

Abstract
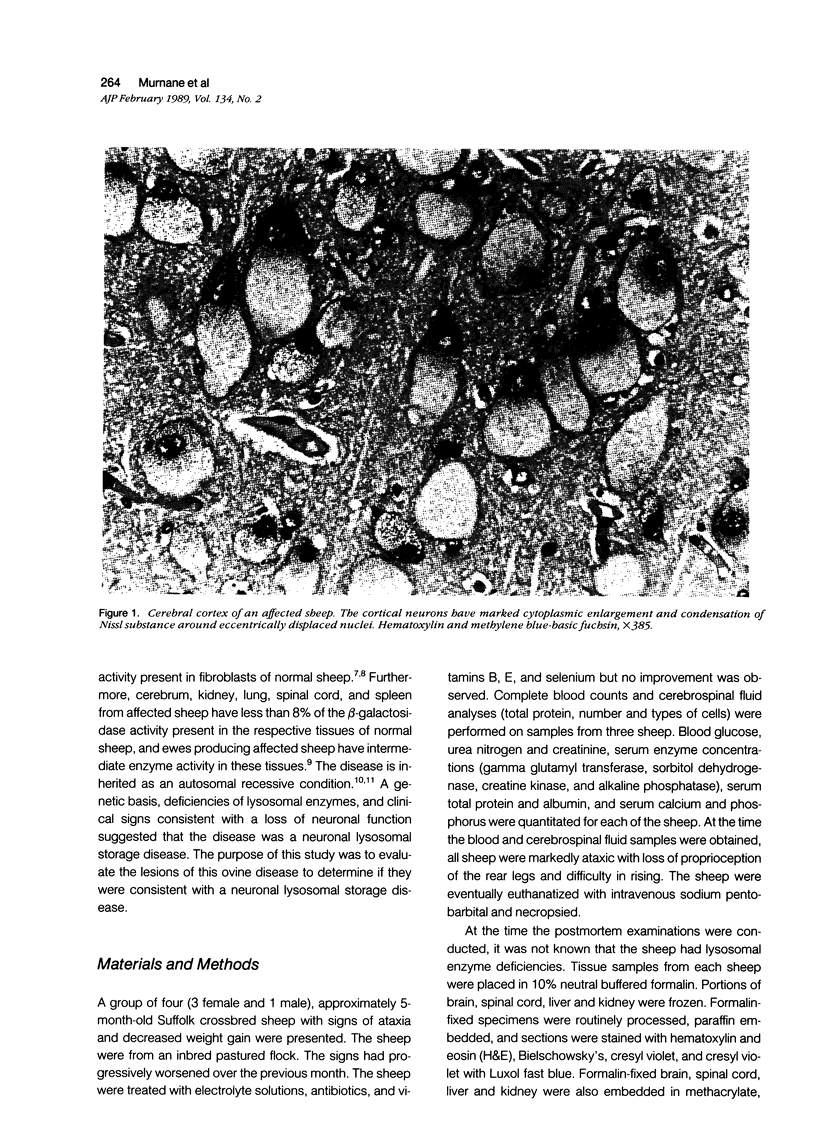
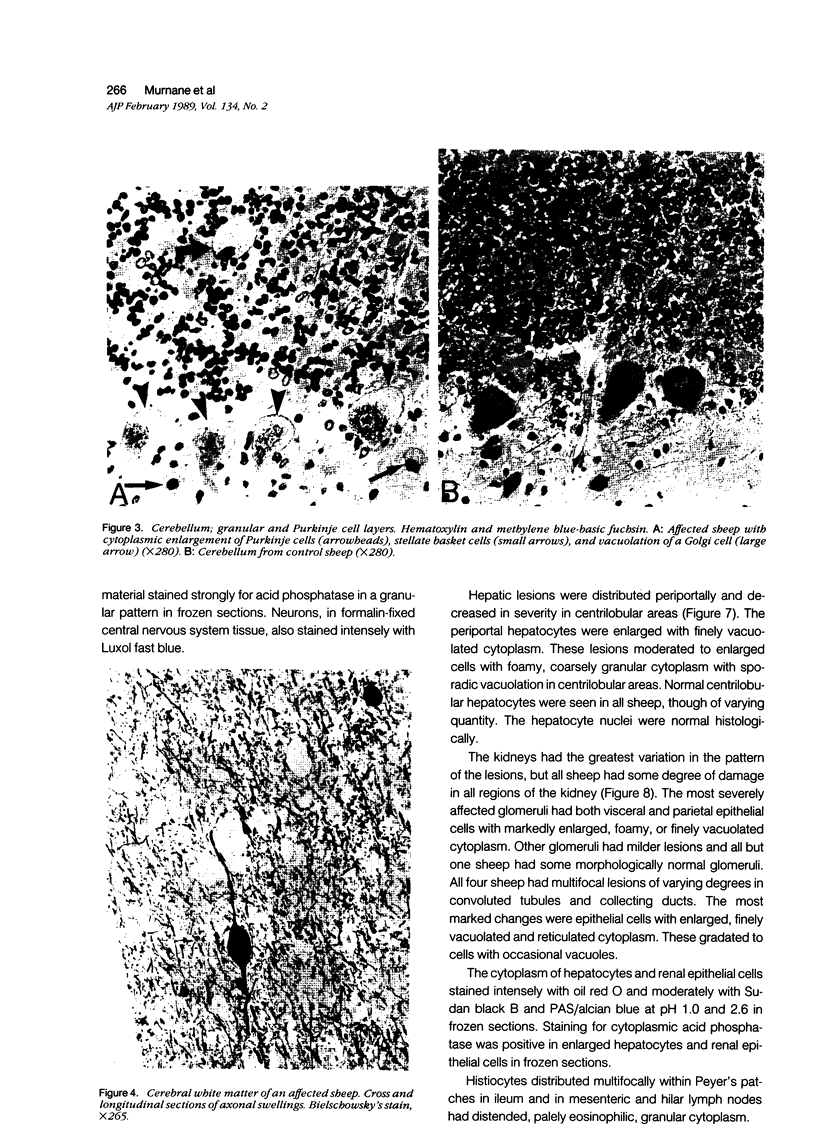
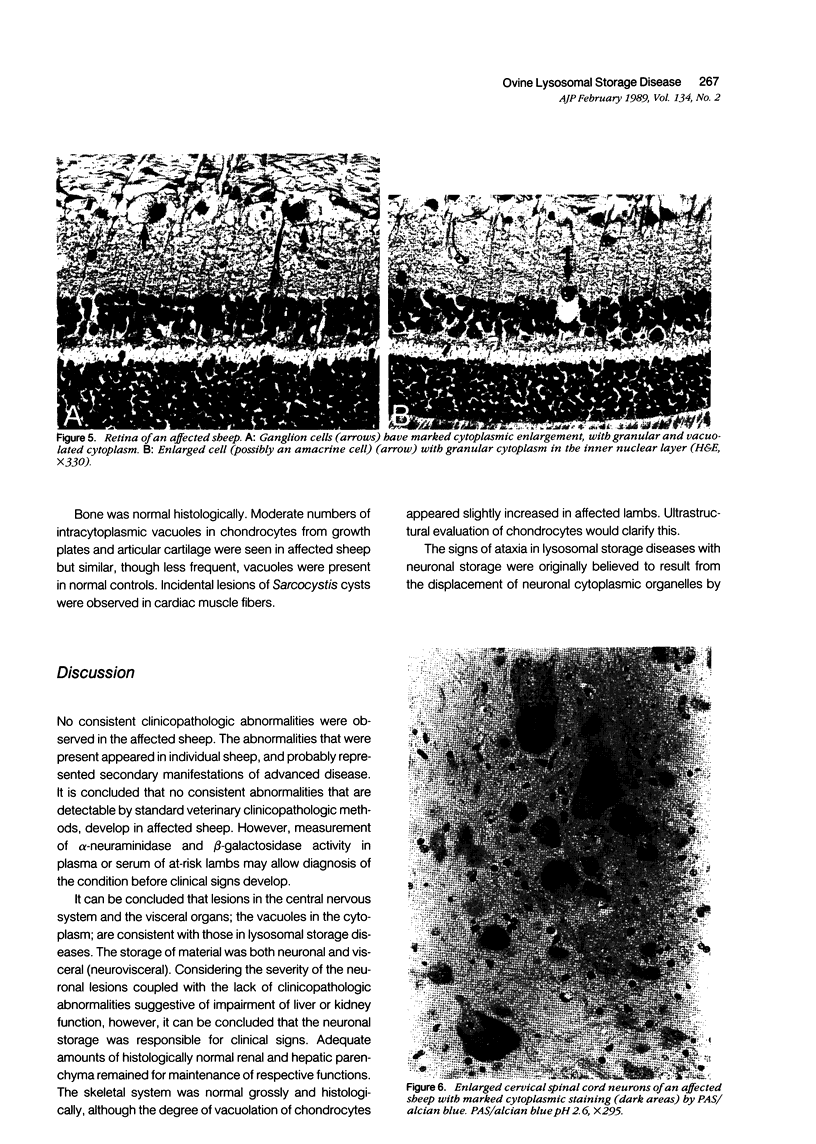
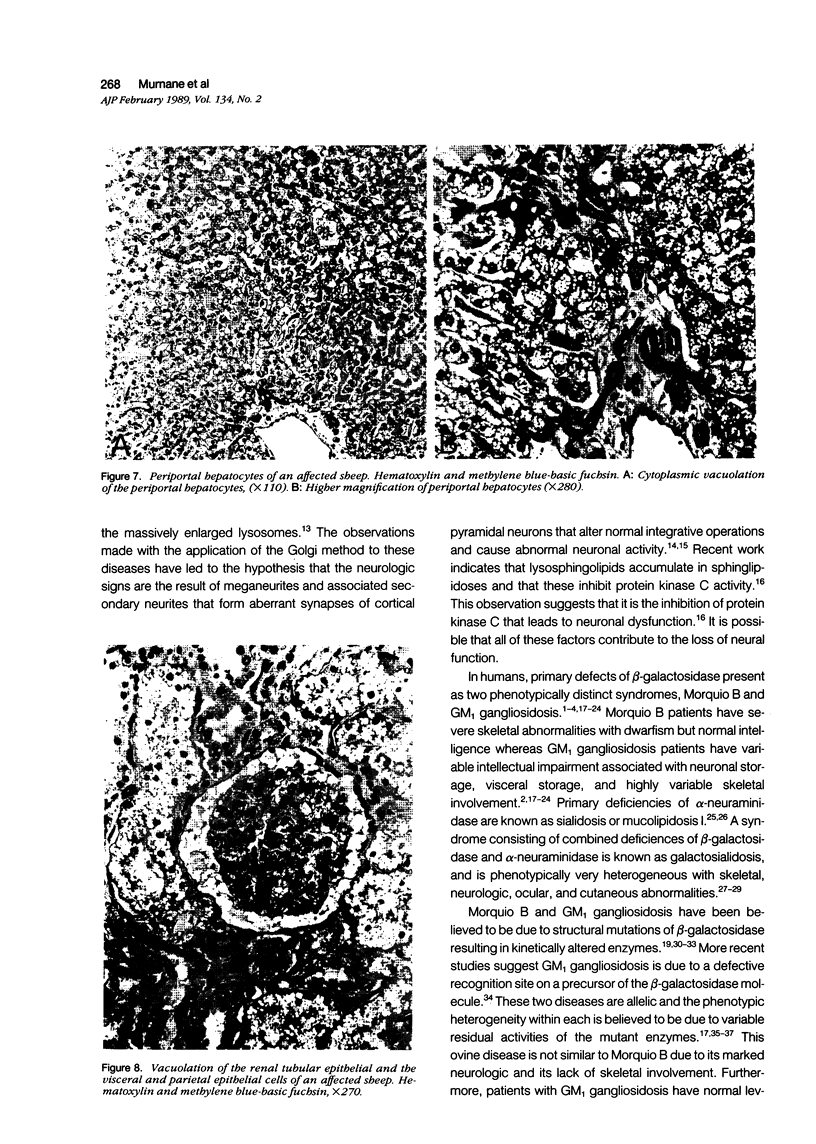
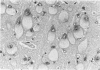
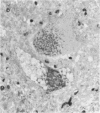
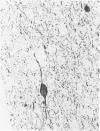
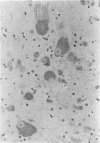
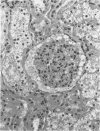
An inherited disease associated with deficiencies of beta-galactosidase and alpha-neuraminidase has been identified recently in sheep. The clinical signs, the deficiency of lysosomal enzymes, and the familial nature of the disorder suggested that the condition was a lysosomal storage disease. Four affected sheep were necropsied and their tissues were examined by histopathologic and histochemical methods to determine if the lesions were consistent with a lysosomal storage disease. Central nervous system neurons were enlarged with finely to coarsely granular cytoplasmic material, or less often, neurons were distended with multiple, variably-sized vacuoles. Loss of neurons without gliosis was evident and the Nissl substance was either dispersed and fragmented or condensed around the nuclei of remaining neurons. Neurons of intestinal and other peripheral ganglia, retinal ganglion cells, and heart Purkinje fibers were enlarged similarly. White matter of the cerebrum and spinal cord had numerous spheroid to ellipsoid axonal enlargements. Periportal hepatocytes and renal epithelial cells were enlarged with marked vacuolation. The neuronal storage material stained intensely with periodic acid-Schiff/alcian blue, with Luxol fast blue, for acid phosphatase, and moderately with oil red O stains. Renal and hepatocyte storage material stained intensely with oil red O and moderately with periodic acid-Schiff/alcian blue and Sudan black B stains. The lesions in these sheep were consistent with those of a lysosomal storage disease. Both neuronal and visceral storage occurred, but the neuronal storage was more severe.
Full text
PDF



Images in this article



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Ahern-Rindell A. J., Murnane R. D., Prieur D. J. Beta-galactosidase activity in fibroblasts and tissues from sheep with a lysosomal storage disease. Biochem Genet. 1988 Dec;26(11-12):733–746. [PubMed] [Google Scholar]
- Ahern-Rindell A. J., Prieur D. J., Murnane R. D., Raghavan S. S., Daniel P. F., McCluer R. H., Walkley S. U., Parish S. M. Inherited lysosomal storage disease associated with deficiencies of beta-galactosidase and alpha-neuraminidase in sheep. Am J Med Genet. 1988 Sep;31(1):39–56. doi: 10.1002/ajmg.1320310108. [DOI] [PubMed] [Google Scholar]
- D'Azzo A., Hoogeveen A., Reuser A. J., Robinson D., Galjaard H. Molecular defect in combined beta-galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci U S A. 1982 Aug;79(15):4535–4539. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnick R. J., Thorpe S. R., Fiddler M. B. Toward enzyme therapy for lysosomal storage diseases. Physiol Rev. 1976 Jan;56(1):57–99. doi: 10.1152/physrev.1976.56.1.57. [DOI] [PubMed] [Google Scholar]
- Galjaard H., Hoogeveen A., de Wit-Verbeek H. A., Reuser A. J., Ho M. W., Robinson D. Genetic heterogeneity in GM1-gangliosidosis. Nature. 1975 Sep 4;257(5521):60–62. doi: 10.1038/257060a0. [DOI] [PubMed] [Google Scholar]
- Glew R. H., Basu A., Prence E. M., Remaley A. T. Lysosomal storage diseases. Lab Invest. 1985 Sep;53(3):250–269. [PubMed] [Google Scholar]
- Gravel R. A., Lowden J. A., Callahan J. W., Wolfe L. S., Ng Yin Kin N. M. Infantile sialidosis: a phenocopy of type 1 GM1 gangliosidosis distinguished by genetic complementation and urinary oligosaccharides. Am J Hum Genet. 1979 Nov;31(6):669–679. [PMC free article] [PubMed] [Google Scholar]
- Hannun Y. A., Bell R. M. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987 Feb 6;235(4789):670–674. doi: 10.1126/science.3101176. [DOI] [PubMed] [Google Scholar]
- Holzgreve W., Gröbe H., von Figura K., Kresse H., Beck H., Mattei J. F. Morquio syndrome: clinical findings in 11 patients with MPS IVA and 2 patients with MPS IVB. Hum Genet. 1981;57(4):360–365. doi: 10.1007/BF00281685. [DOI] [PubMed] [Google Scholar]
- Hoogeveen A. T., Reuser A. J., Kroos M., Galjaard H. GM1-gangliosidosis. Defective recognition site on beta-galactosidase precursor. J Biol Chem. 1986 May 5;261(13):5702–5704. [PubMed] [Google Scholar]
- Hoogeveen A., d'Azzo A., Brossmer R., Galjaard H. Correction of combined beta-galactosidase/neuraminidase deficiency in human fibroblasts. Biochem Biophys Res Commun. 1981 Nov 16;103(1):292–300. doi: 10.1016/0006-291x(81)91692-2. [DOI] [PubMed] [Google Scholar]
- Janckila A. J., Li C. Y., Lam K. W., Yam L. T. The cytochemistry of tartrate-resistant acid phosphatase. Technical considerations. Am J Clin Pathol. 1978 Jul;70(1):45–55. doi: 10.1093/ajcp/70.1.45. [DOI] [PubMed] [Google Scholar]
- Lowden J. A., O'Brien J. S. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet. 1979 Jan;31(1):1–18. [PMC free article] [PubMed] [Google Scholar]
- Meisler M., Rattazzi M. C. Immunological studies of beta galactosidase in normal human liver and in GM1 gangliosidosis. Am J Hum Genet. 1974 Nov;26(6):683–691. [PMC free article] [PubMed] [Google Scholar]
- Norden A. G., O'Brien J. S. An electrophoretic variant of beta-galactosidase with altered catalytic properties in a patient with GM1 gangliosidosis. Proc Natl Acad Sci U S A. 1975 Jan;72(1):240–244. doi: 10.1073/pnas.72.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden A. G., Tennant L. L., O'Brien J. S. GM1 ganglioside beta-galactosidase. A. Purification and studies of the enzyme from human liver. J Biol Chem. 1974 Dec 25;249(24):7969–7976. [PubMed] [Google Scholar]
- O'Brien J. S., Gugler E., Giedion A., Wiessmann U., Herschkowitz N., Meier C., Leroy J. Spondyloepiphyseal dysplasia, corneal clouding, normal intelligence and acid beta-galactosidase deficiency. Clin Genet. 1976 May;9(5):495–504. doi: 10.1111/j.1399-0004.1976.tb01603.x. [DOI] [PubMed] [Google Scholar]
- O'Brien J. S. Molecular genetics of GM1 beta-galactosidase. Clin Genet. 1975 Nov;8(5):303–313. [PubMed] [Google Scholar]
- O'Brien J. S., Norden A. G. Nature of the mutation in adult beta-galactosidase deficient patients. Am J Hum Genet. 1977 Mar;29(2):184–190. [PMC free article] [PubMed] [Google Scholar]
- Palmeri S., Hoogeveen A. T., Verheijen F. W., Galjaard H. Galactosialidosis: molecular heterogeneity among distinct clinical phenotypes. Am J Hum Genet. 1986 Feb;38(2):137–148. [PMC free article] [PubMed] [Google Scholar]
- Pinsky L., Miller J., Shanfield B., Watters G., Wolfe L. S. GM1 gangliosidosis in skin fibroblast culture: enzymatic differences between types 1 and 2 and observations on a third variant. Am J Hum Genet. 1974 Sep;26(5):563–577. [PMC free article] [PubMed] [Google Scholar]
- Purpura D. P., Suzuki K. Distortion of neuronal geometry and formation of aberrant synapses in neuronal storage disease. Brain Res. 1976 Oct 29;116(1):1–21. doi: 10.1016/0006-8993(76)90245-6. [DOI] [PubMed] [Google Scholar]
- Sakuraba H., Suzuki Y., Akagi M., Sakai M., Amano N. beta-Galactosidase-neuraminidase deficiency (galactosialidosis): clinical, pathological, and enzymatic studies in a postmortem case. Ann Neurol. 1983 May;13(5):497–503. doi: 10.1002/ana.410130505. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Fukuoka K., Sakuraba H., Hayashi K., Ko Y. M. Galatosialidosis (beta-galactosidase-neuraminidase deficiency): clinical and biochemical studies on 13 patients. Adv Exp Med Biol. 1982;152:241–251. [PubMed] [Google Scholar]
- Suzuki Y., Nakamura N., Fukuoka K., Shimada Y., Uono M. beta-Galactosidase deficiency in juvenile and adult patients. Report of six Japanese cases and review of literature. Hum Genet. 1977 Apr 15;36(2):219–229. doi: 10.1007/BF00273261. [DOI] [PubMed] [Google Scholar]
- Trojak J. E., Ho C. K., Roesel R. A., Levin L. S., Kopits S. E., Thomas G. H., Toma S. Morquio-like syndrome (MPS IV B) associated with deficiency of a beta-galactosidase. Johns Hopkins Med J. 1980 Feb;146(2):75–79. [PubMed] [Google Scholar]
- Van Diggelen O. P., Schram A. W., Sinnott M. L., Smith P. J., Robinson D., Galjaard H. Turnover of beta-galactosidase in fibroblasts from patients with genetically different types of beta-galactosidase deficiency. Biochem J. 1981 Oct 15;200(1):143–151. doi: 10.1042/bj2000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner T. G., O'Brien J. S. Genetic defects in glycoprotein metabolism. Annu Rev Genet. 1983;17:395–441. doi: 10.1146/annurev.ge.17.120183.002143. [DOI] [PubMed] [Google Scholar]
- Wenger D. A., Goodman S. I., Myers G. G. Letter: Beta-galactosidase deficiency in young adults. Lancet. 1974 Nov 30;2(7892):1319–1320. doi: 10.1016/s0140-6736(74)90173-1. [DOI] [PubMed] [Google Scholar]
- Wenger D. A., Sattler M., Mueller O. T., Myers G. G., Schneiman R. S., Nixon G. W. Adult GM1 gangliosidosis: clinical and biochemical studies on two patients and comparison to other patients called variant or adult GM1 gangliosidosis. Clin Genet. 1980 May;17(5):323–334. doi: 10.1111/j.1399-0004.1980.tb00158.x. [DOI] [PubMed] [Google Scholar]
- van Diggelen O. P., Hoogeveen A. T., Smith P. J., Reuser A. J., Galjaard H. Enhanced proteolytic degradation of normal beta-galactosidase in the lysosomal storage disease with combined beta-galactosidase and neuraminidase deficiency. Biochim Biophys Acta. 1982 Apr 21;703(1):69–76. doi: 10.1016/0167-4838(82)90012-7. [DOI] [PubMed] [Google Scholar]
- van der Horst G. T., Kleijer W. J., Hoogeveen A. T., Huijmans J. G., Blom W., van Diggelen O. P. Morquio B syndrome: a primary defect in beta-galactosidase. Am J Med Genet. 1983 Oct;16(2):261–275. doi: 10.1002/ajmg.1320160215. [DOI] [PubMed] [Google Scholar]