 132.7, 131.6, 130.8, 130.3, 127.9, 126.9, 125.5, 124.2, 113.4, 112.3, 112.1, 110.7; HRMS (FAB) m/z calcd for C21H13O6NSCl3 [M + H]+ 511.9529, found 511.9534.
CDK2-CylinA activity assays
The CDK2-Cyclin A (Invitrogen Corporation, CA, USA) enzyme activity was measured by a fluorescence kinetic assay using the Biosource Omnia Ser/Thr recombinant kit 7 (Invitrogen Corporation, CA,USA). The assay was carried out in a volume of 30 μl at room temperature in 384 well black polystyrene plates. The final concentration of the assay constituents were 1 μM CDK2-Cylin A, 10 μM peptide substrate and 1 mM ATP. The compounds were solubilized in DMSO and added to the reaction mixture at six concentrations with the highest concentration of 100 μM for flavopiridol and 417 μM for the rest of the flavone derivatives. The final concentration of the DMSO in the mixture was 1% for flavopiridol and 17% for the derivatives. The presence of 17% DMSO did not significantly affect the activity of the enzyme. Continuous kinetic monitoring of enzyme activity was performed on Spectramax Gemini reader (λex 355 nm and λem 460 nm) and controlled by the Softmax software. The experiment was carried out in triplicates and the percent inhibition of enzyme activity was calculated for all the compounds at each concentration. The IC50 values were obtained from the non-linear curve fitting of the plot of percent inhibition vs. inhibitor concentration [I] using the equation, %inhibition=100/{1+ (IC50/[I]) k}, where k is the Hill coefficient. For compounds where no inhibition was observed, the IC50 value is reported at the highest concentration tried i.e. 417 μM.
Supplementary Material
Supplementary Information
Supplementary information available: 1H NMR and 13C NMR spectra for compounds 8, 9a, 9b, 10, 12, 14, 15a, 15b, 16, 17a–17e, 18b, 19a–19d, 20a–20c, 21a–21d, 22a–22d, 23, 24a–24c; 69 pages.
Acknowledgments
This investigation was supported by NIH-COBRE award 1 P20 RR15563 (NIH-NCRR) with matching support from the State of Kansas and the University of Kansas. The authors thank Dr. David Vander Velde and Sarah Neuenswander for the assistance with the NMR measurements, Dr. Todd Williams for HRMS analysis and Jacquelyn K. Huff for technical assistance. The authors also thank the National Science Foundation (grant CHE-0079282) and the University of Kansas for funds to purchase the X-ray instrument and computers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Fischer PM, Lane DP. Curr Med Chem. 2000;7:1213. doi: 10.2174/0929867003374048. [DOI] [PubMed] [Google Scholar]; (b) Harper JW, Adams PD. Chem Rev. 2001;101:2511. doi: 10.1021/cr0001030. [DOI] [PubMed] [Google Scholar]; (c) Cohen P. Nat Rev Drug Discov. 2002;1:309. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 2.(a) Rosania GR, Chang YT. Expert Opin Ther Pat. 2000;10:215. [Google Scholar]; (b) Sausville EA, Johnson J, Alley M, Zaharevitz D, Senderowicz AM. Ann N Y Acad Sci. 2000;910:207. doi: 10.1111/j.1749-6632.2000.tb06710.x. [DOI] [PubMed] [Google Scholar]; (c) Sridhar R, Hanson-Painton O, Cooper DR. Pharm Res. 2000;17:1345. doi: 10.1023/a:1007507224529. [DOI] [PubMed] [Google Scholar]; (d) Mani S, Wang C, Wu K, Francis R, Pestell R. Expert Opin Invest Drugs. 2000;9:1849. doi: 10.1517/13543784.9.8.1849. [DOI] [PubMed] [Google Scholar]; (e) Noble MEM, Endicott JA, Johnson LN. Science. 2004;303:1800. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]; (f) Toogood PL. Curr Opin Chem Biol. 2002;6:472. doi: 10.1016/s1367-5931(02)00342-3. [DOI] [PubMed] [Google Scholar]
- 3.(a) Sielecki TM, Boylan JF, Benfield PA, Trainor GL. J Med Chem. 2000;43:1. [PubMed] [Google Scholar]; (b) Crews CM, Mohan R. Curr Opin Chem Biol. 2000;4:47. doi: 10.1016/s1367-5931(99)00050-2. [DOI] [PubMed] [Google Scholar]
- 4.(a) Furusaki A, Hashiba N, Matsumoto T, Hirano A, Iwai Y, Omura S. J Chem Soc, Chem Commun. 1978:800. [Google Scholar]; (b) Furusaki A, Hashiba N, Matsumoto T, Hirano A, Iwai Y, Omura S. Bull Chem Soc Jap. 1982;55:3681. [Google Scholar]; (c) Sausville EA. Curr Med Chem Anti-Cancer Agents. 2003;3:47. doi: 10.2174/1568011033353560. [DOI] [PubMed] [Google Scholar]; (d) Senderowicz AM. Cancer Bio Ther. 2003;24(Suppl 1):S84. [PubMed] [Google Scholar]
- 5.(a) Gray NS, Wodicka L, Thunnissen AMWH, Norman TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L, Kim SH, Lockhart DJ, Schultz PG. Science. 1998;281:533. doi: 10.1126/science.281.5376.533. [DOI] [PubMed] [Google Scholar]; (b) Chang YT, Gray NS, Rosania GR, Sutherlin DP, Kwon S, Norman TC, Sarohia R, Leost M, Meijer L, Schultz PG. Chem Biol. 1999;6:361. doi: 10.1016/S1074-5521(99)80048-9. [DOI] [PubMed] [Google Scholar]
- 6.(a) Losiewicz MD, Carlson BA, Kaur G, Sausville EA, Worland PJ. Biochem Biophys Res Commun. 1994;210:589. doi: 10.1006/bbrc.1994.1742. [DOI] [PubMed] [Google Scholar]; (b) Sedlacek HH, Czech J, Naik R, Kaur G, Worland P, Losiewicz M, Parker B, Carlson B, Smith A. Int J Oncol. 1996;9:1143. doi: 10.3892/ijo.9.6.1143. [DOI] [PubMed] [Google Scholar]; (c) Carlson BA, Dubay MM, Sausville EA, Brizuela L, Worland PJ. Cancer Res. 1996;56:2973. [PubMed] [Google Scholar]; (d) Senderowicz AM. New Drugs. 1999;17:313. doi: 10.1023/a:1006353008903. [DOI] [PubMed] [Google Scholar]; (e) Kelland LR. Expert Opin Invest Drugs. 2000;9:2903. doi: 10.1517/13543784.9.12.2903. [DOI] [PubMed] [Google Scholar]; (f) Wang HK. Expert Opin Invest Drugs. 2000;9:2103. doi: 10.1517/13543784.9.9.2103. [DOI] [PubMed] [Google Scholar]; (g) Filgueira de Azevedo W, Jr, Mueller-Dieckmann H-J, Schulze-Gahmen U, Worland PJ, Sausville E, Kim S-H. Proc Natl Acad Sci USA. 1996;93:2735. doi: 10.1073/pnas.93.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Kim SH. Pure Appl Chem. 1998;70:555. [Google Scholar]; (i) Kim S-H, Schulze-Gahmen U, Brandsen J, Filgueira de Azevedo W., Jr Prog Cell Cycle Res. 1996;2:137. doi: 10.1007/978-1-4615-5873-6_14. [DOI] [PubMed] [Google Scholar]
- 7.(a) Kim KS, Sack JS, Tokarski JS, Qian L, Chao ST, Leith L, Kelly YF, Misra RN, Hunt JT, Kimball SD, Humphreys WG, Wautlet BS, Mulheron JG, Webster KR. J Med Chem. 2000;43:4126. doi: 10.1021/jm000231g. [DOI] [PubMed] [Google Scholar]; (b) Schoepfer J, Fretz H, Chaudhuri B, Muller L, Seeber E, Meijer L, Lozach O, Vangrevelinghe E, Furet P. J Med Chem. 2002;45:1741. doi: 10.1021/jm0108348. [DOI] [PubMed] [Google Scholar]
- 8.Matter WF, Brown RF, Vlahos CJ. Biochem Biophys Res Commun. 1992;31:624. doi: 10.1016/0006-291x(92)90792-j. [DOI] [PubMed] [Google Scholar]
- 9.(a) Naik RG, Kattige SL, Bhat SV, Alreja B, de Souza NJ, Rupp RH. Tetrahedron. 1988;44:2081. [Google Scholar]; (b) Yang DH, Cai SQ, Zhao YY, Liang H. J Asian Nat Prod Res. 2004;6:233. doi: 10.1080/10286020310001608930. [DOI] [PubMed] [Google Scholar]
- 10.Kaur G, Stetler-Stevenson M, Sebers S, Worland P, Sedlacek H, Myers C, Czech J, Naik R, Sausville E. J Nat Cancer Inst. 1992;84:1736. doi: 10.1093/jnci/84.22.1736. [DOI] [PubMed] [Google Scholar]
- 11.(a) Dancey J, Sausville EA. Nat Rev Drug Discovery. 2003;2:296. doi: 10.1038/nrd1066. [DOI] [PubMed] [Google Scholar]; (b) McGovern SL, Shoichet BK. J Med Chem. 2003;46:1478. doi: 10.1021/jm020427b. [DOI] [PubMed] [Google Scholar]
- 12.Aronev AM, Murcko MA. J Med Chem. 2004;47:5616. doi: 10.1021/jm049793g. [DOI] [PubMed] [Google Scholar]
- 13.(a) Riva C, De Toma C, Donadel L, Boi C, Pennini R, Motta G, Leonardi A. Synthesis. 1997:195. [Google Scholar]; (b) Wheeler TS. Org Synth. 1963;Collect IV:478. [Google Scholar]; (c) Adam W, Golsch D, Hadjiiarapoglou L. J Org Chem. 1991;56:7292. [Google Scholar]
- 14.Baker-Venkataraman rearrangement13 using DBU gave by-product 12 as the major product and only a very small amount of cyclized product 14. Our efforts eventually lead to the preparation of the desired product 14 using KOH as a base.
- 15.Shults MD, Imperiali B. J Am Chem Soc. 2003;125:14248. doi: 10.1021/ja0380502. [DOI] [PubMed] [Google Scholar]
- 16.(a) MCF-7 Spletstoser JT, Flaherty PT, Himes RH, Georg GI. J Med Chem. 2004;47:6459. doi: 10.1021/jm030581d. [DOI] [PubMed] [Google Scholar]; (b) ID-8: Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, Persons DL, Smith PG, Terranova PF. Carcinogenesis. 2000;21:585. doi: 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 17.Kramer B, Metz G, Rarey M, Lengauer T. Med Chem Res. 1999;9:463. [Google Scholar]
- 18.(a) Schulze-Gahmen U, Brandsen J, Jones HD, Morgan DO, Meijer L, Vesely J, Kim SH. Proteins. 1995;22:378. doi: 10.1002/prot.340220408. [DOI] [PubMed] [Google Scholar]; (b) Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, Gibson AE, Golding BT, Grant S, Griffin RJ, Jewsbury P, Johnson LN, Lawrie AM, Newell DR, Noble ME, Sausville EA, Schultz R, Yu W. J Med Chem. 2000;43:2797. doi: 10.1021/jm990628o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary information available: 1H NMR and 13C NMR spectra for compounds 8, 9a, 9b, 10, 12, 14, 15a, 15b, 16, 17a–17e, 18b, 19a–19d, 20a–20c, 21a–21d, 22a–22d, 23, 24a–24c; 69 pages.