

Abstract
Background and Purpose
Programmed cell death (pcd) plays a critical role in the development of the nervous system, as well as in its response to insult. Both anti-pcd and pro-pcd modulators play prominent roles in development and disease, including ischemic cerebrovascular disease. The purpose of this article is therefore to review the basics of programmed cell death.
Methods
There have been over 100 000 scientific and clinical publications on the topic of programmed cell death and its most well known form, apoptosis. The principles emerging from these studies are reviewed here.
Results
Programmed cell death is a form of cell death in which the cell plays an active role in its own demise. Apoptosis is the most well-defined form of pcd, but recent studies have begun to characterize an alternative program, autophagic cell death. In addition, there appear to be programmatic cell deaths that do not fit the criteria for either apoptosis or autophagic cell death, arguing that additional programs may also be available to cells.
Conclusion
Constructing a mechanistic taxonomy of all forms of pcd—based on inhibitors, activators, and identified biochemical pathways involved in each form of pcd—should offer new insight into cell deaths associated with cerebrovascular disease and other diseases, and ultimately offer new therapeutic approaches.
Keywords: animal models of human disease, apoptosis, genetically altered mice, ischemic biology, basic studies
“Death is the essential condition of life, not an evil.”
-Charlotte Perkins Gilman
Programmed cell death (pcd) plays a critical role in neural development, and dysregulation of cell death programs features in developmental, neoplastic, neurodegenerative, infectious, traumatic, ischemic, metabolic, and demyelinating disorders of the nervous system. It has been 100 years since the first description of developmental neuronal cell death,1 and over 50 years since the demonstration that such physiological cell death is inhibited by soluble factors such as nerve growth factor.2
In 1964, Lockshin and colleagues introduced the term programmed cell death to describe an apparently endogenous pathway or set of pathways used by cells to commit suicide during insect development.3 In 1966, it was shown that this process requires protein synthesis,2 arguing that it is the result of an active cellular suicide process. Then in 1972, Kerr, Wyllie and Currie coined the term apoptosis to describe a morphologically relatively uniform set of cell deaths that occurs in many different paradigms, from development to insult response to cell turnover.4
Apoptosis has been studied extensively, with well over 100 000 papers published on the subject (www.pubmed.gov). Although pcd has often been equated with apoptosis, it has become increasingly clear that nonapoptotic forms of pcd also exist5-16: for example, certain developmental cell deaths, such as “autophagic” cell death3,5,12-14 and “cytoplasmic” cell death,5,6,9-11,14 do not resemble apoptosis. Furthermore, neurodegenerative diseases such as Huntington disease and amyotrophic lateral sclerosis display neuronal cell death that does not fulfill the criteria for apoptosis7,16 (this is not to say that some classic apoptosis does not occur in these diseases, as well). Ischemia-induced cell death may also display a nonapoptotic morphology, referred to as “oncosis.”8
How many different mammalian cell death programs can be distinguished, and what is their relationship? A number of classifications have been proposed based on morphology, but for the purposes of mechanistic insight and therapeutic intervention, it would be preferable to construct a mechanistic taxonomy of all cell death programs, with special attention to their specific inhibitors and activators. The data required for such a construct are currently far from complete, and thus the present classification will undoubtedly be revised repeatedly over time. Nonetheless, it is informative to consider, based on currently available data, how many programs of cell death can be classified mechanistically (Figure 1).
Figure 1.
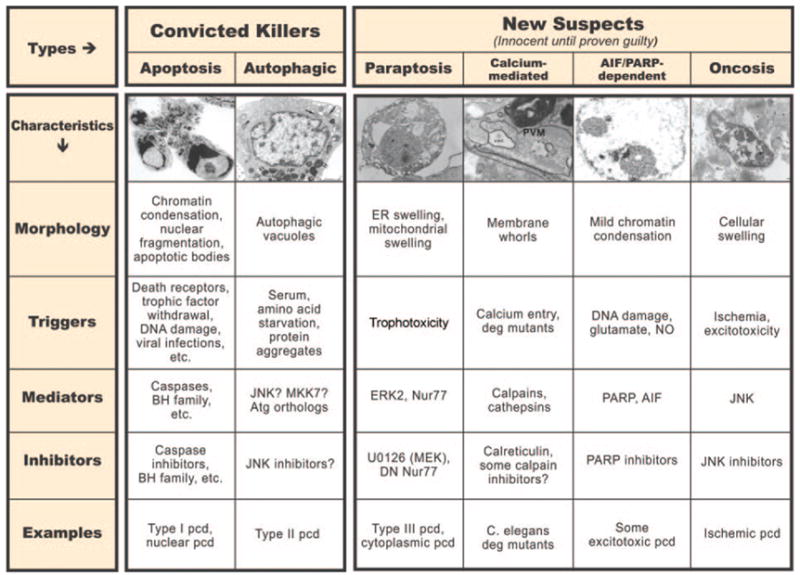
Comparison of different cell death programs. Note the difference in morphology present in each form, as well as the differences in biochemical mediators, inducers, and inhibitors. At the current time, only apoptosis and autophagic pcd are generally accepted as being legitimate forms of programmed cell death; however, ongoing research should reveal which of the additional candidates represent novel pathways of pcd. Photomicrographs are from the following references, used with permission: apoptosis, autophagic cell death, paraptosis, calcium-mediated pcd, AIF/PARP-dependent pcd, and oncosis (from Bredesen et al, 109 with permission).
Cell death has been divided into 2 broad categories: pcd, in which the cell plays an active role; and passive (necrotic) cell death. It is important to note that a semantic issue has arisen with the demonstration that some forms of nonapoptotic cell death previously labeled necrotic, and thus assumed to be passive, have turned out to be programmatic; therefore, some have referred to these as “necrosis-like”,17 whereas others prefer the term “programmed necrosis.”18,19 Based on the traditional view that some term should be reserved for passive (ie, nonprogrammatic) cell death, and that necrosis is the term historically applied to this form of cell death, the term “programmed necrosis” is an oxymoron. However, based on another feature of necrosis—breach of the plasma membrane with resulting initiation of an inflammatory response by leaked cellular contents—“programmed necrosis” is indeed an appropriate term. This phenomenon notwithstanding, reserving the term “necrosis” for nonprogrammatic pcd suggests that such programmatic cell deaths with necrotic morphology and other characteristics should be referred to as “necrosis-like.” As biochemical data accumulate for each form of pcd, it should become clear which paradigms induce necrosis-like pcd and which lead to passive, nonprogrammatic (necrotic) cell death.
Classic developmental studies revealed 3 different morphologies of cell death: type I (nuclear or apoptotic); type II (autophagic); and type III (cytoplasmic).5 These occur reproducibly within specific nuclei and at specific times of nervous system development. However, these physiological cell death pathways may also be activated by various insults, such as ischemia or anoxia.
Apoptosis
Apoptosis (Greek, “falling away”), also referred to as nuclear or type I pcd, is the best characterized type of pcd (Figure 2). Cells round up, form blebs, their chromatin condenses, nuclei fragment, and cellular portions (“apoptotic bodies”) bud off. Phosphatidylserine, which is normally asymmetric in the plasma membrane (facing internally), is exposed externally during apoptosis, providing a signal for phagocytosis.20 These morphological and histochemical changes are largely the result of the activation of caspases, a set of cell-suicide cysteine proteases.21,22
Figure 2.
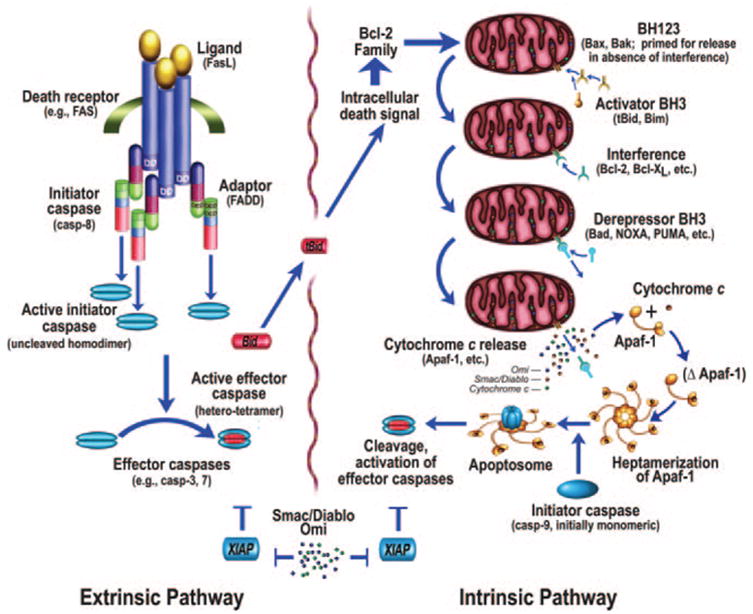
Intrinsic and extrinsic pathways of apoptosis (from Bredesen et al,109 with permission).
The activation of apoptosis is effected via 2 general pathways: the intrinsic pathway, originating from mitochondrial release of cytochrome c and associated activation of caspase-9; and the extrinsic pathway, originating from the activation of cell surface death receptors (eg, Fas), and resulting in the activation of caspase-8 or -10.23 A third general pathway (essentially a second intrinsic pathway) is activated by endoplasmic reticulum stress attributable to unfolded or misfolded proteins, such as may occur with ischemia.24-29 In addition, other organelles, such as the nucleus and Golgi apparatus, also display damage sensors that link to apoptotic pathways.30
Many different pathways, both physiological and pathological, converge on the intrinsic pathway of apoptosis. Whether by transcriptional regulation (eg, via p53) or post-transcriptional regulation, this results in a shift in the balance of a set of related proteins of the Bcl-2 family. These are major determinants of the apostat—the propensity of the cell to undergo apoptosis. Triggers of the intrinsic pathway shift the apostat toward the pro-apoptotic members, which are of 3 types: (i) the multidomain members (BH1-3: proteins that display Bcl-2 homology domains 1 to 3) such as Bax and Bak are capable of permeabilizing mitochondrial outer membranes31,32; (ii) the BH3 only activators such as Bim and tBid (a product of Bid resulting from cleavage by caspases, calpains, or other proteases, and providing communication between the extrinsic and intrinsic systems33) activate Bax and Bak, and may participate in their pore formation; and (iii) the BH3 only derepressors such as Puma, Noxa, and Bad sequester the anti-apoptotic block thrown up by Bcl-2, Bcl-xL, and related anti-apoptotic proteins that display BH1-4 domains, freeing the BH1-3 and activators to permeabilize the mitochondrial outer membrane, thus allowing the release of multiple intermembrane proteins, including cytochrome c, Smac/DIABLO, Omi/HtrA2, AIF, and endonuclease G. An anti-apoptotic modulator peptide, humanin, may also affect this balance, by binding and inhibiting Bax, Bid, and BimEL.34-36 Conversely, the Bax effect may be enhanced by non-Bcl-2 family member modulators such as p53—thus, p53 exhibits both transcriptional and nontranscriptional pro-apoptotic effects, and p53 inhibitors such as pifithrin alpha show promise as potential therapeutic agents for ischemic insults.37,38 Another pro-apoptotic mechanism is exhibited by Nur77/TR3, which binds Bcl-2, exposing its pro-apoptotic BH3 domain, resulting in a pro-apoptotic effect.39
As noted above, the anti-apoptotic members of the Bcl-2 family proteins, such as Bcl-2 and Bcl-xL, display BH1-4 domains. These anti-apoptotic proteins interact with the pro-apoptotic members and prevent the mitochondrial outer membrane permeability (MOMP) that would otherwise be created by the pro-apoptotic members; thus the anti-apoptotic members prevent the mitochondrial release of cytochrome c and other pro-apoptotic mitochondrial proteins.40
The net effect of the interplay of these Bcl-2 family and functionally related proteins is to set, along with other critical determinants, the apostat. Another form of apostat modulation is the inhibition of caspases by iap (inhibitor of apoptosis) proteins such as Xiap (see below), and reversal of this inhibition by Smac/DIABLO and Omi/HtrA2. A candidate that has received increasing attention recently is the fission and fusion of mitochondria.41-43 This may be of special relevance to pcd in neurodegeneration, since mutations in the mitochondrial fusion mediator Opa1 are associated with optic atrophy.44 Fission is mediated by Drp1 and Fis1, and inhibition of these proteins blocks apoptosis induction by staurosporine.43 Pro-apoptotic proteins may be recruited to mitochondria by Drp1, and mitochondrial remodeling may enhance the release of cytochrome c and other mitochondrial proteins. Ongoing work should disclose the roles of mitochondrial fission and fusion in pcd.
After release from the mitochondria, cytochrome c interacts with a cytosolic protein, Apaf-1, via the WD-40 repeats of Apaf-1, resulting in the exposure of a (d)ATP-binding site on Apaf-1, which, when occupied, leads to a conformational change resulting in heptamerization of Apaf-1. The resulting exposure of the Apaf-1 CARD (caspase activation and recruitment domain) recruits caspase-9 into this apoptosomal complex, and the resulting induced proximity of caspase-9 molecules leads to their activation.45 Activation of the apical caspase-9 leads to a cascade of caspase activation, including the effector caspases such as caspase-3 and caspase-7. However, the active caspases-3, -7, and -9 may be inhibited by the iap (inhibitor of apoptosis) proteins, such as Xiap,46 which may function as both direct inhibitors of caspase activity (in the case of caspase-9, by inhibiting dimerization) and as ubiquitin E3 ligases that mediate caspase degradation.47 Xiap and other iap proteins (eg, ciap-1 and ciap-2) display three BIR (Baculovirus iap repeat) domains, BIR-1, -2, and -3, which interact with caspases as noted above, but may also interact with other proteins.48 The iap-mediated caspase block may be released by additional mitochondrial proteins that, themselves, interact with iaps—Smac/DIABLO49,50 and Omi/HtrA251,52 (Figure 2).
In contrast to the intrinsic pathway, which features caspase-9 as its apical caspase, the extrinsic pathway features caspase-8 or caspase-10. In the best characterized example, Fas (probably trimerized before ligand binding53,54) is bound by trimeric Fas ligand, resulting in the recruitment of FADD (Fas-associated death domain protein) through its death domain, and then caspase-8 through FADD’s death effector domain (DED).55 The induced proximity of the apical caspase again leads to activation, as is the case for caspase-9, and subsequent activation of effector caspases such as caspase-3 and caspase-7. In addition, FLIP(L) (FLICE-like inhibitory protein, long form), which may function as an inhibitor of extrinsic pathway activation, may also act as a caspase-8 activator by being a higher affinity dimeric partner of caspase-8 than caspase-8 itself, resulting in activation by heterodimerization in preference to homodimerization.56
Both the intrinsic and extrinsic pathways of apoptosis thus converge on the activation of effector caspases. Caspases are cysteine aspartyl-specific proteases that cleave with high specificity at a small subset of aspartic acid residues in proteins. Their substrates (which total somewhere between a few hundred and one thousand) contribute to the apoptotic phenotype via proteolytic cascade activation, structural alterations, repair inactivation, internucleosomal DNA cleavage, phagocytic uptake signaling, mitochondrial permeabilization, and other effects. Caspases are synthesized as zymogens, but differ markedly in their activation. The apical caspases (caspase-8, -9, and -10) exist in the cytosol as monomers until dimerization is effected by adaptor molecules such as heptameric Apaf-1 (as part of the apoptosome) or trimeric FADD.
Contrary to earlier belief, cleavage of apical caspases has turned out to be neither required nor sufficient for activation.57 Their zymogenicity, which is the ratio of activity of the active form to that of the zymogen, is relatively low—from 10 (caspase-9) to 100 (caspase-8)57—and thus the (monomeric) zymogens themselves are relatively active. These caspases display large prodomains that are used in the protein-protein interactions that mediate activation—CARD (caspase activation and recruitment domain) in caspase-9, and DED (death effector domain) in caspase-8 and -10. The substrates of the apical caspases typically display I/L/V-E-X-D in the P4-P1 positions, with preference for small or aromatic residues in the P1′ position.57
Effector caspases such as caspase-3 and -7 are activated by the apical caspases. In contrast to the apical caspases, the effector caspases exist as cytosolic dimers, display high zymogenicity (>10 000 for caspase-3) and short prodomains, and are activated by cleavage rather than induced proximity. Cleavage produces a heterotetramer with two large subunits of 17 to 20 kilodaltons and two small subunits of 10 to 12 kilodaltons. Because of a difference in the S4 pocket structure of these caspases (in comparison to the apical caspases), their substrate preference is D-E-X-D, with a significant preference for Asp over Glu in the P4 position.57
Other caspases do not fit neatly within these two groups: caspase-2 has a long prodomain like an initiator caspase but has a substrate preference similar to effector caspases (with the exception that, unlike other caspases, it has a P5 preference [for small hydrophobic residues]); caspase-6 has a short prodomain like an effector caspase but has a substrate preference similar to apical caspases; the inflammatory caspases (-1, -4, -5) are involved in the processing of interleukin-1β and interleukin-18. The inflammatory caspases are thought not to play a role in pcd; however, inhibition in some paradigms such as cerebral ischemia has been associated with a reduction in infarct size.58
Caspase-12 is anomalous: in mice, it appears to play a role in apoptosis induced by endoplasmic reticulum (ER) stress.25,27,59 However, murine caspase-12 lacks Arg-341 (having a Lys instead), which in other caspases underlies the Asp specificity in the P1 position.57 Nonetheless, caspase-12 has been claimed to exhibit proteolytic activity,25 catalytically inactive caspase-12 inhibits ER stress-induced apoptosis,27 caspase-uncleavable caspase-12 also inhibits ER stress-induced apoptosis, and mice null for caspase-12 are less susceptible to amyloid-β toxicity than wild-type mice.59
In contrast to the murine system, in the great majority of humans a nonsense mutation is present in the caspase-12 gene, preventing expression of an active caspase.60 People without such a mutation are at increased risk for sepsis, attributable to the attenuation of the immune response to endotoxins such as lipopolysaccharide.61
Apoptosis Induced by Unfolded, Misfolded, or Alternatively Folded Proteins
As noted above, ischemia may induce ER stress; furthermore, ischemic preconditioning induces GRP78/Bip (glucose-regulated protein 78), which inhibits apoptosis induced by ER stress.62 In addition to the intrinsic and extrinsic pathways of apoptosis, ER stress may induce caspase activation. Mis-folded proteins and other activators of endoplasmic reticulum stress trigger an alternative intrinsic pathway that leads to caspase-9 activation, apparently via caspase-12 (at least in the murine system), and includes both Apaf-1-independent and Apaf-1-dependent activation of pcd.26,27 Misfolded proteins elicit cellular stress responses, including an ER stress response that protects cells against the toxic accumulation of misfolded proteins. This ER stress response is activated by the exposure of hydrophobic protein regions that bind GRP78/Bip, preventing its inhibition of UPR-activating proteins PERK, ATF6, and Ire1.63-66 Accumulation of these proteins in excessive amounts ultimately overwhelms the cellular systems that induce folding, translational, degradative, and aggresomal protection, finally triggering cellular suicide pathways.67-72
Because the degradation of cellular proteins is coupled, via the proteasomal degradation pathway, to ER dislocation of proteins,69,70,72 any condition that blocks the ER retrotranslocation of proteins or proteasome function may lead to the accumulation of misfolded protein substrates within the ER. Thus misfolded proteins, whether within or outside the ER, may trigger the ER stress response.
A number of mediators of pcd induced by misfolded or unfolded proteins have recently been identified. As for the classic intrinsic pathway, Bcl-2 family proteins play a critical role in the cellular suicide decision process,73 with communication between the ER and the mitochondria. Bax/Bak double knock-out cells fail to activate caspases after ER stress, arguing that these are required mediators.74 Bik may function to activate Bax and Bak in this pathway,75 whereas BI-1 inhibits Bax activation and translocation to the ER.76 Other Bcl-2 family proteins are also involved: for example, the BH3 protein Puma interacts with an hsp90 (heat shock protein of 90 kilodaltons)-independent fraction of p23, which, when cleaved by caspases, releases Puma (a BH3-only derepressor protein), leading to Bax interaction, oligomerization, and pcd.77 Noxa and p53 have also been implicated in this pathway.78
Part of the resulting apoptotic pathway is Apaf-1-dependent; however, part is also Apaf-1-independent yet caspase-9-dependent.26,78 Caspase-7 is recruited to the ER by an unknown mechanism, where it interacts with caspase-12. Caspase-12 is cleaved and released, leading to interaction with caspase-9.27 GRP78/Bip interacts with caspase-7 (requiring the ATP-binding domain79) and -12, preventing activation, but this inhibition is reversed by (d)ATP.28 Although the upstream activation of this pathway is not certain, one candidate is the triggering of JNK activation by IRE1, via TRAF2 and ASK1.80
A caspase-8-dependent pathway also mediates ER stress-induced apoptosis: Bap31, an ER membrane protein, binds Bcl-2 (or Bcl-xL) and a caspase-8-containing pro-apoptotic complex.81 When Bap31 is cleaved, a pro-apoptotic p20 fragment is derived, which, among other effects, induces mitochondrial fission, enhancing cytochrome c release.82 Conversely, BAR (bifunctional apoptosis regulator), which is expressed primarily in neurons of the CNS, also bridges Bcl-2 and caspase-8, but functions as an anti-apoptotic protein.83,84
Autophagic Programmed Cell Death
Autophagy (Greek, “self eating”)—which includes macroautophagy, microautophagy, and chaperone-mediated autophagy—is a regulated lysosomal pathway of degradation that complements the proteasomal pathway in that long-lived proteins, protein aggregates, and organelles are degraded. Targets for degradation, such as damaged mitochondria or aggregates of misfolded proteins, are encircled by an autophagosome, which then fuses with a lysosome, resulting in the degradation of the contents of the autophagosome. The molecular details of this process have been characterized best in yeast, in which a number of Atg (autophagy) genes have been identified, most with orthologs in higher eukaryotes.
Because the degradation of molecules and organelles by autophagy results in energy and amino acids for protein synthesis, it is a cellular protective pathway that, although active constitutively at low level, can be upregulated markedly by nutrient starvation. Nutrient withdrawal inactivates TOR (target of rapamycin), activating a complex of Atg proteins.85 Although the roles of the autophagic process in protein and organellar degradation, and in cellular protection during nutrient starvation, are well accepted, the role of autophagy in programmed cell death is more controversial.85-87 This is in part because the term “autophagic cell death” has been applied to two distinct observations: cell death associated with autophagy and cell death requiring autophagy. The vast majority of examples of autophagic cell death represent the former rather than the latter. However, increasing evidence suggests that the autophagic process is indeed required for at least some of what have been referred to as autophagic cell deaths. For example, mouse embryo fibroblasts (MEFs) that are null for both Bax and Bak, when treated with either of two frequently used apoptosis inducers—staurosporine and etoposide—undergo autophagic pcd dependent on autophagy genes Atg5 and beclin-1, and inhibited by the autophagy/class III PI3 kinase inhibitor, 3-methyladenine.86 This autophagic pcd may, however, be a slower process than apoptosis,88 or may somehow be triggered by an apoptosis block. Indeed, caspase inhibition by zVAD.fmk in L929 cells results in autophagy-dependent pcd,89 which has been proposed to be mediated by the selective degradation of catalase.90 On the one hand, this may serve as an admonishment that anti-apoptotic therapies carry the potential risk of inducing nonapoptotic pcd; on the other hand, it may argue that therapeutics directed at multiple cell death pathways will be required for optimal efficacy in diseases involving pcd.
Although much less is known about the mediators of autophagic pcd than about those of apoptotic pcd, JNK (c-Jun N-terminal kinase) has been implicated in a number of studies: for example, NMDA (N-methyl-D-aspartate) induced excitotoxic neuronal death in hippocampal slice cultures was shown to pursue an autophagic path, with selective phosphorylation of c-Jun in regions CA1, CA3, and the dentate gyrus.91 JNK inhibition prevented the neuronal death as well as the autophagy, suggesting that the requirement for JNK may lie upstream of the activation of autophagy. However, many questions about autophagic pcd remain unanswered: if autophagy is indeed a cellular protective program that—like the UPR—at some point activates pcd, what is the signal for the “switch” to pcd initiation? How important is the role of autophagic pcd in ischemic neuronal cell death? Does autophagic pcd occur in vivo in the absence of apoptosis inhibition? Are there “executioners” analogous to caspases in autophagic pcd?
Other Cell Death Programs
In comparison to apoptosis, relatively little is known about autophagic pcd, and even less is known about other nonapoptotic forms of pcd. Furthermore, most of what is known is based on morphological descriptions. Mechanistic requirements within type I include two general groups: caspase-dependent apoptosis (extrinsic and intrinsic, as noted above), and caspase-independent apoptosis. Types II (autophagic pcd) and III do not require caspase activation.
Type III pcd, which was subdivided by Clarke5 into type A and B, is a “necrosis-like” form of pcd that includes swelling of ER and mitochondria, and an absence of typical apoptotic features such as apoptotic bodies and nuclear fragmentation. Recently, it was reported that the hyperactivation of the tyrosine kinase receptor insulin-like growth factor I receptor (IGFR) induces a nonapoptotic form of cell death dubbed paraptosis.15 This form of pcd was found to require transcription and translation, to be indistinguishable morphologically from type III pcd, with swelling of the endoplasmic reticulum and mitochondria and an absence of apoptotic features. Admittedly, this morphology is observed in many cell deaths labeled necrotic, so the mechanistic implications of this particular morphological pattern remain an open question. Caspases are not activated during paraptosis, and therefore it is not surprising that neither Bcl-2 nor caspase inhibitors block this form of pcd. However, inhibitors of ERK2 (but not ERK1) were found to inhibit paraptosis,92 as was AIP-1/Alix. In addition, antisense oligonucleotides directed against JNK1 were partially inhibitory.
Induction of pcd by hyperactivation of a trophic factor receptor—essentially “trophotoxicity”—as observed in this paraptosis paradigm, is compatible with earlier observations that some trophic factors may increase neuronal cell death, for example induced by excitotoxicity.93 Such an effect might conceivably be protective against neoplasia, in that it may eliminate cells that would otherwise undergo autocrine loop-stimulated oncogenesis. The resulting program would understandably be nonapoptotic, because trophic factor signaling inactivates apoptotic signaling.
Other forms of pcd have been described that do not fit the criteria for type I, II, or III pcd. A nonapoptotic, caspase-independent form of cell death that does not resemble type II or type III developmental pcd has been described by Driscoll et al94,95 in Caenorhabditis elegans that express mutant channel proteins such as mec-4(d). A uniform, necrosis-like cell death is induced, characterized morphologically by membranous whorls lacking in other cell death types, triggered by calcium entry, mediated by specific calpains and cathepsins, and inhibited by calreticulin. Although it is possible that this alternative form of pcd will ultimately turn out to proceed via one of the previously described pathways (eg, given the cathepsin requirement, lysosomes may be involved, suggesting a relationship to autophagic pcd, perhaps as a final common pathway), the morphological characteristics suggest that it is indeed a distinct form of pcd.
A fifth apparent form of pcd has been described by the Dawsons and their colleagues, who demonstrated that a nonapoptotic form of cell death ensues after the activation of poly-(ADP-ribose) polymerase (PARP) and the consequent translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus.96 AIF is a flavoprotein that is involved with DNA fragmentation, along with endonuclease G and DNA fragmentation factor. This form of pcd was found to be activated by agents that induce DNA damage, such as hydrogen peroxide, N-methyl-D-aspartate, and N-methyl-N′-nitro-N-nitrosoguanidine. PARP-dependent pcd displays a morphology and biochemistry (to the extent that it is currently known) that is distinct from types I, II, and III pcd.
A form of cell death activated by ischemia has been dubbed oncosis. An extensive literature on the morphological criteria for oncosis exists, but the biochemical pathway(s) of oncosis have not yet been described. Oncosis is thought to be mediated by the failure of plasma membrane ionic pumps. One potential mediator of oncosis is a calpain-family protease (possibly a mitochondrial calpain97), which suggests that oncosis may turn out to be related to, or synonymous with, the calcium-activated necrosis-like cell death described by Driscoll et al.
“Aponecrosis” is a term applied to a combination of apoptosis and necrosis.98 Many cytotoxins induce pcd at low concentrations, but apparently necrotic cell death at higher concentrations, presumably attributable to overwhelming the cellular homeostatic processes before the completion of cell death programs. In fact, this a common pattern seen with cellular toxins, from hydrogen peroxide and other oxidants to mitochondrial toxins such as antimycin A.98 However, the necrotic morphology associated with aponecrosis has not been proven to be nonprogrammatic, so it is still unknown whether aponecrosis represents a combination of apoptosis and a nonapoptotic form of pcd as opposed to a combination of apoptosis and nonprogrammatic cell death.
What of the biochemical pathways common to these different forms of pcd? In the intrinsic pathway of apoptosis, holocytochrome c and other pcd mediators are released from the intermembrane space of mitochondria secondary to outer membrane permeability that is induced by pro-apoptotic members of the Bcl-2 family such as Bax and Bak, in concert with BH3 proteins Bim or tBid. However, mitochondrial proteins may also be released in association with the mitochondrial membrane permeability transition (MPT).99 Whether by consequent swelling and rupture of the mitochondrial outer membrane or by another mechanism, activation of the MPT by calcium, oxidants, or other activators offers a Bcl-2-independent (or at least partially independent: Bax may interact with components of the MPT such as the adenine nucleotide translocator100 and the voltage-dependent anion channel101), at least partially cyclophilin-D-dependent, route for the release of mitochondrially-derived pro-apoptotic factors.
Beyond these two general categories of mitochondrial pro-apoptotic factor release, novel scenarios are beginning to be defined: for example, recent work from Polster et al showed that the release of AIF (apoptosis-inducing factor102) from mitochondria requires a combination of mitochondrial membrane permeabilization (eg, by tBid or by the MPT) and active calpain. This work implicated an endogenous mitochondrial calpain in AIF release.103
These results suggest that combinatorial paths to pcd may be dissected: for example, Bcl-2 inhibitable (ie, BH1-3 mediated) versus independent (presumably related to MPT); caspase-dependent versus -independent; calpain-dependent versus -independent; AIF-dependent versus -independent; PARP-dependent versus -independent; and so on for other critical factors such as cathepsins JNK, and the autophagy-mediating gene products (Atg6, etc). Using such a dissection, classic apoptosis would fall predominantly into three groups: caspase-dependent and Bcl-2 inhibitable (intrinsic pathway, and extrinsic pathway with amplification via the intrinsic pathway), caspase-dependent and Bcl-2 resistant (some extrinsic pathway paradigms without amplification, and some MPT activators that lead to caspase-dependent pcd), and caspase-independent, Bcl-2 inhibitable (eg, some paradigms of endoplasmic reticulum stress104 and intracellular pathogen-induced pcd105).
In contrast, toxins that inactivate caspases directly or indirectly, such as diethylmaleate and buthionine sulfoximine, would induce pcd that is Bcl-2 inhibitable and caspase-independent.106 On the other hand, an increase in cytosolic calcium, such as occurs with the mec-4(d) mutants of C elegans95 could induce MPT—which would explain the Bcl-2 (ced-9) independence—and activate calpains, which would potentially inactivate caspases,107 compatible with the caspase independence of this form of pcd. As noted above, the cathepsin dependence suggests lysosomal involvement, and thus a potential relationship with type II pcd. Adding DNA damage to the calcium entry, such as occurs with excitotoxic damage, should trigger a similar scenario with the addition of PARP activation, with the combination of calcium-activated MPT and calpain activation explaining the AIF activation.103,108
All of these alternative pathways share the common feature of caspase inhibition, whether direct (eg, by zVAD.fmk or diethylmaleate) or indirect (eg, via receptor tyrosine kinase or calpain activation). For the prevention of cell death in ischemia, it is likely to be important to identify the specific molecular linchpins of each available pathway of pcd, because it is likely that blocking a single path will simply shunt the cell to an alternative route to pcd.
Acknowledgments
I thank Molly Susag, Loretta Sheridan, and Rowena Abulencia for manuscript preparation.
Footnotes
Disclosures
None.
Sources of Funding
This work was supported in part by National Institutes of Health grants AG12282, NS45093, and NS33376, a grant from the Joseph Drown Foundation, and a grant from American Bioscience, Inc.
References
- 1.Studnicka FK. Die parietalorgane. In: Oppel A, editor. Lehrbuch der vergleichende mikroskopischen anatomie der wirbeltiere. Jena: S.G. Fischer Verlag; 1905. [Google Scholar]
- 2.Levi-Montalcini R. The nerve growth factor: its mode of action on sensory and sympathetic nerve cells. Harvey Lect. 1966;60:217–259. [PubMed] [Google Scholar]
- 3.Lockshin RA, Williams CM. Programmed cell death. II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol. 1964;10:643–649. [Google Scholar]
- 4.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 6.Cunningham TJ. Naturally occurring neuron death and its regulation by developing neural pathways. Int Rev Cytol. 1982;74:163–186. doi: 10.1016/s0074-7696(08)61172-9. [DOI] [PubMed] [Google Scholar]
- 7.Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- 8.Majno G, Joris I. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [see comments] [PMC free article] [PubMed] [Google Scholar]
- 9.Oppenheim RW. Naturally occurring cell death during neural development. Trends Neurosci. 1985;17:487–493. [Google Scholar]
- 10.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 11.Pilar G, Landmesser L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J Cell Biol. 1976;68:339–356. doi: 10.1083/jcb.68.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz LM. The role of cell death genes during development. Bioessays. 1991;13:389–395. doi: 10.1002/bies.950130805. [DOI] [PubMed] [Google Scholar]
- 13.Schweichel JU. electron microscopic studies on the degradation of the apical ridge during the development of limbs in rat embryos. Z Anat Entwicklungsgesch. 1972;136:192–203. [PubMed] [Google Scholar]
- 14.Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 15.Sperandio S, de Belle I, Bredesen DE. An alternative, non-apoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2000;97:8093–8097. doi: 10.1073/pnas.110078997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. Bnip3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [in process citation] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niquet J, Liu H, Wasterlain CG. Programmed neuronal necrosis and status epilepticus. Epilepsia. 2005;46 (Suppl 5):43–48. doi: 10.1111/j.1528-1167.2005.01025.x. [DOI] [PubMed] [Google Scholar]
- 19.Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev. 2006;20:1–15. doi: 10.1101/gad.1376506. [DOI] [PubMed] [Google Scholar]
- 20.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 21.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 22.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 23.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 24.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving chop. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 25.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis: cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 26.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, Del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program: an apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 27.Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 28.Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone grp78. FEBS Lett. 2002;514:122–128. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yuan J, Yankner BA. Caspase activity sows the seeds of neuronal death. Nat Cell Biol. 1999;1:E44–E45. doi: 10.1038/10037. [DOI] [PubMed] [Google Scholar]
- 30.Green DR, Kroemer G. Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest. 2005;115:2610–2617. doi: 10.1172/JCI26321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. Bh3 domains of Bh3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 33.Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, Ellerby LM, Bredesen D, Freeze H, Abrahamson M, Bromme D, Krajewski S, Reed JC, Yin XM, Turk V, Salvesen GS. Lysosomal protease pathways to apoptosis: cleavage of Bid, not pro- caspases, is the most likely route. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 34.Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423:456–461. doi: 10.1038/nature01627. [DOI] [PubMed] [Google Scholar]
- 35.Zhai D, Luciano F, Zhu X, Guo B, Satterthwait AC, Reed JC. Humanin binds and nullifies Bid activity by blocking its activation of Bax and Bak. J Biol Chem. 2005;280:15815–15824. doi: 10.1074/jbc.M411902200. [DOI] [PubMed] [Google Scholar]
- 36.Luciano F, Zhai D, Zhu X, Bailly-Maitre B, Ricci JE, Satterthwait AC, Reed JC. Cytoprotective peptide humanin binds and inhibits proapoptotic Bcl-2/Bax family protein bimel. J Biol Chem. 2005;280:15825–15835. doi: 10.1074/jbc.M413062200. [DOI] [PubMed] [Google Scholar]
- 37.Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- 38.Leker RR, Aharonowiz M, Greig NH, Ovadia H. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin alpha. Exp Neurol. 2004;187:478–486. doi: 10.1016/j.expneurol.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B, Chen G, Lu J, Lin F, Xie Z, Fontana JA, Reed JC, Zhang X. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor tr3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- 40.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [see comments] [DOI] [PubMed] [Google Scholar]
- 41.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 42.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum Bik initiates Drp1-regulated remodelling of mitochondrial cristae during apoptosis. Embo J. 2005;24:1546–1556. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. Opa1, encoding a dynamin-related GTpase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. doi: 10.1038/79944. [DOI] [PubMed] [Google Scholar]
- 45.Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 46.Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked iap is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–304. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 47.Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S. Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol. 2002;4:439–444. doi: 10.1038/ncb798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurakin A, Bredesen DE. An unconventional IAP-binding motif revealed by target-assisted iterative screening (TAIS) of the Bir3-ciap1 domain. J Mol Recognit. 2006;20:39–50. doi: 10.1002/jmr.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 50.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of diablo, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 51.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/Htra2 regulates apoptosis by binding Xiap through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, Htra2, is released from the mitochondria and interacts with Xiap, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 53.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 54.Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- 55.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME, Dixit VM. Flice, a novel Fadd-homologous ice/ced-3-like protease, is recruited to the cd95 (fas/apo-1) death-inducing signaling complex. Cell. 1996;85:817–827. doi: 10.1016/s0092-8674(00)81266-0. [DOI] [PubMed] [Google Scholar]
- 56.Boatright KM, Deis C, Denault JB, Sutherlin DP, Salvesen GS. Activation of caspases-8 and -10 by flip(l) Biochem J. 2004;382:651–657. doi: 10.1042/BJ20040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 60.Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun. 2002;293:722–726. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- 61.Saleh M, Vaillancourt JP, Graham RK, Huyck M, Srinivasula SM, Alnemri ES, Steinberg MH, Nolan V, Baldwin CT, Hotchkiss RS, Buchman TG, Zehnbauer BA, Hayden MR, Farrer LA, Roy S, Nicholson DW. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature. 2004;429:75–79. doi: 10.1038/nature02451. [DOI] [PubMed] [Google Scholar]
- 62.Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Chan PH. Induction of grp78 by ischemic preconditioning reduces endoplasmic reticulum stress and prevents delayed neuronal cell death. J Cereb Blood Flow Metab. 2003;23:949–961. doi: 10.1097/01.WCB.0000077641.41248.EA. [DOI] [PubMed] [Google Scholar]
- 63.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 64.Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000;10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 65.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 66.Dobson CM, Ellis RJ. Protein folding and misfolding inside and outside the cell. Embo J. 1998;17:5251–5254. doi: 10.1093/emboj/17.18.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Selkoe DJ. Folding proteins in fatal ways. Nature. 2003;426:900–904. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 68.Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science. 2002;296:1991–1995. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- 69.Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 70.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 71.Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 72.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 73.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. Bax and Bak regulation of endoplasmic reticulum CA2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 74.Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ. Proapoptotic Bax and Bak control multiple initiator caspases. EMBO Rep. 2005;6:379–385. doi: 10.1038/sj.embor.7400375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mathai JP, Germain M, Shore GC. Bh3-only Bik regulates Bax, Bak-dependent release of CA2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–23836. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- 76.Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, Kitada S, Monosov E, Thomas M, Kress CL, Babendure JR, Tsien RY, Lipton SA, Reed JC. Bi-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell. 2004;15:355–366. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 77.Rao RV, Niazi K, Mollahan P, Mao X, Crippen D, Poksay KS, Chen S, Bredesen DE. Coupling endoplasmic reticulum stress to the cell-death program: a novel hsp90-independent role for the small chaperone protein p23. Cell Death Differ. 2006;13:415–425. doi: 10.1038/sj.cdd.4401761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (puma) and noxa by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 79.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. Endoplasmic reticulum chaperone protein grp78 protects cells from apoptosis induced by topoisomerase inhibitors: role of atp binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–20924. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 80.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of Jnk protein kinases by transmembrane protein kinase ire1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 81.Ng FW, Nguyen M, Kwan T, Branton PE, Nicholson DW, Cromlish JA, Shore GC. P28 bap31, a bcl-2/bcl-xl- and procaspase-8-associated protein in the endoplasmic reticulum. J Cell Biol. 1997;139:327–338. doi: 10.1083/jcb.139.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC. Caspase cleavage product of bap31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol. 2003;160:1115–1127. doi: 10.1083/jcb.200212059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roth W, Kermer P, Krajewska M, Welsh K, Davis S, Krajewski S, Reed JC. Bifunctional apoptosis inhibitor (bar) protects neurons from diverse cell death pathways. Cell Death Differ. 2003;10:1178–1187. doi: 10.1038/sj.cdd.4401287. [DOI] [PubMed] [Google Scholar]
- 84.Zhang H, Xu Q, Krajewski S, Krajewska M, Xie Z, Fuess S, Kitada S, Pawlowski K, Godzik A, Reed JC. Bar: an apoptosis regulator at the intersection of caspases and bcl-2 family proteins. Proc Natl Acad Sci U S A. 2000;97:2597–2602. doi: 10.1073/pnas.97.6.2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 87.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 89.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an atg7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 90.Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Borsello T, Croquelois K, Hornung JP, Clarke PG. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci. 2003;18:473–485. doi: 10.1046/j.1460-9568.2003.02757.x. [DOI] [PubMed] [Google Scholar]
- 92.Sperandio S, Poksay K, de Belle I, Lafuente MJ, Liu B, Nasir J, Bredesen DE. Paraptosis: mediation by MAP kinases and inhibition by aip-1/alix. Cell Death Differ. 2004;11:1066–1075. doi: 10.1038/sj.cdd.4401465. [DOI] [PubMed] [Google Scholar]
- 93.Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 94.Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- 95.Bianchi L, Gerstbrein B, Frokjaer-Jensen C, Royal DC, Mukherjee G, Royal MA, Xue J, Schafer WR, Driscoll M. The neurotoxic mec-4(d) deg/enac sodium channel conducts calcium: implications for necrosis initiation. Nat Neurosci. 2004;7:1337–1344. doi: 10.1038/nn1347. [DOI] [PubMed] [Google Scholar]
- 96.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(adp-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 97.Liu X, Van Vleet T, Schnellmann RG. The role of calpain in oncotic cell death. Annu Rev Pharmacol Toxicol. 2004;44:349–370. doi: 10.1146/annurev.pharmtox.44.101802.121804. [DOI] [PubMed] [Google Scholar]
- 98.Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE, Capaccioli S, Orlandini SZ. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182:41–49. doi: 10.1002/(SICI)1097-4652(200001)182:1<41::AID-JCP5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 99.Novgorodov SA, Gudz TI. Permeability transition pore of the inner mitochondrial membrane can operate in two open states with different selectivities. J Bioenerg Biomembr. 1996;28:139–146. doi: 10.1007/BF02110644. [DOI] [PubMed] [Google Scholar]
- 100.Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, Leber B, Andrews D, Duclohier H, Reed JC, Kroemer G. Bcl-2 and bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- 101.Adachi M, Higuchi H, Miura S, Azuma T, Inokuchi S, Saito H, Kato S, Ishii H. Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2004;287:G695–G705. doi: 10.1152/ajpgi.00415.2003. [DOI] [PubMed] [Google Scholar]
- 102.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 103.Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 104.Egger L, Schneider J, Rheme C, Tapernoux M, Hacki J, Borner C. Serine proteases mediate apoptosis-like cell death and phagocytosis under caspase-inhibiting conditions. Cell Death Differ. 2003;10:1188–1203. doi: 10.1038/sj.cdd.4401288. [DOI] [PubMed] [Google Scholar]
- 105.Perfettini JL, Reed JC, Israel N, Martinou JC, Dautry-Varsat A, Ojcius DM. Role of Bcl-2 family members in caspase-independent apoptosis during chlamydia infection. Infect Immun. 2002;70:55–61. doi: 10.1128/IAI.70.1.55-61.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kane DJ, Ord T, Anton R, Bredesen DE. Expression of bcl-2 inhibits necrotic neural cell death. J Neurosci Res. 1995;40:269–275. doi: 10.1002/jnr.490400216. [DOI] [PubMed] [Google Scholar]
- 107.Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 108.Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- 109.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kerr JFR, Harmon BV. Definition and incidence of apoptosis: an historical perspective. In: Tomei LD, Cope FO, editors. Apoptosis: The molecular basis of cell death. Plainview, New York: Cold Spring Harbor Laboratory Press; 1991. p. 321. [Google Scholar]
- 111.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113(Pt 7):1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 112.Hall IH, Elkins AL, Karthikeyan S, Spielvogel BF. The cytotoxicity of 1-(phenylmethyl)-4,7,10-tris-[(4′ methylphenyl) sulfonyl]-1,4,7,10-tetraazacyclododecane in human tmolt3 t leukemic cells. Anticancer Res. 1997;17:1195–1198. [PubMed] [Google Scholar]
- 113.Susin SA, Daugas E, Ravagnan L, Samejima K, Zamzami N, Loeffler M, Costantini P, Ferri KF, Irinopoulou T, Prevost MC, Brothers G, Mak TW, Penninger J, Earnshaw WC, Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ohno M, Takemura G, Ohno A, Misao J, Hayakawa Y, Minatoguchi S, Fujiwara T, Fujiwara H. “Apoptotic” Myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with in situ nick end-labeling. Circulation. 1998;98:1422–1430. doi: 10.1161/01.cir.98.14.1422. [DOI] [PubMed] [Google Scholar]