

Abstract
The levels of amyloid-β40 (Aβ40) and Aβ42 peptides were quantified in temporalis muscles and brain of neuropathologically diagnosed Alzheimer disease (AD) and of nondemented individuals. This was achieved by using a novel analytical approach consisting of a combination of fast-performance liquid chromatographic (FPLC) size exclusion chromatography developed under denaturing conditions and europium immunoassay on the 4.0- to 4.5-kd fractions. In the temporalis muscles of the AD and nondemented control groups, the average values for Aβ42 were 15.7 ng/g and 10.2 ng/g (P = 0.010), and for Aβ40 they were 37.8 ng/g and 29.8 ng/g (P = 0.067), respectively. Multiple regression analyses of the AD and control combined populations indicated that 1) muscle Aβ40 and muscle Aβ42 levels were correlated with each other (P < 0.001), 2) muscle Aβ40 levels were positively correlated with age (P = 0.036), and 3) muscle Aβ42 levels were positively correlated with Braak stage (P = 0.042). Other forms of the Aβ peptide were discovered by mass spectrometry, revealing the presence of Aβ starting at residues 1, 6, 7, 9, 10, and 11 and ending at residues 40, 42, 44, 45, and 46. It is possible that in AD the skeletal muscle may contribute to the elevated plasma pool of Aβ and thus indirectly to the amyloid deposits of the brain parenchyma and cerebral blood vessels. The increased levels of Aβ in the temporalis muscles of AD patients suggest that alterations in AβPP and Aβ metabolism may be manifested in peripheral tissues.
The profuse accumulation of amyloid-β (Aβ) peptide in the extracellular space of the cerebral cortex and walls of cerebral and leptomeningeal blood vessels represents one of the major histopathological lesions of Alzheimer disease (AD). The Aβ peptide is derived from the normal proteolytic processing of a larger Aβ precursor protein (AβPP), which in the brain is expressed in neurons, glial cells, and vascular myocytes. 1 However, peripheral tissues such as heart, liver, pancreas, thyroid, lymph nodes, spleen, skeletal muscle, salivary and adrenal glands, skin, intestine, leukocytes, and platelets also express AβPP. 2-6 In addition, the presence of AβPP has also been confirmed in the skeletal muscle neuromuscular junctions by immunofluorescence confocal microscopy and immunoelectron microscopy. 6 The exact biochemical roles of AβPP and Aβ remain unknown, although some important functions for these molecules have been suggested. 1 It is interesting that, during the course of acute phase reactions such as traumatic brain injury, the AβPP is up-regulated and associated with transient elevation of Aβ level. 7,8 Although it has been largely accepted that the origin of the Aβ deposited in the brain is the brain tissue itself, recent investigations have demonstrated that circulating Aβ is capable of crossing the blood brain-barrier (BBB), implying a potentially hematogenous source for the brain’s parenchymal and vascular Aβ peptides. 9 One possible source for the circulating Aβ is the skeletal muscle. Transgenic mice overexpressing the last 99 amino acids of human AβPP in peripheral tissues produce high circulating levels of human Aβ in the plasma. 10 Moreover, individuals suffering from inclusion body myositis, the most common muscle disease observed in the elderly, exhibit prominent intracytoplasmic vacuoles containing Aβ immunoreactive filaments. 11 A systematic study comparing the levels of Aβ in brain and skeletal muscle in AD and control individuals has not been performed. In this investigation we quantified for the first time the Aβ40 and Aβ42 in the temporalis muscles of demented individuals neuropathologically confirmed to have AD. These values were compared with those obtained from a cohort of elderly nondemented control individuals.
Materials and Methods
Materials
The equipment and materials used in the separation and immunoassay of Aβ were from the following sources. Fast-performance liquid chromatography (FPLC) equipped with fraction collector and Superose 12 size exclusion column were from Pharmacia Biotech (Uppsala, Sweden). Tris(hydroxymethyl)aminomethane (Tris), acetonitrile, NaOH, Tween 20, NaCl, and Na2CO3 were from Sigma Chemical Co. (St Louis, MO). Paraformaldehyde was from Fisher Scientific (St. Louis, MO). Hydrochloric acid and formic acid (98%) were from Fluka Chemie AG (Buchs, Switzerland). The formic acid was in all instances further purified in our laboratory by glass distillation. Oligonucleotides, restriction enzyme HhaI, and all of the reagents used for ApoE genotyping were obtained from Genosys (The Woodlands, TX) and GIBCO/BRL (Gaithersburg, MD). The monoclonal antibody 4G8, raised against residues 17–24 of the Aβ peptide (Senetek, Maryland Heights, MO), was labeled with europium (Eu) by the procedures given by the manufacturer (Wallac Inc. Gaithersburg, MD). The Eu, enhancement solution, and fluorimeter were from Wallac Inc. The polyclonal antibodies R163 and R165, specifically raised against Aβ40 and Aβ42, 12 respectively, were obtained from P. Mehta (New York Institute for Basic Research, New York, NY). These antibodies do not exhibit cross-recognition of the two forms of Aβ molecules at concentrations >100 ng/ml. Microtiter plates were purchased from Nalge Nunc International (Denmark). The antibody 22C11, raised against the N terminus of AβPP, was obtained from Roche Molecular Biochemicals (Indianapolis, IN).
Human Tissues
The brains from 43 elderly individuals, 23 AD (mean age, 85.6; range 79–96 years) and 20 nondemented (mean age, 77.7; range 63–91 years), were removed and coronally sectioned into 1-cm slices. The left hemispheres were fixed in 4% buffered paraformaldehyde and used for histopathological analyses. The right hemispheres were immediately frozen at −85°C. Postmortem delay (time from death to freezing) averaged 2.5 hours (range, 1–5 hours).
All AD (cases 1–23) and nondemented control (cases 101–120) brains were obtained from the brain bank at the Sun Health Research Institute (Table 1) ▶ . All brains were bequeathed on a strictly voluntary basis, with the expectation that the tissue would be used for research in AD and other neurodegenerative diseases. AD cases were diagnosed neuropathologically according to the criteria established by the Consortium to Establish a Registry for Alzheimer Disease (CERAD). 13 The Braak stage classification was also considered. 14 Dementia with Lewy bodies (DLB) cases and progressive supranuclear palsy (PSP) cases were diagnosed according to published consensus criteria. 15-17 Control cases, including neuropathologic conditions other than AD as well as nondemented cases free of neuropathologic abnormalities, were also rated by Braak stage and CERAD neuritic plaque density.
Table 1.
Neuropathological Diagnosis
| Case | Age/Sex | ApoE | Neuropathology | BRAAK stage | Neuritic plaques |
|---|---|---|---|---|---|
| AD | |||||
| 1 | 79 /M | 3 /3 | CERAD + | III | Moderate |
| 2 | 80 /M | 2 /3 | CERAD+/PD | IV | Frequent |
| 3 | 82 /M | 3 /4 | CERAD+ | V | Moderate/Frequent |
| 4 | 82 /M | 3 /3 | CERAD+ | V | Moderate/Frequent |
| 5 | 82 /F | 3 /4 | CERAD+/DLB | V | Moderate/Frequent |
| 6 | 82 /F | 3 /4 | CERAD+/PSP | III | Sparse/Moderate |
| 7 | 83 /M | 3 /3 | CERAD+/DLB | IV | Moderate/Frequent |
| 8 | 83 /F | 3 /3 | CERAD+/PD | IV | Frequent |
| 9 | 84 /M | 3 /4 | CERAD+/PSP | V | Moderate/Frequent |
| 10 | 84 /F | 3 /3 | CERAD+/DLB | VI | Moderate |
| 11 | 84 /M | 4 /4 | CERAD+ | VI | Moderate/Frequent |
| 12 | 85 /F | 3 /4 | CERAD+/MID | VI | Frequent |
| 13 | 86 /F | 3 /3 | CERAD+/Hip. Sc. | VI | Moderate |
| 14 | 86 /F | 3 /3 | CERAD+/Hip. Sc. | V | Moderate |
| 15 | 86 /F | 3 /4 | CERAD+/PD | IV | Moderate |
| 16 | 88 /M | 3 /3 | CERAD+ | V | Moderate/Frequent |
| 17 | 88 /M | 3 /4 | CERAD+ | V | Frequent |
| 18 | 88 /M | 3 /3 | CERAD+/MID | V | Moderate/Frequent |
| 19 | 88 /M | 2 /3 | CERAD+/Inc. LB | V | Sparse/Moderate |
| 20 | 90 /M | 3 /3 | CERAD+ | III | Sparse/Moderate |
| 21 | 91 /F | 3 /3 | CERAD+ | III | Sparse/Moderate |
| 22 | 91 /F | 3 /4 | CERAD+ | VI | Moderate/Frequent |
| 23 | 96 /F | 3 /3 | CERAD+/MID | II | Sparse/Moderate |
| Control | |||||
| 101 | 63 /M | 3 /3 | CERAD −/ACH | I | None |
| 102 | 64 /M | 2 /4 | CERAD −/PD | II | None |
| 103 | 64 /M | 3 /3 | CERAD −/PD | I | None |
| 104 | 68 /M | 3 /3 | CERAD− | I | None |
| 105 | 72 /M | 3 /3 | CERAD−/Hip. Sc. | I | None |
| 106 | 74 /M | 3 /3 | CERAD−/PD | IV | None |
| 107 | 76 /F | 3 /3 | CERAD+/− | I | Sparse/Moderate |
| 108 | 76 /M | 3 /3 | CERAD−/PD/PSP | III | Sparse |
| 109 | 77 /M | 3 /3 | CERAD− | II | Sparse |
| 110 | 77 /F | 3 /3 | CERAD −/PD | II | None |
| 111 | 78 /M | 3 /4 | CERAD +/− | III | Moderate |
| 112 | 79 /M | 3 /3 | CERAD−/Hip. Sc./AGD | II | Sparse |
| 113 | 81 /M | 2 /3 | CERAD −/PD | I | None |
| 114 | 82 /F | 3 /3 | CERAD − | I | Sparse |
| 115 | 83 /M | 3 /3 | CERAD −/LB | III | Sparse |
| 116 | 83 /M | 3 /3 | CERAD −/PS/PSP/Hip. Sc. | III | Sparse |
| 117 | 86 /M | 3 /3 | CERAD − | II | None |
| 118 | 89 /F | 3 /3 | CERAD +/− | IV | Moderate |
| 119 | 91 /F | 3 /3 | CERAD −/PSP | II | None |
| 120 | 91 /M | 3 /4 | CERAD +/− | III | Sparse/Moderate |
CERAD, Consortium to Establish a Registry for Alzheimer’s Disease; MID, multiple infarct dementia; Hip. Sc., hippocampal sclerosis; PSP, progressive supranuclear palsy; DLB, dementia with Lewy bodies; Inc. LB, inclusion Lewy bodies; ACH, acute cerebral hemorrhage; PD, Parkinson’s disease; AGD, argyrophilic grain disease.
Sections (40 μm each) were stained by hematoxylin-eosin (H&E), thioflavine-S, Gallyas, and Campbell-Switzer silver techniques. All individuals were ApoE genotyped by the technique of Hixson and Vernier, 18 using blood obtained by cardiac puncture in the immediate postmortem. Skeletal muscles (right and left temporalis) were carefully removed at autopsy and kept frozen at −85°C until the moment of use. The temporalis muscles were studied because they are easily accessible during the removal of the brain. The temporalis muscles have a mixed population of fiber types 19 and are similar to most of the skeletal muscles in the human body. 20
Isolation and Quantification of Aβ Peptides
Temporalis muscle (200 mg), free of adipose and connective tissue, was finely minced with a razor blade. The tissue was thoroughly disrupted in 12 ml of 90% formic acid by using a Dounce glass homogenizer. The specimens were loaded into 12-ml polyallomer tubes and centrifuged for 30 minutes at 250,000 × g in a Sorvall TH-641 rotor at 5°C. A sample of 500 μl was carefully taken from the middle of the tube and loaded onto a Superose 12 size exclusion column. The column was equilibrated, and the chromatography was developed with 80% glass distilled formic acid. Fractions corresponding to the retention time of 4.5 kd (defined by the synthetic Aβ reverse sequence 40–1) were collected, pooled, and mixed with 30 μl of 10% betaine, and the acid was immediately eliminated by vacuum centrifugation. The dried specimens were dissolved in 50 μl of 80% formic acid, which were then diluted with 250 μl of 0.5 mol/L Tris/HCl, pH 8.0, 1.37 mol/L NaCl, 27 mmol/L KCl, and 0.5% Tween 20. The volume was brought to 1 ml with distilled water, and the pH was adjusted to 7.4 with 10 N NaOH with a pH meter equipped with a microelectrode. 21 The final volume was brought to 2.5 ml by the addition of distilled water. To prepare the microtiter plate, 50 μl of the capture antibody, either R163 or R165, at a concentration of 10 μg/ml in 10 mmol/L Na2CO3, pH 9.6, was added to the wells of microtiter plates and left at room temperature for 2 h. A blocking solution of 1% bovine serum albumin in TTBS (0.05% Tween 20 in 50 mmol/L Tris/HCl, 137 mmol/L NaCl, 2.7 mmol/L KCl, pH 7.4) was added to each well and incubated at room temperature for 1 hour. Either 100 μl of the specimens under investigation or of the Aβ40 and Aβ42 standards were applied in triplicate to the wells and allowed to stand at room temperature for 2 hours on a rocking platform. The unbound materials were removed by washing the plate 3 times with TTBS. Fifty μl of Eu-labeled 4G8 antibody (4 μg/ml) were added to the wells and incubated for 1 hour, followed by four washes with TTBS and three washes with distilled water. Finally, 50 μl of Enhancement solution were added to each well, and the plates were read in a fluorimeter by using excitation and emission wavelengths of 320 and 615 nm, respectively. A standard curve was plotted for each plate, and sample Aβ concentrations were calculated with reference to these standard curves. The Aβ40 and Aβ42 peptides used to construct the standard curves were dissolved in dimethylsulfoxide (DMSO) at a concentration of 1 mg/ml and subsequently diluted with TTBS to the required concentrations (from 25 to1000 pg/ml).
Superior frontal gyrus (250 mg) was thoroughly homogenized in the presence of 12 ml of 98% formic acid. The brain specimens were centrifuged as described for the muscle tissue. This permitted the separation of an insoluble pellet and of a small amount of lipid material that floated at the top of the supernatant. An aliquot of 500 μl from each specimen was chromatographically separated, and the 4.5-kd fraction was isolated, dried, suspended in formic acid, pH adjusted to neutrality, and europium immunoassayed as previously described for the temporalis muscle.
Mass Spectrometry
Mass spectra were acquired on a Kratos Kompact MALDI IV time-of-flight mass spectrometer (Manchester, UK) in a positive linear mode with a 337-nm N2 laser and a 20-kV extraction potential. An aliquot of the relevant FPLC fractions was lyophilized and suspended in 10 μl of glass-distilled formic acid, of which 0.3 μl was deposited on a sample probe, followed by the addition of 0.3 μl of matrix consisting of a saturated solution of α-cyano-4-hydroxy-cinnamic acid in 50% ethanol. Reported spectra were the average of 50 laser shots. Each spectrum was calibrated using external standards.
AβPP Western Blot Quantification
Four hundred μg of muscle or 300 μg of brain tissue were homogenized in 1.6 ml of buffer: 20 mmol/L Tris-HCl, pH 8.4, 0.2% Triton X-100, and a protease inhibitor cocktail. Homogenates were centrifuged at 12,000 × g for 15 minutes at 4°C, and the supernatants were submitted to Western blotting. The homogenates were diluted with sample buffer (Novex, San Diego, CA) to equal protein concentrations and loaded onto 16% tricine or 10% Tris-Glycine precast gels (Novex). The electrophoretically separated proteins were transferred onto nitrocellulose membranes and blocked with 5% nonfat milk in Tris-buffered saline (TBS; 20 mmol/L Tris-HCl, pH 7.4. 0.5 mol/L NaCl). The primary antibody 22C11 (10 μg/ml) made to the N terminus of AβPP was incubated with blots overnight in 1% milk-TBS and blots were washed with TTBS. Secondary anti-mouse antibodies (Amersham; 1:750) for 22C11, diluted in milk-TBS, were added to the blots and incubated for 1 hour. The washed blots were developed using the Pierce (Rockford, IL) chemiluminescent detection system and exposed to film, and the band densities were analyzed with a Kodak Digital Imaging System.
Aβ Western Blot Detection
FPLC-separated, 4.5-kd fractions from six runs of temporalis muscle samples were pooled and dried by vacuum centrifugation. The samples were dissolved in tricine-sodium dodecyl sulfate (SDS) sample buffer (Novex) and separated in a 10% to 20% tricine gel (Novex). The peptides were transferred onto a polyvinylidene difluoride (PVDF) membrane (BioRad) and reacted with a mixture of Aβ antibodies 4G8 and 6E10 as previously published. 21 The membranes were developed as described for AβPP blots.
Immunocytochemistry
Temporalis muscles from four cases with high levels of Aβ on the EuIA were stained immunocytochemically for Aβ with an antibody that recognizes residues 1–16 (monoclonal antibody 10D5, Athena Neurosciences, South San Francisco, CA). Sections were also stained with the antibodies R163 and R165, which recognize Aβ peptides ending in residues 40 and 42, respectively. Paraffin-embedded muscle tissues were cut at 8 μm, deparaffinized, and treated with 90% formic acid for 5 minutes. Sections were incubated in primary antibodies at 1:1000 overnight at 4°C and followed by a biotinylated secondary antibody and an avidin-biotin-peroxidase complex (ABC, Vector Laboratories, Burlingame, CA) using 3,3′-diaminobenzidine as the substrate, as previously described. 22 Control sections were treated identically except for the omission or preabsorption of the primary antibody with 100 μg/ml synthetic Aβ 1–40. A section of frontal cortex from an AD case was used as a positive control.
Statistical Analyses
The two-tailed Student’s t-test was applied when variable means were compared between AD and control subjects. Multiple stepwise (backward) regression was used to analyze the relationships among the specified dependent variables and independent variables in the combined control and AD populations (n = 43). At each step the variable that contributed least to the result was removed, as determined by t-tests of the regression coefficients. The regression was stopped at the point in which all of the probabilities of the variables were smaller than 0.05. The apoE genotypes were assigned the following numerical scale: 1 = no apoE ε4; 2 = apoE ε2/ε4 or apoE ε3/ε4; 3 = apoE ε4/ε4. Neuritic plaque densities were converted to numerical scores as follows: none = 0; sparse = 1; sparse/moderate = 2; moderate = 3; moderate/frequent = 4; frequent = 5.
Results
Neuropathological examination established that, in this study, 21 individuals met the CERAD criteria for AD (Table 1) ▶ . Two cases (19 and 20) were, as far as we could investigate, nondemented, but met the neuropathological criteria for AD and therefore were included in the CERAD-positive group. These two cases probably belong to the cohort of high-pathology individuals, who, despite having considerable numbers of neuritic plaques and neurofibrillary tangles, do not exhibit the symptoms of dementia. 23 Within the CERAD-positive group, 11 subjects presented supplementary pathological changes such as multiple infarcts, Lewy bodies, hippocampal sclerosis, or signs of progressive supranuclear palsy. In addition, three more patients had the neuropathological and clinical symptoms of Parkinson’s disease. In the control nondemented group, none of the 20 individuals exhibited symptoms of dementia or met the CERAD standards for AD, with the exception of four cases that had borderline CERAD values and therefore could be considered as possible AD. In four cases no pathological changes were observed. Parkinson’s disease was present in seven individuals. In addition, three cases had hippocampal sclerosis, three cases had progressive supranuclear palsy, and one case each exhibited Lewy body disease and argyrophilic grain disease (Table 1) ▶ . The allele frequencies for Apo ε2, ε3, and ε4 in the AD group were 4%, 74%, and 22%, respectively, whereas in the nondemented group they represented 5%, 87%, and 8%, respectively. The AD group had a significantly higher ε4 allele frequency than the nondemented control group (χ 2 = 7.69, P = 0.006).
The chromatographic separation of the 4.5-kd fractions containing the Aβ peptides, under denaturing conditions, allows for the partition of Aβ peptides from other molecules with higher and lower Mr that interact with Aβ (Figure 1) ▶ . Western blotting of the 4.5-kd chromatographic fractions from AD and control temporalis muscles demonstrated the presence of three Aβ-immunopositive bands that may represent monomers, dimers, trimers, or tetramers of Aβ (Figure 2) ▶ . The quantification of the Aβ peptides by EuIA in the brains of AD and control individuals clearly demonstrated that, on average, the levels of these peptides were much higher in AD (Table 2) ▶ . The amounts of Aβ40 in the AD and control brains were 608 ng/g and 209 ng/g, respectively (P = 0.022). The levels of Aβ42 in the AD and control brains were 6096 ng/g in the former and 784 ng/g in the latter (P < 0.001). The differences in Aβ40 and Aβ42 in the AD brain approximate those observed in chemically purified AD plaque cores, in which about 90% of the Aβ ends at residue 42. 24
Figure 1.

Chromatographic profile of the temporalis muscle proteins. The solid trace depicts the chromatographic contour of the muscle proteins produced by a size exclusion Superose 12 column (1 × 30 cm) developed with 80% glass-distilled formic acid. The separation was carried out using a FPLC apparatus (Pharmacia Biotechnology) at a flow rate of 15 ml per hour at room temperature and monitored at 280 nm. Fractions were automatically collected every 2 minutes. To define the retention time of the Aβ-containing fractions in the muscle preparation, an unused column was calibrated with synthetic Aβ reverse amino acid sequence 40–1 (molecular mass, 4331) which is indicated by the hyphened trace. After this calibration, extreme care was taken to decontaminate the injection system by multiple formic acid injections until, in a mock run, there were no traces of Aβ in the eluant as detected by EuIA. The Aβ eluted between 56 and 62 minutes was collected, pooled, and mixed with betaine (see Materials and Methods). The addition of this zwitterion prevents undesirable adsorption of Aβ to the glass during the drying process. The arrows indicate the elution times of calibration markers separated under the same denaturing conditions (in order from left to right): bovine serum albumin, ovalbumin, chymotrypsinogen, cytochrome C, Aβ dimer, Aβ monomer, and bacitracin.
Figure 2.

Western blot of the chromatographically separated 4.5-kd fraction from AD and control temporalis muscles. In each case, six chromatographic separations were pooled, totally dried by vacuum centrifugation, dissolved in tricine-SDS sample buffer (Novex), and separated in a 10% to 20% tricine gel. The peptides were transferred onto a PVDF membrane (BioRad) and reacted with a mixture of Aβ antibodies 4G8 and 6E10. The membranes were developed as previously published. 21 The numbers on the right margin indicate the position of molecular weight markers.
Table 2.
Muscle and Brain Aβ Concentrations in AD and Control Subjects
| Muscle (ng/g) | Brain (ng/g) | |||||
|---|---|---|---|---|---|---|
| Aβ40 | Aβ42 | Total | Aβ40 | Aβ42 | Total | |
| AD | ||||||
| 1 | 53.2 | 21.5 | 74.7 | 321 | 15125 | 15446 |
| 2 | 24.3 | 8.2 | 32.5 | 455 | 1854 | 2310 |
| 3 | 35.2 | 19.8 | 55.0 | 805 | 6916 | 7722 |
| 4 | 32.1 | 30.1 | 62.2 | 80 | 2460 | 2541 |
| 5 | 50.5 | 19.3 | 69.8 | 317 | 16031 | 16349 |
| 6 | 25.6 | 8.2 | 33.9 | 1259 | 4049 | 5307 |
| 7 | 23.2 | 15.1 | 38.3 | 26 | 4634 | 4660 |
| 8 | 35.4 | 9.3 | 44.7 | 400 | 2134 | 2535 |
| 9 | 46.5 | 23.5 | 70.0 | 149 | 19680 | 19829 |
| 10 | 30.0 | 9.1 | 39.1 | 1241 | 3531 | 4773 |
| 11 | 31.1 | 11.7 | 42.7 | 3434 | 8180 | 11614 |
| 12 | 25.8 | 17.3 | 43.1 | 123 | 3724 | 3847 |
| 13 | 48.9 | 19.9 | 68.8 | 278 | 2960 | 3238 |
| 14 | 20.5 | 11.1 | 31.7 | 1238 | 2469 | 3707 |
| 15 | 67.4 | 9.6 | 77.0 | 998 | 3283 | 4281 |
| 16 | 18.6 | 8.2 | 26.9 | 784 | 4208 | 4992 |
| 17 | 29.5 | 13.5 | 43.0 | 281 | 2374 | 2656 |
| 18 | 55.9 | 19.1 | 75.0 | 145 | 5432 | 5578 |
| 19 | 36.2 | 13.5 | 49.8 | 911 | 4719 | 5631 |
| 20 | 30.9 | 17.6 | 48.5 | 359 | 15773 | 16132 |
| 21 | 39.6 | 15.7 | 55.3 | 66 | 5944 | 6010 |
| 22 | 27.9 | 16.4 | 44.3 | 76 | 3677 | 3753 |
| 23 | 80.4 | 23.5 | 104.0 | 241 | 1047 | 1288 |
| Mean | 37.8 | 15.7 | 53.5 | 608.2 | 6095.8 | 6704.2 |
| SE* | 3.2 | 1.2 | 3.9 | 154.1 | 1098.8 | 1102.4 |
| Control | ||||||
| 101 | 21.7 | 3.2 | 24.8 | 63 | 87 | 150 |
| 102 | 30.3 | 12.1 | 42.4 | 731 | 614 | 1345 |
| 103 | 15.1 | 7.5 | 22.5 | 20 | 463 | 483 |
| 104 | 27.2 | 11.5 | 38.7 | 117 | 859 | 975 |
| 105 | 45.8 | 12.1 | 57.9 | 510 | 49 | 559 |
| 106 | 52.3 | 24.3 | 76.5 | 17 | 155 | 172 |
| 107 | 21.8 | 2.2 | 23.9 | 0 | 231 | 231 |
| 108 | 16.8 | 13.9 | 30.7 | 41 | 316 | 357 |
| 109 | 21.2 | 2.7 | 23.9 | 38 | 148 | 186 |
| 110 | 28.8 | 8.0 | 36.8 | 525 | 849 | 1374 |
| 111 | 21.0 | 10.7 | 31.7 | 56 | 1742 | 1799 |
| 112 | 29.8 | 12.7 | 42.5 | 22 | 44 | 67 |
| 113 | 15.8 | 14.2 | 29.9 | 161 | 3402 | 3563 |
| 114 | 33.2 | 9.2 | 42.4 | 36 | 92 | 128 |
| 115 | 52.1 | 23.1 | 75.2 | 22 | 89 | 111 |
| 116 | 36.2 | 8.7 | 44.9 | 644 | 2306 | 2950 |
| 117 | 25.6 | 1.3 | 26.9 | 377 | 206 | 583 |
| 118 | 27.3 | 1.2 | 28.6 | 46 | 1162 | 1208 |
| 119 | 45.0 | 24.4 | 69.4 | 40 | 172 | 212 |
| 120 | 30.0 | 0.8 | 30.7 | 705 | 2686 | 3390 |
| Mean | 29.8 | 10.2 | 40.0 | 208.6 | 783.6 | 992.2 |
| SE | 2.5 | 1.7 | 3.8 | 58.7 | 221.0 | 250.1 |
| P† | 0.067 | 0.010 | 0.019 | 0.022 | <0.001 | <0.001 |
*SE, Standard error of mean.
†Two-tailed Student’s t-test (unequal variance) probability.
The presence of formic acid may alter Aβ structure in such a way that, in the immunoassay, the binding of antibodies is enhanced. To investigate this possibility, the Aβ1–40 and Aβ1–42 standard curves for the EuIA were constructed using either DMSO or formic acid (80%) as initial dissolving agents. Before dilution with TTBS, the formic acid was neutralized using the same procedure as used for the tissue samples. No statistically significant differences in the amount of Aβ detected were found between the two procedures: Aβ40, r = 0.960; Aβ42, r = 0.968.
In the temporalis muscles of the AD and nondemented control groups, the average values for Aβ42 were 15.7 ng/g and 10.2 ng/g (P = 0.010), and for Aβ40 were 37.8 ng/g and 29.8 ng/g (P = 0.067), respectively. The amounts of Aβ were distributed in a wide range: Aβ40, AD 18.6–80.4 ng/g, versus control, 15.1–52.3 ng/g; Aβ42, AD 8.2–30.1 ng/g, versus control, 0.8–24.4 ng/g (Table 2) ▶ .
Several multiple regressions were used to analyze the relationships among the variables shown in Tables 1 and 2 ▶ ▶ . When apoE was chosen as a dependent variable, the resulting regression demonstrated that neuritic plaque density score (P = 0.024) and brain Aβ40 (P < 0.001) were significantly correlated at the 95% confidence level (r = 0.608). Likewise, there was a strong positive interaction (r = 0.843) between the Braak stage and neuritic plaque density score (P < 0.001), brain Aβ40 (P = 0.006), and a somewhat less pronounced effect correlated with the muscle Aβ42 (P = 0.015). When muscle Aβ42 was chosen as the dependent variable, muscle Aβ40 and Braak stage showed significant positive correlations (P < 0.001 and P = 0.042, respectively). It is interesting that the regression analysis revealed that there were positive correlations between age and neuritic plaque density score (P < 0.001) and age and muscle Aβ40 (P = 0.036). There were no apparent associations between gender and the muscle or brain Aβ levels in the AD and control populations.
Mass spectrometry of the isolated Aβ from the temporalis muscles produced a serendipitous finding: the presence of Aβ peptides ending at residues 44, 45, and 46 (Aβ notation) in both the nondemented control cases and AD individuals (Table 3) ▶ . It is unlikely, given the specificity of our antibodies, that these Aβ forms were detected in our immunoassays. Quantification of these longer Aβ peptides awaits further investigation because the relative intensities of the molecular ion signals in the mass spectrum of a single sample do not necessarily reflect their relative abundance. In addition, mass spectrometry revealed the presence of Aβ peptides beginning at positions 6, 7, 9, 10, or 11.
Table 3.
Mass Spectrometry of Aβ Peptides from Skeletal Muscle
| Expected Mr | Observed Mr | Aβ sequence |
|---|---|---|
| AD muscle | ||
| 3500.1 | 3499.7 | 10–42 |
| 3557.2 | 3559.8 | 9–42 |
| 3749.5 | 3751.7 | 11–46 |
| 3896.1 | 3896.6 | 6–42 |
| 4330.9 | 4330.9 | 1–40 |
| 4715.4 | 4715.4 | 1–44 |
| 4926.7 | 4926.7 | 1–46 |
| Control muscle | ||
| 3759.4 | 3758.4 | 7–42 |
| 381835 | 3813.5 | 10–45 |
| 3896.5 | 3896.7 | 6–42 |
| 4330.9 | 4331.4 | 1–40 |
| 4926.7 | 4926.7 | 1–46 |
The determination of muscle and brain relative levels of AβPP in both populations demonstrated no significant differences between the two cohorts in terms of relative units of density (Figure 3) ▶ . Last, temporalis muscle sections from AD and nondemented cases, stained for Aβ using three different specific antibodies (10D5, R163, and R165), revealed no differences between the two groups.
Figure 3.
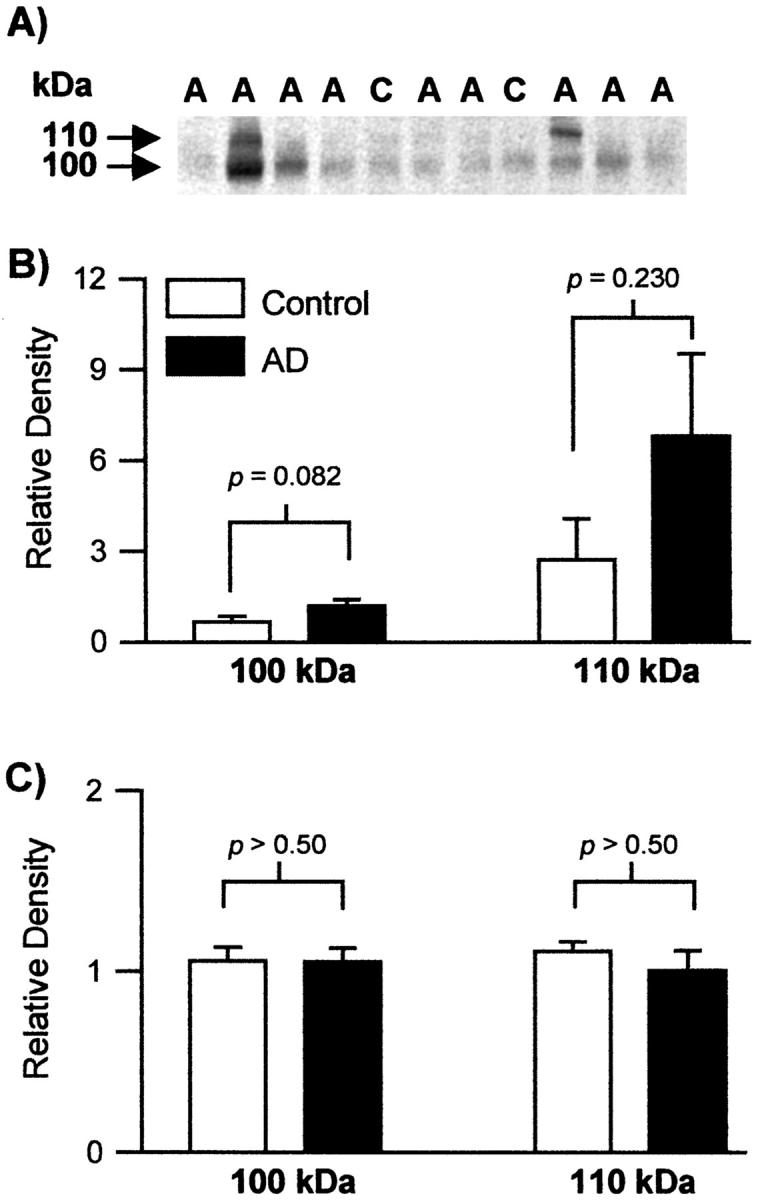
The relative levels of AβPP in the temporalis muscle and brain. A: A representative Western blot of AβPP stained by 22C11 from muscle obtained from control (C) and Alzheimer (A) tissues is shown. B and C: Histograms showing the relative levels of the 100- and 110-kd AβPP in the temporalis muscle (B) and the brain (C). The Western blots were densitometrically scanned, and the relative band intensities of the AD and control groups were averaged. There was no statistically significant difference between the two cohorts.
Discussion
A milestone in understanding the pathophysiology of AD was the discovery of Aβ40 and Aβ42 peptides in the brain and biological fluids of normal individuals. 25-27 For AD, these observations unseated the dogma that Aβ accumulation in this dementia was the result of an anomalous AβPP proteolytic degradation. This insight also ushered in alternative explanations for the pathogenic mechanisms of amyloidosis, such as Aβ overproduction, Aβ conformational misfolding, and defective Aβ clearance. In addition, it reopened the door for an earlier view postulating that a hematogenous source of Aβ could serve as a substrate for the vascular and parenchymal amyloid deposits seen in the AD brain. 28,29
The current results indicate that in AD there is a significant elevation of Aβ in the temporalis muscles relative to that in nondemented individuals. In the former, the mean value for total Aβ was 53.5 ng/g, whereas in the latter it was only 40.0 ng/g. This observation leads to some interesting prospects, the most striking of which is that the peripheral tissue metabolism of AβPP may be disturbed as reflected in the elevated muscle Aβ levels present in AD patients. This is an unprecedented finding because it provides an indication that AD may be a systemic disease rather than exclusively a disease of the central nervous system. The increased levels of plasma Aβ 30 might result from skeletal muscle abnormalities, much as serum Aβ levels are elevated in transgenic mice that overexpressed the AβPP C-99 transgene only in peripheral tissues. 10
Our study reveals for the first time that human skeletal muscle tissue possesses measurable amounts of Aβ40 and Aβ42. Because skeletal muscle represents about one quarter of body weight in humans, the pool of muscle Aβ could, over time, contribute to a substantial build up of Aβ in plasma and ultimately the brain. Alternatively, Aβ produced in the brain could be released into circulation and taken up by skeletal muscle. A series of recent observations supports the contention that peripherally circulating Aβ is a potential contributor to the cerebral amyloidosis of AD. 31,32 After intravenous injections of Aβ in aged primates, small amounts of this peptide have been recovered from the brains of these animals. 32 The uptake of peripheral Aβ by the brain may be dependent on a compromised BBB, and aging and neurodegenerative diseases may contribute to BBB impairment. 33 Disturbances in the BBB also occur in head trauma 34 and vascular diseases such as atherosclerosis, 35 hypertension, 36 and stroke 37 all of which have been found to represent risk factors for AD. Interestingly, Aβ42 chronically infused into the circulation of rats, in which the BBB was breached, was localized in the brain parenchyma. 38
In agreement with previous studies, 39,40 a positive relationship between apoE ε4 and neuritic plaque density was also seen in our multiple regression analysis model. Our results support earlier findings that indicated a strong positive correlation between the concentration of brain Aβ40, but not Aβ42, and apoE ε4 allele dosage. 41-43 As expected, the Braak stage strongly correlated with the neuritic plaque density. The brain Aβ40 levels also correlated with the Braak stage. This trend, which reflects the gravity of the disease, is in agreement with recent observations demonstrating that the higher the level of Aβ40 grows, the lower the level of synaptic density falls. 23
Our finding of Aβ peptides ending in residues 44, 45, and 46 by mass spectrometry in the temporalis muscles suggests that the site of γ-secretase cleavage could be closer to the cytosolic leaflet of the membrane. This possibility was previously suggested by cell culture studies using mutated forms of Aβ. 44 It also suggests that there are multiple sites for γ-secretase hydrolysis. 45 Alternatively, γ-secretase hydrolysis may only occur at residue 46, with the Aβ thereafter shortened by carboxypeptidases to peptides ending in residues 45 and 44 and more frequently terminating at residues 42 and 40. The potential significance of the cleavage of Aβ at residue 46, which corresponds to residue 717 of the AβPP770, is underscored by mutations at this position that lead to AD. 46 The degradation of the C terminus of Aβ by carboxypeptidases deserves further investigation in view of the apparent effectiveness by which the N terminus of Aβ is degraded by proteolytic enzymes. 24
A long-standing question in AD is why Aβ does not accumulate in tissues other than the brain, despite the fact that several peripheral tissues express AβPP. Our efforts to immunocytochemically detect deposits of Aβ in muscle met with frustration. This is not surprising, because the Aβ peptide is promptly sequestered by a large number of proteins such as albumin that are present in high quantities in plasma and in the extracellular spaces of the body. We and others have recently found that albumin readily interacts with soluble Aβ, masking its antigenic determinants and, in addition, inhibiting Aβ fibrillogenesis. 21,47,48 Histological examination of temporalis sections stained by H&E only demonstrated mild nonspecific myopathic changes consistent with normal aging. A more detailed and extensive histological investigation of the temporalis muscle is underway.
In summary, the temporalis muscles of AD individuals on the average contain significantly higher levels of Aβ relative to the nondemented group. This suggests that the skeletal muscle may participate as a contributor to the plasma Aβ pool as well as to the amyloid deposits of the brain and its vasculature observed in AD. The elevated levels of Aβ in the temporalis muscles suggest that alterations in AβPP metabolism might be a systemic problem in AD and not a feature unique to the central nervous system.
Acknowledgments
This paper is dedicated to the memory of our dear colleague W. Harold Civin, MD.
Footnotes
Address reprint requests to Dr. Alex E. Roher, Haldeman Laboratory for Alzheimer Disease Research, Sun Health Research Institute, 10515 West Santa Fe Dr., Sun City, AZ 85351. E-mail: aroher@mail.sunhealth.org.
Supported in part by the State of Arizona Center for Alzheimer’s Disease Research.
References
- 1.Selkoe DJ: Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci 1994, 17:489-517 [DOI] [PubMed] [Google Scholar]
- 2.Joachim CL, Mori H, Selkoe DJ: Amyloid β-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 1989, 341:226-230 [DOI] [PubMed] [Google Scholar]
- 3.Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG: Expression of β amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron 1990, 4:253-267 [DOI] [PubMed] [Google Scholar]
- 4.Li QX, Whyte S, Tanner JE, Evin G, Beyreuther K, Masters CL: Secretion of Alzheimer’s disease Aβ amyloid peptide by activated human platelets. Lab Invest 1998, 78:461-469 [PubMed] [Google Scholar]
- 5.Nordstedt C, Naslund J, Thyberg J, Messamore E, Gandy SE, Terenius L: Human neutrophil phagocytic granules contain a truncated soluble form of the Alzheimer β/A4 amyloid precursor protein (APP). J Biol Chem 1994, 269:9805-9810 [PubMed] [Google Scholar]
- 6.Schubert W, Prior R, Weidemann A, Dircksen H, Multhaup G, Masters CL, Beyreuther K: Localization of Alzheimer βA4 amyloid precursor protein at central and peripheral synaptic sites. Brain Res 1991, 563:184-194 [DOI] [PubMed] [Google Scholar]
- 7.Gentleman SM, Greenberg BD, Savage MJ, Noori M, Newman SJ, Roberts GW, Griffin WS, Graham DI: Aβ 42 is the predominant form of amyloid β-protein in the brains of short-term survivors of head injury. Neuroreport 1997, 8:1519-1522 [DOI] [PubMed] [Google Scholar]
- 8.Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR: Traumatic brain injury increases β-amyloid peptide 1–42 in cerebrospinal fluid. J Neurochem 1998, 71:2505-2509 [DOI] [PubMed] [Google Scholar]
- 9.Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, McComb JG, Frangione B, Ghiso J: Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer’s amyloid β. Biochem Biophys Res Commun 1994, 205:1431-1437 [DOI] [PubMed] [Google Scholar]
- 10.Fukuchi K, Ho L, Younkin SG, Kunkel DD, Ogburn CE, LeBoeuf RC, Furlong CE, Deeb SS, Nochlin D, Wegiel J, Wisniewski HM, Martin GM: High levels of circulating β-amyloid peptide do not cause cerebral β-amyloidosis in transgenic mice. Am J Pathol 1996, 149:219-227 [PMC free article] [PubMed] [Google Scholar]
- 11.Askanas V, Engel WK: Sporadic inclusion-body myositis and its similarities to Alzheimer disease brain: recent approaches to diagnosis and pathogenesis, and relation to aging. Scand J Rheumatol 1998, 27:389-405 [DOI] [PubMed] [Google Scholar]
- 12.Mehta PD, Dalton AJ, Mehta SP, Kim KS, Sersen EA, Wisniewski HM: Increased plasma amyloid β protein 1–42 levels in Down syndrome. Neurosci Lett 1998, 241:13-16 [DOI] [PubMed] [Google Scholar]
- 13.Mirra SS, Hart MN, Terry RD: Making the diagnosis of Alzheimer’s disease: a primer for practicing pathologists. Arch Pathol Lab Med 1993, 117:132-144 [PubMed] [Google Scholar]
- 14.Braak H, Braak E: Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997, 18:351-357 [DOI] [PubMed] [Google Scholar]
- 15.McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, Ballard C, de Vos RA, Wilcock GK, Jellinger KA, Perry RH: Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996, 47:1113-1124 [DOI] [PubMed] [Google Scholar]
- 16.Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I: Preliminary NINDS neuropathologic criteria for Steele-Richardson- Olszewski syndrome (progressive supranuclear palsy). Neurology 1994, 44:2015-2019 [DOI] [PubMed] [Google Scholar]
- 17.Litvan I, Hauw JJ, Bartko JJ, Lantos PL, Daniel SE, Horoupian DS, McKee A, Dickson D, Bancher C, Tabaton M, Jellinger K, Anderson DW: Validity and reliability of the preliminary NINDS neuropathologic criteria for progressive supranuclear palsy and related disorders. J Neuropathol Exp Neurol 1996, 55:97-105 [DOI] [PubMed] [Google Scholar]
- 18.Hixson JE, Vernier DT: Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res 1990, 31:545-548 [PubMed] [Google Scholar]
- 19.Korfage JA, Van Eijden TM: Regional differences in fibre type composition in the human temporalis muscle. J Anat 1999, 194:355-362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banker BQ, Engel AG: Basic reactions of muscle. 2nd ed. Engel AG Franzini-Armstrong C eds. Myology: Basic and Clinical, 1994, :pp. 832-888 McGraw-Hill, New York [Google Scholar]
- 21.Kuo YM, Emmerling MR, Lampert HC, Hempelman SR, Kokjohn TA, Woods AS, Cotter RJ, Roher AE: High levels of circulating Aβ42 are sequestered by plasma proteins in Alzheimer disease. Biochem Biophys Res Commun 1999, 257:787-791 [DOI] [PubMed] [Google Scholar]
- 22.Beach TG, Tago H, Nagai T, Kimura H, McGeer PL, McGeer EG: Perfusion-fixation of the human brain for immunohistochemistry: comparison with immersion-fixation. J Neurosci Methods 1987, 19:183-192 [DOI] [PubMed] [Google Scholar]
- 23.Lue L-F, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel R, Rogers J: Soluble amyloid peptide concentration as a predictor of synaptic change in Alzheimer’s diseases. Am J Pathol 1999, 155:853-862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ: Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem 1993, 268:3072-3083 [PubMed] [Google Scholar]
- 25.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE: Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem 1996, 271:4077-4081 [DOI] [PubMed] [Google Scholar]
- 26.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C: Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 1992, 359:325-327 [DOI] [PubMed] [Google Scholar]
- 27.Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, Wisniewski T, Frangione B: Alzheimer’s soluble amyloid β is a normal component of human urine. FEBS Lett 1997, 408:105-108 [DOI] [PubMed] [Google Scholar]
- 28.Selkoe DJ: Molecular pathology of amyloidogenic proteins and the role of vascular amyloidosis in Alzheimer’s disease. Neurobiol Aging 1989, 10:387-395 [DOI] [PubMed] [Google Scholar]
- 29.Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B: Blood-brain barrier transport of circulating Alzheimer’s amyloid β. Biochem Biophys Res Commun 1993, 197:1034-1040 [DOI] [PubMed] [Google Scholar]
- 30.Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, Merchant C, Small SA, Stern Y, Wisniewski HM, Mehta PD: Plasma amyloid β-peptide 1–42 and incipient Alzheimer’s disease. Ann Neurol 1999, 46:412-416 [DOI] [PubMed] [Google Scholar]
- 31.Zlokovic BV, Martel CL, Matsubara E, McComb JG, Zheng G, McCluskey RT, Frangione B, Ghiso J: Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid β at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA 1996, 93:4229-4234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV: Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid β peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 1998, 70:210-215 [DOI] [PubMed] [Google Scholar]
- 33.Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM: Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994, 87:469-480 [DOI] [PubMed] [Google Scholar]
- 34.van Duijn CM, Tanja TA, Haaxma R, Schulte W, Saan RJ, Lameris AJ, Antonides-Hendriks G, Hofman A: Head trauma and the risk of Alzheimer’s disease. Am J Epidemiol 1992, 135:775-782 [DOI] [PubMed] [Google Scholar]
- 35.Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, van Duijn CN, Van Broeckhoven C, Grobbee DE: Atherosclerosis, apolipoprotein E, and prevalence of dementia, and Alzheimer’s disease in the Rotterdam Study. Lancet 1997, 349:151-154 [DOI] [PubMed] [Google Scholar]
- 36.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, Persson G, Oden A, Svanborg A: 15-year longitudinal study of blood pressure and dementia. Lancet 1996, 347:1141-1145 [DOI] [PubMed] [Google Scholar]
- 37.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR: Brain infarction and the clinical expression of Alzheimer disease: the Nun Study. J Am Med Assoc 1997, 277:813-817 [PubMed] [Google Scholar]
- 38.Pluta R, Misicka A, Januszewski S, Barcikowska M, Lipkowski AW: Transport of human β-amyloid peptide through the rat blood-brain barrier after global cerebral ischemia. Acta Neurochir Suppl (Wien) 1997, 70:247-249 [DOI] [PubMed] [Google Scholar]
- 39.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD: Increased amyloid β-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci USA 1993, 90:9649-9653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pirttila T, Soininen H, Mehta PD, Heinonen O, Lehtimaki T, Bogdanovic N, Paljarvi L, Kim KS, Kosunen O, Winblad B, Riekkinen PS, Wisniewski HM: Apolipoprotein E genotype, and amyloid load in Alzheimer disease, and control brains. Neurobiol Aging 1997, 18:121-127 [DOI] [PubMed] [Google Scholar]
- 41.Gearing M, Mori H, Mirra SS: Aβ peptide length and apolipoprotein E genotype in Alzheimer’s disease. Ann Neurol 1996, 39:395-399 [DOI] [PubMed] [Google Scholar]
- 42.Ishii K, Tamaoka A, Mizusawa H, Shoji S, Ohtake T, Fraser PE, Takahashi H, Tsuji S, Gearing M, Mizutani T, Yamada S, Kato M, St. George-Hyslop PH, Mirra SS, Mori H: Aβ1–40 but not A β1–42 levels in cortex correlate with apolipoprotein E ε4 allele dosage in sporadic Alzheimer’s disease. Brain Res 1997, 748:250-252 [DOI] [PubMed] [Google Scholar]
- 43.Mann DM, Iwatsubo T, Pickering-Brown SM, Owen F, Saido TC, Perry RH: Preferential deposition of amyloid β protein (Aβ) in the form Aβ40 in Alzheimer’s disease is associated with a gene dosage effect of the apolipoprotein E ε4 allele. Neurosci Lett 1997, 221:81-84 [DOI] [PubMed] [Google Scholar]
- 44.Tischer E, Cordell B: β-Amyloid precursor protein: location of transmembrane domain and specificity of γ-secretase cleavage. J Biol Chem 1996, 271:21914-21919 [DOI] [PubMed] [Google Scholar]
- 45.Lichtenthaler SF, Wang R, Grimm H, Uljon SN, Masters CL, Beyreuther K: Mechanism of the cleavage specificity of Alzheimer’s disease γ-secretase identified by phenylalanine-scanning mutagenesis of the transmembrane domain of the amyloid precursor protein. Proc Natl Acad Sci USA 1999, 96:3053-3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hardy J: Amyloid, the presenilins, and Alzheimer’s disease. Trends Neurosci 1997, 20:154-159 [DOI] [PubMed] [Google Scholar]
- 47.Bohrmann B, Tjernberg L, Kuner P, Poli S, Levet-Trafit B, Naslund J, Richards G, Huber W, Dobeli H, Nordstedt C: Endogenous proteins controlling amyloid β-peptide polymerization: possible implications for β-amyloid formation in the central nervous system and in peripheral tissues. J Biol Chem 1999, 274:15990-15995 [DOI] [PubMed] [Google Scholar]
- 48.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ: Amyloid β-peptide is transported on lipoproteins, and albumin in human plasma. J Biol Chem 1996, 271:32916-32922 [DOI] [PubMed] [Google Scholar]