

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder that results from a loss of synaptic transmission and ultimately cell death. The presenting pathology of AD includes neuritic plaques composed of beta-amyloid peptide (Aβ) and neurofibrillary tangles composed of hyperphosphorylated tau, with neuronal loss in specific brain regions. However, the mechanisms that induce neuronal cell loss remain elusive. Focal adhesion (FA) proteins assemble into intracellular complexes involved in integrin-mediated communication between the extracellular matrix and the actin cytoskeleton, regulating many cell physiological processes including the cell cycle. Interestingly, recent studies report that integrins bind to Aβ fibrils, mediating Aβ signal transmission from extracellular sites of Aβ deposits into the cell and ultimately to the nucleus[1]. In this review, we will discuss the Aβ induced integrin/FA signaling pathways that mediate cell cycle activation and cell death.
Keywords: Integrin, FAK, Paxillin, Cyclin D1, Alzheimer’s disease, cell cycle
1. Introduction
In tissue, the ECM regulates many aspects of cellular function. Typically, specific ECM molecules bid to integrin cell surface receptors and activates downstream FA CAMs involved in the regulation of anchorage-dependent cell survival signals [2–4, 13, 15]. Prior studies indicate that FAK plays an important role in cell cycle progression at the G1/S transition by regulating the expression and activity of cell cycle proteins [39]. Integrins and numerous FA CAMs are expressed in all cells throughout the brain and numerous studies indicate a role for integrin signaling in neurite outgrowth during differentiation and in response to the toxic effects associated with neurodegeneration [3, 8, 10, 14, 16, 20]. Integrins have now been shown to bind Aβ and activate FAs through integrin clustering, CAM mobilization, and/or cooperatively with growth factor signaling through cell surface growth factor receptors [16–19]. Specifically, if fibrillar Aβ is added to cells in culture, both FAK and paxillin are rapidly phosphorylated leading to downstream signaling events that can regulate cell viability. Neuronal degeneration in AD occurs in specific regions of the brain and these cell type specificities are most likely dependent on gene expression of cell cycle regulatory proteins. Recent data indicates that fully differentiated and mature neurons in the adult brain exhibit evidence of cell cycle activation including DNA synthesis upon oxidative stress or exposure to Aβ fibrils [5, 21, 35, 69, 70, 71]. Therefore, we propose that alterations in the integrin/FAK/FA signaling pathway by fibrillar Aβ induces cell death within neurons that concurrently exhibit activation of cell cycle proteins. Neuronal viability and synaptic loss during the course of AD and potentially other neurodegenerative disorders may be solely mediated through FA signaling. Attention to focal adhesion proteins, and downstream pathways warrant further investigation in AD and highlight new targets for therapy.
2. Aβ toxicity through integrin signaling
2.1: Integrins
Cells adhere to the extracellular matrix (ECM), basement membrane or connective tissues, to regulate various cellular processes including growth, proliferation, survival, differentiation, morphology, migration, and death [2, 3]. The ECM signals through the cell surface integrins, a family of transmembrane subunits including 18 alpha and 9 beta subunits which generate at least 24 different integrins that function as heterodimeric receptors [2–4]. Integrins mediate both cell/ECM and cell/cell adhesions, although they do not contain intrinsic enzymatic activities [2, 5–7]. Instead, they associate with numerous intracellular effector cell adhesion molecules (CAMs). Activated integrins induce CAM activation by tyrosine phosphorylation during the initial stages of cell adhesion [8–11]. These CAMs assemble into immature less dense peripherially located focal adhesion (FA) complexes and mature more dense centrally located FA complexes associated with actin stress fibers [11, 12]. More than 50 CAMs are localized to FAs coupling to the actin cytoskeleton and regulating the structural components of the FAs to efficiently organize multiple signaling pathways (Fig. 2). Structural CAMs include actin, α-actinin, α-tubulin, hydrogden peroxide inducible clone 5 (hic-5), paxillin, Crk associated substrate (p130cas), talin, tensin, vinculin, and zyxin. Signaling CAMs include focal adhesion kinase (FAK), Fyn, phosphoinositide-3 (PI-3) kinase, c-Abl, Crk, Csk, Grb-2, Nck, and PYK2 [2–4, 6, 7, 13]. Many of these signaling CAMs are tryrosine kinases known to be upstream of serine/threonine kinases including members of the mitogen activated protein kinase (MAPK) pathway, cyclin dependent kinase 5 (CDK5), and glycogen synthase kinase-3β (GSK-3β) [2–4, 6, 11, 14]. In addition, protein tyrosine phosphatases (PTP-1D, PTP-PEST, and PTP-1B) have been shown to dephosphorylate CAMs upon cell detachment and regulate FA turnover [2, 3, 6, 13]. CAMs localized to FAs have been shown to be involved in bidirectional signaling including the compartmentalization of integrin activated downstream signaling molecules regulating “outside-in-signaling” and the localization and affinity of integrin receptors to regulate “inside-out-signaling” [2–4, 7, 15]. Ultimately, cell adhesion signaling through integrins and FAs can impact the viability of the cell.
Figure 2. Focal adhesions regulate cell cycle progression.
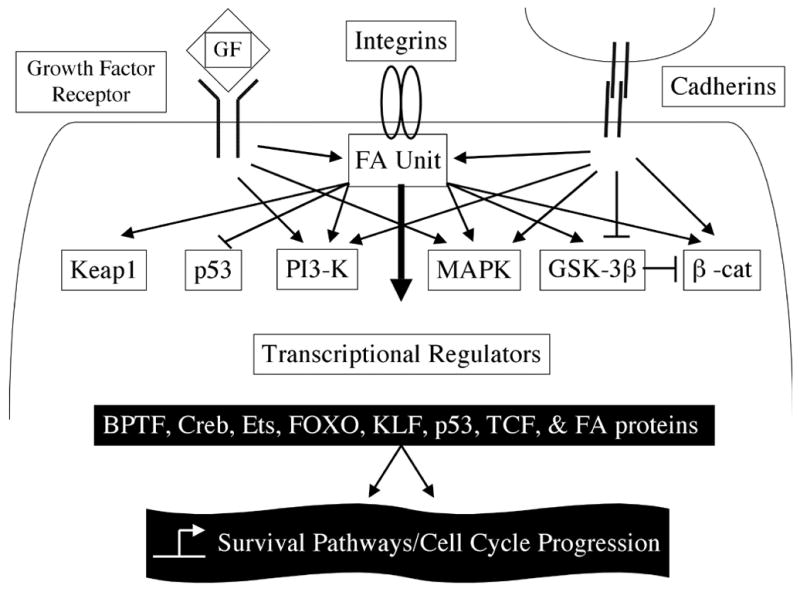
Fibrillar Aβ binds to integrin and activates FA signaling pathway to regulate cell cycle through different pathways including MAPK, PI3-K, and GSK-3β kinases (see text for details). The FA also receives input by growth factor receptors and cell-cell adhesions (cadherins). The sum of all inputs determines gene expression for cell survival and/or cell cycle progression through numerous transcriptional regulators. Signaling inputs from the ECM, growth factor receptors, and cadherins to the FA unit in the cytoplasm regulates cell cycle progression. Transcriptional regulators and nuclear events are denoted in black boxes.
Integrins are expressed in all cells throughout the brain and influence long-term potentiation (LTP) in hippocampal neurons, regulate neurite outgrowth induced by growth factors, and regulate survival and death signals [3, 8, 10, 14, 16]. Not surprisingly, numerous FA CAMs are also expressed in brain and numerous studies indicate a role for integrin signaling in neurite outgrowth during differentiation and in response to the toxic effects associated with neurodegeneration [14, 16–20]. Neurite outgrowth is promoted by the growth of neuronal cells on laminin, an integrin substrate [8, 10, 20, 21]. Specific inhibiters of the Src-family kinases can block neurite outgrowth [8, 10, 20]. When SH-SY5Y (human), N1E-115 (rat), and Neuro-2a (mouse) cells were plated on laminin and treated with retinoic acid (RA) to induce neuronal differentiation, both FAK and paxillin were shown to have increased tyrosine phosphorylation compared to adhesion during active neurite outgrowth [8, 17, 22].
2.2: FA unit and its function
FAK is a non-receptor tyrosine kinase responsible for integrin clustering and signaling [23–25]. When cells adhere to the ECM, integrins are activated and FAK is autophosphorylated at position Y397, creating a high affinity-binding site for Src-homology 2 (SH2) domain containing proteins [4, 13, 23–25]. Src family kinases (Src and Fyn) contain SH2 domains and recognize phosphorylated FAK-Y397 [4, 13, 23–25]. This complex can further phosphorylate FAK and other FA structural and signaling CAMs including paxillin and p130cas [4, 13, 23–25]. The assembly of FAK/Src family/paxillin/p130cas creates the essential four CAMs necessary for FA stabilization and integrin signaling that has been termed “quartenary signaling and structural FA-unit” [4, 26] (Fig. 1). The FA-unit connects the various signaling CAMs mobilized to FAs to coordinate integrin clustering, cell-cell adhesion, and growth factor signaling [2–4, 6, 7, 15, 23, 25]. In fact, this ECM/integrin/FA-unit pathway is involved in the regulation of anchorage-dependent cell survival [2–4, 13, 15]. Disruption of this pathway, either through the loss of cell adhesion or microinjection of anti-FAK antibodies, results in the activation of an anchorage-dependent cell death pathway called anoikis (greek for “homelessness”) [2–4, 7, 13, 15, 25]. Expression of constitutively active FAK can prevent anoikis in anchorage independent cells, including cancer cells [13, 15, 23–25]. Cell adhesion to the ECM and over-expression of FAK protects cells from oxidative stress by H2O2 or ionizing radiation (IR) induced apoptosis [4, 13, 25]. The anti-apoptotic effect is dependent on FAK Y397-autophosphorylation and the PI3-K/Akt survival pathway [4, 13, 25]. Recently, FAK has also been shown to directly interact with and inhibit the tumor suppressor protein p53 induced apoptotic signaling and induced the expression of cdk inhibitor p21 [27, 28]. Interestingly, p53 also has the ability to directly regulate the expression of FAK and provide a negative feedback mechanism for p53-induced apoptosis leading towards cell survival [27]. Therefore, FAK has the ability to regulate multiple survival pathways involved in cell death or survival. These findings support the important role for cell attachment in cell adhesion-mediated drug resistance and resistance to IR. Such pathways may also contribute to neuronal cell death/survival pathways during AD.
Figure 1. Early signaling pathways mobilized at focal adhesion formation.
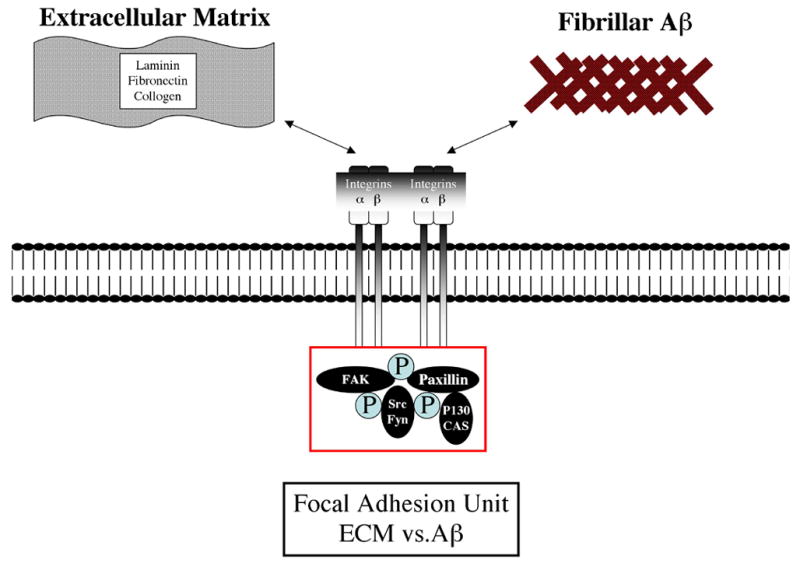
Integrin activation/adhesion promotes FAK autophosphorylation site recruits SH2 domain proteins including Src and Fyn leading to further FAK phosphorylation and other FA proteins including paxillin. Phosphorylated paxillin then binds to p130CAS forming the FA unit. Integrin receptor binding to fibrillar Aβ mimics early ECM/integrin signaling.
In addition to regulating cell adhesion and survival pathways, FAK and paxillin also function during neuronal differentiation [8, 10, 20]. Nerve growth factor (NGF) increases the level of paxillin tyrosine phosphorylation during neuronal differentiation in PC-12 cells [20]. Two members of the FAK family (FAK and PYK2) regulate neurite outgrowth in both PC-12 and SHSY-5Y cells [29]. The c-Jun N-terminal kinase (JNK) pathway has also been shown to activate paxillin during cell migration in PC-12 cells [30]. Finally, during NGF induced neuronal differentiation p38 MAPK and CDK5/p35 were shown to activate paxillin [20]. The role of FA CAMs in neurite outgrowth and neuronal differentiation is now appreciated and investigators are interested in how FAs function in neurodegenerative diseases such as AD.
2.3: Aβ binding to integrins and activation of intracellular pathways
Integrins can bind Aβ and activate FAs through integrin clustering, CAM mobilization, and/or cooperatively with growth factor signaling through cell surface growth factor receptors [16–19] (Fig. 2). The phosphorylation of tyrosine residues has been shown to be involved in numerous cell signaling pathways leading to Aβ toxicity which have stimulated studies examining how integrins modulate the toxic effects of Aβ [16–19]. Both FAK and paxillin were found to be tyrosine phosphorylated after exposure to fibrillar Aβ, well in advance of cell death [16–19]. Aβ induced cell death was found to be dependent on cell anchorage and required the treated cells to have a neuronal phenotype, actin cytoskeleton, and Aβ induction of tyrosine phosphorylation [16–19]. In a recent report, when SHSY-5Y cells treated with fibrillar Aβ, FAK or paxillin phosphorylation status was not altered until they were differentiated by retinoic acid (RA) [8, 17, 22]. RA differentiated SHSY-5Y cells treated with cytochalasin D to disrupt actin polymerization or monomer Aβ did not alter tyrosine phosphorylation of FAK or paxillin nor induce cell death [8]. Fibrillar Aβ exposure did not induce changes in the overall protein levels of FAK or paxillin but induces a substantial increase in their tyrosine phosphorylation (FAK-Y397 and paxillin-Y31/118) and evidence of protein mobilization from the cytosolic fraction to the membrane fraction [16–18]. Specifically, FA size and numbers that are positive for paxillin were found to be elevated in fibrillar Aβ treated neurons [18]. These results were specific to FAK and paxillin because no changes in the mobilization of another FA protein, vinculin, were found [18]. It has been suggested that in cell culture the early effects of fibrillar Aβ are the induction of FAK and paxillin tyrosine phosphorylation, which mimic early events in integrin signaling during adhesion and/or growth factor signaling [16]. Therefore, FAK and paxillin are rapidly phosphorylated by fibrillar Aβ, leading to downstream signaling events that can regulate cell viability.
To investigate the downstream pathways activated by FAK in primary cortical neurons, Williamson et al. used a variety of kinase inhibitors and demonstrated that FAK activated by fibrillar Aβ induces both ERK2 and GSK-3 serine/threonine kinase signaling pathways [16]. FAK activation relied on both PI3-K and the Src family member Fyn for downstream signaling. Interestingly, FAK activation did not rely on ERK1, Src, PYK2, PKC, or Ca+2 signaling. Fibrillar Aβ causes an increase in the tyrosine phosphorylation of MAP2c and tau that could be blocked with inhibitors specific for Src family kinases and PI3-K. ERK2 activity was sustained for at least 1 hour and tau phosphorylation occurred on serine residues recognized by the paired-helical-filament-1 (PHF-1) antibody. Thus, it is likely that FAK/Fyn/PI3-K activation induces ERK2 and GSK-3 signaling and phosphorylation of tau and MAP2c. In this model system FAK also induced anti-apoptotic signaling via the sequential activation of Fyn/FAK/PI3-K/MAPK pathways [16]. However, incomplete activation of these pathways fails to enhance cell survival and may lead to cell death. Fibrillar Aβ does not activate the complete Fyn/FAK/PI3-K/MAPK pathways and therefore the incomplete mimicking of this survival pathway may contribute to neuronal cell death during AD.
Paxillin is a member of the group III LIM domain family, closely related to hic-5, localizes in FAs and implicated in AD pathology [11, 18, 19] [Caltagarone, J. and Bowser, R., unpublished data]. Paxillin was detected in dystrophic neurites surrounding amyloid plaques and co-localized to hyperphosphorylated tau in primary neuronal cultures and human AD brain [19]. The c-terminal LIM domains of paxillin that are responsible for many protein-protein interactions are required for Aβ induced neuronal dystrophy in primary neuronal cultures [19]. While hic-5 and paxillin share many interacting partners, they are believed to act competitively in many signal transduction pathways [9]. The potential interactions between hic-5 and paxillin have not been studied in any neuronal system and warrant further investigation. Therefore, hic-5 and paxillin localization and subsequent interactions could lead to downstream signaling events that regulate neuronal dystrophy and cell death.
3. Integrin mediated cell cycle activation in AD
3.1: Cell cycle activation in AD
The neuronal degeneration of AD occurs in specific regions of the brain, including the hippocampus, frontal cortex and temporal cortex, that function in learning, memory and language. Mature neurons are arrested in the cell cycle G0 phase to permit the ongoing morphological alteration and synaptic remodeling. Thus, downregulation of G0 arrest proteins or induction of G1 entry proteins will result in the unbalance between cell cycle and differentiation control.
Cell cycle progression is controlled by regulating activity of a series of CDKs through the coordinated expression of cyclins and cyclin dependent kinase inhibitors (CDKI) [31]. Cyclin D1 is a critical regulator of the cell cycle transition from G0 to G1 phase. Accumulation of the cyclin D1/cdk4 complex leads to activation of kinases that phosphorylate and inactivate retinoblastoma protein (Rb), releasing its inhibition of E2F and allowing the transcription of E2F-target genes, such as cyclin E, cyclin A and cdc2. Cyclin E forms a complex with cdc2 and promotes G1 to S phase transition. Both cyclinE and cdc2 have been shown to be upregulated in cell culture models of neuronal cell death [32]. The activity of cyclin D1/cdk4 complex and cyclin E/cdc2 complex is regulated by CDKI, p27 and p21, respectively. Increased levels of p27 and phosphorylated p27 (Thr187) are present in the cytoplasm of vulnerable neuronal populations in AD brain [33]. Phosphorylated p27 (Thr187) co-localizes with dystrophic neurites, neurofibrillary tangles and neuropil threads.
In AD, cyclin D and cdk4 show aberrant expression in the hippocampus and subiculum, but not in the cerebellum [34]. We have demonstrated increased phosphorylated Rb protein (ppRb) and E2F1 immunoreactivity in AD brain compared to control brain, and ppRb was predominantly localized within the nucleus while E2F1 was in the cytoplasm [35]. Both ppRb and E2F1 were found in the cells surrounding a subset of Aβ-containing plaques. In vitro cell models exhibit similar ppRb and E2F1 distributions in PC12 cells upon Aβ treatment [36]. Increased levels of Cyclin D1, cyclin E and ppRb have been demonstrated in the early stage of AD, however, their levels are decreased by late stage AD [37].
MAPK is phosphorylated by its upstream kinase called MAPKK. Both MAPK and MAPKK exhibit increased expression in potentially vulnerable neurons still unaffected by neurofibrillary degeneration during the early stages of AD [38]. The subcellular redistribution of MAPK from cytoplasm to nucleus also indicates that MAPK might contribute to AD pathology via activating downstream nuclear signal pathways regulating protein transcription and/or translation. It is well known that MAPK signaling pathway contributes to neuronal apoptosis by directly phosphorylating cytoskeleton protein tau or indirectly activating downstream kinases that consequently phosphorylates tau [39]. However, additional evidence in cancer biology indicates that MAPK signaling pathway plays a role in the balance between cell survival and death through its influence on cell cycle progression, as discussed above. Since abnormal upregulation of cell cycle regulators and MAPK activation have been found in AD and MAPK signaling pathway is also activated in AD, it remains to be determined if the MAPK signaling pathway promotes cell cycle re-entry in AD.
3.2: Integrin/FAK activation of the cell cycle
Prior studies indicate that FAK plays an important role in cell cycle progression at the G1/S transition by regulating the expression and activity of cell cycle proteins including cyclins, cyclin dependent kinases (CDKs) and CDK inhibitors [40]. As discussed above, activation of FAK leads to autophosphorylation and binding to intracellular signal molecules including Src and PI3-kinase [23]. FAK/Src association activates ERK through two different signal pathways, whose phosphorylation and activation was associated with neuronal cell bodies and dystrophic neurites around plaques in late AD [41]. The first pathway phosphorylates FA proteins paxillin and p130cas, and leads to activation of MAPK signaling pathway through recruiting FA adaptor proteins Crk and Nck [42]. P130cas has been shown to be functional in central neuronal system disorders, such as hypoxia-ischemia induced neuronal degeneration [43]. The other pathway is activated upon binding with FA adaptor Grb2/SOS and Ras dependent phosphorylation, a family of small G protein including Ras, Rac and Rho proteins, linking growth factor signaling and tyrosine kinases with MAP Kinase, JNK, and p38 MAP kinases [44, 45]. After activation of the ERK signaling pathway by FAK, Cyclin D1 is upregulated at the transcriptional level [24].
FAK also interacts with PI3-K to activate the PI3-K signaling pathway, regulating cell survival and cell progression by Akt, Rac and PKC [40]. A recent study reported that in tumor cell lines, FAK induced PI3-K/PKC/Akt activation resulted in an induction of cyclin D3 expression and promotion of cell cycle progression from G1 to S phase [46]. The induction of cyclin D3, but not cyclin D1, also has been demonstrated in endothelin treated astrocytes [47]. However, cyclin D3 induction in astrocytes is via the small G-protein Rho, and is not dependent on PI3-K and Src. This difference indicates that FAK regulates cell cycle progression through different signal pathways in different cell types. The signal pathways involved in the function of FAK on cell cycle regulation in neurons remain unclear. However, FAK activated by amyloid beta peptide 1–42 induced MAPK signaling pathways through its association with Fyn, instead of Src in primary cultured cortical neurons [16]. As noted above, Aβ leads to incomplete activation of the FAK/Fyn/PI3-K/MAP kinase pathways that can induce cell death. In addition, FAK activation of ERK kinase pathways may upregulate cyclin D1 and induce cell cycle progression.
However, Guan and his colleagues found that FAK/PI3-K association is insufficient for cell cycle progression but FAK/Src complex plays an essential role [48]. Additionally, Src-dependent association of FAK with p130cas and Grb2 are both required for the cell cycle regulation by FAK. Grb2 regulates the cell cycle as an adaptor for cytoskeleton proteins. Recently, it has been reported to bind with c-terminal of amyloid protein precursor (APP) and this interaction increases in reactive astrocytes of AD, resulting in Ras/MAPK activation [49, 50].
Cyclin D1, a key component of the cell cycle, is regulated by FAK by multiple pathways [23]. A recent study demonstrated that FAK upregulates cyclin D1 expression through ERK activation in fibroblast cell lines [24]. After the Ras/Raf/MEK/ERK signal pathway is activated by FAK, ERK phosphorylates its downstream target Ets. Ets then binds to the Ets binding consensus site B, EtsB, in the promoter of the cyclin D1 gene to activate its transcription. However, the MEK/ERK pathway not only acts to induce cyclin D1 gene transcription but functions post-translationally to enhance cyclin D1 assembly with CDK4 and sequester p27Kip1 from inhibiting the cyclin E/CDK2 complex [51]. In addition, the PI3-K/Akt signal pathway, another downstream target of FAK, regulates the phosphorylation and turnover of cyclin D1 [52]. Finally, Rac increases cyclin D1 protein level through stabilizing cyclin D1 mRNA [7]. Integrin mediated FAK activation also results in a decrease of p21 and p27Kip, key CDK inhibitors [23].
In a summary, FAK/Src or FAK/Fyn association upregulates cyclin D1 expression at the transcriptional level through ERK, PI3-K or Rac signaling pathway and can stabilize cyclin D1 mRNA through Rac. FAK also regulates the turnover of cyclin D1 through PI3-K/Akt signaling pathway, and modulates the level of key CDK inhibitors (Fig. 2).
4. Integrin Mediated Transcription
Cell type specificities in the control of the cell cycle by the ECM-integrin-FAK-FA pathway are most likely dependent on gene expression of cell cycle regulatory proteins. As discussed above, FAK signaling is directly involved in the regulation of cyclin D1 [23, 24]. Activation of these transcription factors is dependent on the activities of FAK/Src and FAK/PI3-K complexes and directly targets the promoter of cyclin D1. However, FAK activation can also regulate the expression of transcription factors that can indirectly regulate the expression of cell cycle regulators. Recently FAK was shown to regulate the expression of a member of the Kruppel-like factor (KLF) family of transcription factors [53]. The KLF family contains at least 25 members, many of which are important regulators of cell cycle regulators and have been shown to both positively and negatively regulate the cyclin D1 promoter in various cells [53–55]. In mouse fibroblast cells, KLF8 was shown to positively regulate cyclin D1 expression and cell cycle progression that is dependent on both FAK/Src and FAK/PI3-K integrin mediated signaling pathways [53]. FAK activities were found to coordinate the expression of cyclin D1 promoter element through a “temporal differential manner” of the cell cycle with an early induction through Ets binding followed by a later induction after FAK up-regulated the expression of KLF8. Numerous types of human cancer have elevated FAK and KLF8 expression levels and their activities are implicated in cell cycle progression during tumorigenesis [53–56]. However, other members of the KLF family including KLF1, 4, 6, and 7 have all been shown to be negative regulators of cell cycle progression by decreasing cyclin D1 promoter activity and increasing cdk inhibitor p21 expression levels [54, 55]. KLF7 is also believed to control neuronal differentiation and exit from the cell cycle by increasing expression of TrkA, a receptor for NGF [54]. How integrin signaling mediated by FAK regulates the cell cycle progression dynamically either positively (Ets and KLF8) or negatively mediated by members of KLF family in neuronal cells and AD remains to be determined.
The complex balance of both positive and negative signals for cyclin D1 expression during cell cycle progression at multiple layers as described for FAK is conserved in other well-known pathways that utilize FA’s. Wnt signaling through membrane bound frizzled protein family members and β-catenin mediates cell-cell adhesion signaling to the actin cytoskeleton similar to how FAK mediates ECM-cell adhesion signaling [6, 57–59]. GSK-3β is an important negative regulator in β-catenin signaling through phosphorylating and targeting β-catenin to the proteosome for removal [58]. Not surprisingly, β-catenin is also an important mediator in the signaling of cell cycle events [58]. The β-catenin interaction and regulation of T cell factor (TCF) transcription factor has been well characterized in development and regeneration in multiple tissue types [57, 58]. Enhanced β-catenin signaling and activation of TCF has been shown to induce cyclin D, bcl-2, and engrailed-1/2 (en-1/2) expression [58, 60, 61]. These proteins function during development and favor cell survival and cell cycle progression. In AD, the Wnt/β-catenin/TCF pathway is disrupted with subsequent loss of β-catenin expression and TCF survival signaling [58, 60, 61]. However, activation of the Wnt/β-catenin/TCF survival pathway by insulin, protein kinase C (PKC), agonists for PPARγ, and inactivation of GSK-3β all have the ability to protect primary cortical neurons to Aβ induced cell death [56, 58, 60, 61]. β-catenin can also mediate cell cycle arrest through enhanced Forkhead box O (FOXO) transcriptional activity [59, 62, 63]. Under conditions of oxidative stress, β-catenin interacts with FOXO for translocation into the nucleus to induce the expression of cell cycle inhibitor p27, leading to cell cycle arrest in late G1 phase [59]. Thus, CAMs associated with at least two distinctly different adhesion complexes (ECM/integrin and cell/cell) dynamically regulate the progression and arrest of the cell cycle. The mechanism of cell adhesion complexes regulating the cell cycle is redundant yet specific for unique signaling inputs. Determining how focal adhesion complexes regulate neuronal viability in AD will lead to novel therapeutic approaches to modulate neuronal survival and synaptic remodeling during AD.
A number of CAMs have been shown to shuttle between FA’s and the nucleus to regulate gene expression, including c-Abl, β-cat, CASK, CDK5, ERK, FAK, GSK3β, ILK, JAB1, PINCH, PYK2, and members of the LIM family of proteins (hic-5, paxillin, Trip6, leupaxin, and zyxin) [6, 11]. Many of which have been implicated in the molecular pathology of neurodegenerative diseases, though continued investigations are required to determine the functional role for each of these proteins in neurodegeneration.
Our lab and others have shown that Kelch-like Ech Associated Protein (Keap1) is an actin binding protein that has the ability to regulate the subcellular distribution of transcription factors including Fetal ALZ-50 Reactive Clone 1 (FAC1) and Nrf2 in neurons [64–67]. In normal conditions, Keap1 is localized to periphery FAs containing paxillin, vinculin, and tyrosine phosphylated proteins [65]. Keap1 bound to Nrf2 targets their destruction by ubiquitin-mediated pathways [66, 67]. However, under conditions of oxidative stress both Keap1 and Nrf2 translocate to the nucleus and nuclear Nrf2 positively regulates promoters containing the anti-oxidant response elements (AREs) [65–67]. The induction of Nrf2 mediated survival pathways at the transcription level is considered a conserved function in every cell type [67]. FAC1 is a developmentally regulated transcription factor that modulates expression of numerous genes [36, 68]. In AD brain and PC-12 cells treated with Aβ, FAC1 exhibits altered expression and subcellular distribution from the cytoplasm to the perinuclear region in cells [36]. FAC1 is believed to be an important regulator in Rb and E2F cell cycle activities in response to neurotoxic signals [68]. Recently, FAC1 was shown to be an alterantive splice form from a much larger bromodomain PHD transcription factor (BPTF) that is implicated in chromatin remodeling [69]. En-1 gene expression was positively regulated by BPTF [69]. Since en-1 and en-2 gene expression is required for normal neuronal development, altered expression and subcelluar distribution of factors that associate with their promoter elements could disrupt engrailed expression and activities in AD. How integrin signaling regulates the FA/Keap1/Nrf2/ARE survival pathway and FA/Keap1/FAC1/BPTF transcriptional activities in response to toxic neuronal stimuli remains to be determined but supports the role of altered distribution of CAMs in regulating numerous cellular responses to toxic stimuli (Fig. 2).
5. Integrins Modulate Neuronal Survival
During early steps of the cell cycle, the ECM/integrin/FAK/FA pathway regulates anchorage-dependent cell survival [2, 3]. Disruption of this cell attachment pathway results in a specific cell death called anoikis and this ECM/integrin/FAK/FA pathway may actually limit neuronal mitosis [13, 15]. Fully differentiated and mature neurons in the adult brain exhibit evidence of cell cycle activation upon oxidative stress or exposure to Aβ fibrils [5, 21, 36, 70, 71]. A recent study used a genomic probe for chromosome 11 to identify a pyramidal neuron in the hippocampus of AD brain with four copies of chromosome 11 indicating that this neuron synthesized DNA [71, 72].
In vitro models of neuronal injury using Aβ and oxidative stress have reproduced the induction of integrin signaling, cell cycle regulators, DNA damage, and apoptosis. Recent models of S-phase induction in neurons include two mouse mutants (stagger and lurcher), overexpression of viral oncogenes and E2F1, targeted disruption of Rb, and a model of stroke show increased levels of BrdU incorporation in neuronal cells indicative of DNA synthesis [21, 70, 73]. However, BrdU incorporation is not specific to DNA replication and may also be incorporated during DNA repair [71]. Post-mitotic cells have increased vulnerablility to DNA damage and repair [71]. Growth factor and ECM/integrin signaling appear to inhibit DNA synthesis in neuronal but not in other cell types including glia [21]. We propose that activation of the integrin/FAK/FA pathway by fibrillar Aβ induces cell death within neurons that concurrently exhibit activation of cell cycle proteins. Loss of ECM/integrin signaling (i.e. loss of cell attachment signaling) may induce anoikis mediated cell death with concurrent disinhibition of DNA systhesis. Therefore, the appearance of DNA synthesis in some neurons during AD may result from altered focal adhesion signaling.
6. Conclusion
Activation of FAK/paxillin by Aβ can induce tau phosphorylation, cell cycle activation, and the loss of cell adhesions can lead to subsequent cell death. The alteration of expression and function of FAK signaling pathway related proteins in AD brains supports our hypothesis that integrin/FAK signal pathway induces neuronal cell cycle re-entry and neuronal cell death. Given the data relating focal adhesion activation by Aβ and neuronal cell death pathways, attention to focal adhesion proteins, and downstream pathways warrant further investigation in AD and highlight new targets for therapy.
Acknowledgments
This study was supported by NIH grant NS042724 to RB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Verdier Y, Penke B. Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer's disease. Curr Protein Pept Sci. 2004;5:19–31. doi: 10.2174/1389203043486937. [DOI] [PubMed] [Google Scholar]
- 2.Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signaling and cell growth control. Curr Opin Cell Biol. 1998;10:220–31. doi: 10.1016/s0955-0674(98)80144-0. [DOI] [PubMed] [Google Scholar]
- 3.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–32. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 4.Cordes N, Meineke V. Integrin signalling and the cellular response to ionizing radiation. J Mol Histol. 2004;35:327–37. doi: 10.1023/b:hijo.0000032364.43566.3a. [DOI] [PubMed] [Google Scholar]
- 5.Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:343–7. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aplin AE. Cell adhesion molecule regulation of nucleocytoplasmic trafficking. FEBS Lett. 2003;534:11–4. doi: 10.1016/s0014-5793(02)03840-1. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–60. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- 8.Leventhal PS, Feldman EL. Tyrosine phosphorylation and enhanced expression of paxillin during neuronal differentiation in vitro. J Biol Chem. 1996;271:5957–60. doi: 10.1074/jbc.271.11.5957. [DOI] [PubMed] [Google Scholar]
- 9.Nishiya N, Tachibana K, Shibanuma M, Mashimo JI, Nose K. Hic-5-reduced cell spreading on fibronectin: competitive effects between paxillin and Hic-5 through interaction with focal adhesion kinase. Mol Cell Biol. 2001;21:5332–45. doi: 10.1128/MCB.21.16.5332-5345.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Curtis I, Malanchini B. Integrin-mediated tyrosine phosphorylation and redistribution of paxillin during neuronal adhesion. Exp Cell Res. 1997;230:233–43. doi: 10.1006/excr.1996.3423. [DOI] [PubMed] [Google Scholar]
- 11.Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84:1315–39. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 12.Wehrle-Haller B, Imhof B. The inner lives of focal adhesions. Trends Cell Biol. 2002;12:382–9. doi: 10.1016/s0962-8924(02)02321-8. [DOI] [PubMed] [Google Scholar]
- 13.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24:425–39. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 14.Sonoda Y, Watanabe S, Matsumoto Y, Aizu-Yokota E, Kasahara T. FAK is the upstream signal protein of the phosphatidylinositol 3-kinase-Akt survival pathway in hydrogen peroxide-induced apoptosis of a human glioblastoma cell line. J Biol Chem. 1999;274:10566–70. doi: 10.1074/jbc.274.15.10566. [DOI] [PubMed] [Google Scholar]
- 15.Gilmore AP. Anoikis. Cell Death Differ. 2005;12(Suppl 2):1473–7. doi: 10.1038/sj.cdd.4401723. [DOI] [PubMed] [Google Scholar]
- 16.Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci. 2002;22:10–20. doi: 10.1523/JNEUROSCI.22-01-00010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang C, Lambert MP, Bunch C, Barber K, Wade WS, Krafft GA, Klein WL. Focal adhesion kinase expressed by nerve cell lines shows increased tyrosine phosphorylation in response to Alzheimer's A beta peptide. J Biol Chem. 1994;269:25247–50. [PubMed] [Google Scholar]
- 18.Berg MM, Krafft GA, Klein WL. Rapid impact of beta-amyloid on paxillin in a neural cell line. J Neurosci Res. 1997;50:979–89. doi: 10.1002/(SICI)1097-4547(19971215)50:6<979::AID-JNR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 19.Grace EA, Busciglio J. Aberrant activation of focal adhesion proteins mediates fibrillar amyloid beta-induced neuronal dystrophy. J Neurosci. 2003;23:493–502. doi: 10.1523/JNEUROSCI.23-02-00493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang C, Borchers CH, Schaller MD, Jacobson K. Phosphorylation of paxillin by p38MAPK is involved in the neurite extension of PC-12 cells. J Cell Biol. 2004;164:593–602. doi: 10.1083/jcb.200307081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith DS, Leone G, DeGregori J, Ahmed MN, Qumsiyeh MB, Nevins JR. Induction of DNA replication in adult rat neurons by deregulation of the retinoblastoma/E2F G1 cell cycle pathway. Cell Growth Differ. 2000;11:625–33. [PubMed] [Google Scholar]
- 22.Lambert MP, Stevens G, Sabo S, Barber K, Wang G, Wade W, Krafft G, Snyder S, Holzman TF, Klein WL. Beta/A4-evoked degeneration of differentiated SH-SY5Y human neuroblastoma cells. J Neurosci Res. 1994;39:377–85. doi: 10.1002/jnr.490390404. [DOI] [PubMed] [Google Scholar]
- 23.Zhao JH, Reiske H, Guan JL. Regulation of the cell cycle by focal adhesion kinase. J Cell Biol. 1998;143:1997–2008. doi: 10.1083/jcb.143.7.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao J, Pestell R, Guan JL. Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell. 2001;12:4066–77. doi: 10.1091/mbc.12.12.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasahara T, Koguchi E, Funakoshi M, Aizu-Yokota E, Sonoda Y. Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid Redox Signal. 2002;4:491–9. doi: 10.1089/15230860260196290. [DOI] [PubMed] [Google Scholar]
- 26.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–25. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 29.Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth BU, Dikic I. Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol. 2000;2:574–81. doi: 10.1038/35023515. [DOI] [PubMed] [Google Scholar]
- 30.Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K. JNK phosphorylates paxillin and regulates cell migration. Nature. 2003;424:219–23. doi: 10.1038/nature01745. [DOI] [PubMed] [Google Scholar]
- 31.Bowser R, Smith MA. Cell cycle proteins in Alzheimer's disease: plenty of wheels but no cycle. J Alzheimers Dis. 2002;4:249–54. doi: 10.3233/jad-2002-4316. [DOI] [PubMed] [Google Scholar]
- 32.Yu X, Caltagarone J, Smith MA, Bowser R. DNA damage induces cdk2 protein levels and histone H2B phosphorylation in SH-SY5Y neuroblastoma cells. J Alzheimers Dis. 2005;8:7–21. doi: 10.3233/jad-2005-8102. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa O, Lee HG, Zhu X, Raina A, Harris PL, Castellani RJ, Perry G, Smith MA. Increased p27, an essential component of cell cycle control, in Alzheimer's disease. Aging Cell. 2003;2:105–10. doi: 10.1046/j.1474-9728.2003.00042.x. [DOI] [PubMed] [Google Scholar]
- 34.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–7. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jordan-Sciutto KL, Malaiyandi LM, Bowser R. Altered distribution of cell cycle transcriptional regulators during Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:358–67. doi: 10.1093/jnen/61.4.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jordan-Sciutto K, Rhodes J, Bowser R. Altered subcellular distribution of transcriptional regulators in response to Abeta peptide and during Alzheimer's disease. Mech Ageing Dev. 2001;123:11–20. doi: 10.1016/s0047-6374(01)00334-7. [DOI] [PubMed] [Google Scholar]
- 37.Hoozemans JJ, Stieler J, van Haastert ES, Veerhuis R, Rozemuller AJ, Baas F, Eikelenboom P, Arendt T, Scheper W. The unfolded protein response affects neuronal cell cycle protein expression: implications for Alzheimer's disease pathogenesis. Exp Gerontol. 2006;41:380–6. doi: 10.1016/j.exger.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 38.Arendt T, Holzer M, Grossmann A, Zedlick D, Bruckner MK. Increased expression and subcellular translocation of the mitogen activated protein kinase kinase and mitogen-activated protein kinase in Alzheimer's disease. Neuroscience. 1995;68:5–18. doi: 10.1016/0306-4522(95)00146-a. [DOI] [PubMed] [Google Scholar]
- 39.Haddad JJ. Mitogen-activated protein kinases and the evolution of Alzheimer's: a revolutionary neurogenetic axis for therapeutic intervention? Prog Neurobiol. 2004;73:359–77. doi: 10.1016/j.pneurobio.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 40.Cary LA, Guan JL. Focal adhesion kinase in integrin-mediated signaling. Front Biosci. 1999;4:D102–13. doi: 10.2741/cary. [DOI] [PubMed] [Google Scholar]
- 41.Webster B, Hansen L, Adame A, Crews L, Torrance M, Thal L, Masliah E. Astroglial activation of extracellular-regulated kinase in early stages of Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:142–51. doi: 10.1097/01.jnen.0000199599.63204.6f. [DOI] [PubMed] [Google Scholar]
- 42.Guan JL. Focal adhesion kinase in integrin signaling. Matrix Biol. 1997;16:195–200. doi: 10.1016/s0945-053x(97)90008-1. [DOI] [PubMed] [Google Scholar]
- 43.Zalewska T, Makarewicz D, Janik B, Ziemka-Nalecz M. Neonatal cerebral hypoxia-ischemia: involvement of FAK-dependent pathway. Int J Dev Neurosci. 2005;23:657–62. doi: 10.1016/j.ijdevneu.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 44.Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J Biol Chem. 1997;272:13189–95. doi: 10.1074/jbc.272.20.13189. [DOI] [PubMed] [Google Scholar]
- 45.Kacimi R, Gerdes AM. Alterations in G protein and MAP kinase signaling pathways during cardiac remodeling in hypertension and heart failure. Hypertension. 2003;41:968–77. doi: 10.1161/01.HYP.0000062465.60601.CC. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto D, Sonoda Y, Hasegawa M, Funakoshi-Tago M, Aizu-Yokota E, Kasahara T. FAK overexpression upregulates cyclin D3 and enhances cell proliferation via the PKC and PI3-kinase-Akt pathways. Cell Signal. 2003;15:575–83. doi: 10.1016/s0898-6568(02)00142-0. [DOI] [PubMed] [Google Scholar]
- 47.Koyama Y, Yoshioka Y, Shinde M, Matsuda T, Baba A. Focal adhesion kinase mediates endothelin-induced cyclin D3 expression in rat cultured astrocytes. J Neurochem. 2004;90:904–12. doi: 10.1111/j.1471-4159.2004.02546.x. [DOI] [PubMed] [Google Scholar]
- 48.Reiske HR, Zhao J, Han DC, Cooper LA, Guan JL. Analysis of FAK-associated signaling pathways in the regulation of cell cycle progression. FEBS Lett. 2000;486:275–80. doi: 10.1016/s0014-5793(00)02295-x. [DOI] [PubMed] [Google Scholar]
- 49.Russo C, Dolcini V, Salis S, Venezia V, Violani E, Carlo P, Zambrano N, Russo T, Schettini G. Signal transduction through tyrosine-phosphorylated carboxy-terminal fragments of APP via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. Ann N Y Acad Sci. 2002;973:323–33. doi: 10.1111/j.1749-6632.2002.tb04660.x. [DOI] [PubMed] [Google Scholar]
- 50.Russo C, Venezia V, Repetto E, Nizzari M, Violani E, Carlo P, Schettini G. The amyloid precursor protein and its network of interacting proteins: physiological and pathological implications. Brain Res Brain Res Rev. 2005;48:257–64. doi: 10.1016/j.brainresrev.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 51.Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci U S A. 1998;95:1091–6. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walker JL, Assoian RK. Integrin-dependent signal transduction regulating cyclin D1 expression and G1 phase cell cycle progression. Cancer Metastasis Rev. 2005;24:383–93. doi: 10.1007/s10555-005-5130-7. [DOI] [PubMed] [Google Scholar]
- 53.Zhao J, Bian ZC, Yee K, Chen BP, Chien S, Guan JL. Identification of transcription factor KLF8 as a downstream target of focal adhesion kinase in its regulation of cyclin D1 and cell cycle progression. Mol Cell. 2003;11:1503–15. doi: 10.1016/s1097-2765(03)00179-5. [DOI] [PubMed] [Google Scholar]
- 54.Suske G, Bruford E, Philipsen S. Mammalian SP/KLF transcription factors: bring in the family. Genomics. 2005;85:551–6. doi: 10.1016/j.ygeno.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Zhao J. KLF8 transcription factor participates in oncogenic transformation. Oncogene. 2006 doi: 10.1038/sj.onc.1209796. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, Wang SM, Wu JC, Huang SH. Effects of PPARgamma agonists on cell survival and focal adhesions in a Chinese thyroid carcinoma cell line. J Cell Biochem. 2006;98:1021–35. doi: 10.1002/jcb.20839. [DOI] [PubMed] [Google Scholar]
- 57.Micsenyi A, Tan X, Sneddon T, Luo JH, Michalopoulos GK, Monga SP. Beta-catenin is temporally regulated during normal liver development. Gastroenterology. 2004;126:1134–46. doi: 10.1053/j.gastro.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 58.Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-beta-peptide neurotoxicity: role in Alzheimer disease. Brain Res Brain Res Rev. 2004;47:275–89. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 59.Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–4. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- 60.Alvarez AR, Godoy JA, Mullendorff K, Olivares GH, Bronfman M, Inestrosa NC. Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res. 2004;297:186–96. doi: 10.1016/j.yexcr.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 61.Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. Faseb J. 2002;16:1982–4. doi: 10.1096/fj.02-0327fje. [DOI] [PubMed] [Google Scholar]
- 62.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 63.Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A. 2004;101:2975–80. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strachan GD, Morgan KL, Otis LL, Caltagarone J, Gittis A, Bowser R, Jordan-Sciutto KL. Fetal Alz-50 clone 1 interacts with the human orthologue of the Kelch-like Ech-associated protein. Biochemistry. 2004;43:12113–22. doi: 10.1021/bi0494166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Velichkova M, Hasson T. Keap1 in adhesion complexes. Cell Motil Cytoskeleton. 2003;56:109–19. doi: 10.1002/cm.10138. [DOI] [PubMed] [Google Scholar]
- 66.Velichkova M, Hasson T. Keap1 regulates the oxidation-sensitive shuttling of Nrf2 into and out of the nucleus via a Crm1-dependent nuclear export mechanism. Mol Cell Biol. 2005;25:4501–13. doi: 10.1128/MCB.25.11.4501-4513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JM, Li J, Johnson DA, Stein TD, Kraft AD, Calkins MJ, Jakel RJ, Johnson JA. Nrf2, a multi-organ protector? Faseb J. 2005;19:1061–6. doi: 10.1096/fj.04-2591hyp. [DOI] [PubMed] [Google Scholar]
- 68.Jordan-Sciutto KL, Dragich JM, Rhodes JL, Bowser R. Fetal Alz-50 clone 1, a novel zinc finger protein, binds a specific DNA sequence and acts as a transcriptional regulator. J Biol Chem. 1999;274:35262–8. doi: 10.1074/jbc.274.49.35262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R. Isolation of human NURF: a regulator of Engrailed gene expression. Embo J. 2003;22:6089–100. doi: 10.1093/emboj/cdg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuan CY, Schloemer AJ, Lu A, Burns KA, Weng WL, Williams MT, Strauss KI, Vorhees CV, Flavell RA, Davis RJ, Sharp FR, Rakic P. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24:10763–72. doi: 10.1523/JNEUROSCI.3883-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herrup K, Neve R, Ackerman SL, Copani A. Divide and die: cell cycle events as triggers of nerve cell death. J Neurosci. 2004;24:9232–9. doi: 10.1523/JNEUROSCI.3347-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001;21:2661–8. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–95. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]