

Abstract
To explore the role of glucocorticoids in regulation of kinase pathways during innate immune responses, we generated mice with conditional deletion of glucocorticoid receptor (GR) in macrophages (MGRKO). Activation of toll-like receptor 4 (TLR4) by lipopolysaccharide (LPS) caused greater mortality and cytokine production in MGRKO mice than in controls. Ex vivo, treatment with dexamethasone (Dex) markedly inhibited LPS-mediated induction of inflammatory genes in control but not GR-deficient macrophages. We show that Dex inhibits p38 MAPK, but not PI3K/Akt, ERK, or JNK, in control macrophages. Associated with p38 inhibition, Dex induced MAP kinase phosphatase-1 (MKP-1) in control, but not MGRKO, macrophages. Consistent with the ex vivo studies, treatment with a p38 MAPK–specific inhibitor resulted in rescue of MGRKO mice from LPS-induced lethality. Taken together, we identify p38 MAPK and its downstream targets as essential for GR-mediated immunosuppression in macrophages.
Introduction
Pathogen invasion, mechanical trauma, and chemical irritation activate the host innate immune system to maintain physiologic homeostasis.1–3 This inflammatory response is characterized by a temporally restricted cascade of cytokines and other signaling molecules that limit ongoing infection or compartmentalize damaged tissue.2 Excessive activation or failure to efficiently terminate these innate responses, however, contributes to common acute human disease states such as septic shock and chronic disorders such as arthritis, asthma, inflammatory bowel disease, osteolysis in periodontal disease and otitis media, and atherosclerosis.4–6 Members of the nuclear-receptor superfamily of transcription factors have emerged as important modulators of innate immunity through their effects on macrophage gene expression.7–9
Amongst nuclear-receptor superfamily members, the glucocorticoid receptor (GR),10–12 peroxisome-proliferator–activated receptors (PPARs),8,13 and liver X receptors (LXRs)14,15 have been most robustly associated with regulation of macrophage function. Recent evidence suggests combinatorial interaction of these receptors to provide integration of responses.6 The relative contribution of or requirement for each of these factors is likely to depend upon the nature of the macrophage-activating stimulus. However, for no single mechanism of macrophage activation have the key molecular interactions of these nuclear receptors with the complex network of proinflammatory intracellular signaling pathways been completely elucidated.
Toll-like receptors (TLRs) on macrophages recognize pathogen-associated molecular patterns (PAMPs) to generate immunologically relevant responses.16–19 Recently, it has been shown that glucocorticoids acting through GR efficiently suppress cytokine induction downstream of toll-like receptor 4 (TLR4) and MyD88 activation by lipopolysaccharide (LPS).6 Normally, engagement of TLR4 triggers a cascade of signaling events leading to induction of mitogen-activated protein kinases (MAPKs; eg, ERK, JNK, and p38, phosphatidylinositol-3-hydroxykinase [PI3K]/Akt, activator protein-1 [AP-1], and NFκB), resulting in enhanced transcription and stabilization of the mRNAs for several proinflammatory cytokines including IL-6, IL-1β, TNF-α, and IL-12.20–23 Particular attention has been given to glucocorticoids acting through GR to impair NFκB-mediated cytokine induction, with recent evidence implicating an important role for GR mitigation of IRF-3–p65 interaction.6 GR has also been suggested to block activation of MAPK/Akt-mediated gene induction, but the relative importance of specific kinase pathways in GR-mediated macrophage suppression of cytokine production remains unclear.24–26 Further, ligand-dependent GR action can result in transactivation of transcription by binding to glucocorticoid response elements and recruitment of coactivators, transrepression of transcription by protein-protein interaction with other components of transcription complexes either directly or through recruitment of corepressors, or nongenomic effects with action outside the cell nucleus.10,27–30 Mice harboring a dimerization-deficient GR (GRdim) retain the capacity to efficiently repress cytokine responses, suggesting that DNA binding and classic transactivation of new gene expression are not essential for GR-mediated suppression of inflammation.12,31 However, the GRdim mutation engineered has been suggested to not completely abrogate GR transactivation.32 Global deletion of one novel target of GR transactivation, MKP-1, has recently shown this molecule to be important for survival after TLR4 expression by limiting p38 and JNK signaling.33,34 A recent report demonstrates that MKP-1–deficient macrophages also show resistance to dexamethasone-mediated cytokine suppression.35 Interestingly, GRdim mice normally up-regulate MKP-1 expression with glucocorticoid treatment.35
Here, we test the hypothesis that macrophage glucocorticoid receptors are essential to limit cytokine production and morbidity after TLR4 activation in vivo. Further, using in vivo and ex vivo analyses of macrophages from control and macrophage GR–deficient (MGRKO) mice, we find that glucocorticoids acting through the glucocorticoid receptor specifically impact upon p38 MAPK, but not ERK, JNK, or Akt, activation. This effect on p38 MAPK is associated with induction of MKP-1 in control but not MGRKO macrophages. Specific inhibition of p38 MAPK results in substantial down-regulation of cytokine production in LPS-stimulated macrophages, though not to the extent of dexamethasone in GR-containing cells. The importance of p38 MAPK induction was confirmed in vivo, as MGRKO mice are rescued from LPS-induced mortality by pharmacologic p38 MAPK inhibition.
Materials and methods
Animal handling
Mouse protocols were approved by the Animal Care and Use Committee of Washington University. Mice were housed on a 12-hour light and 12-hour dark cycle. Blood was collected by retro-orbital phlebotomy into heparinized capillary tubes, with the time from first handling the animal to completion of the bleeding not exceeding 30 seconds. Mice used for the experimentation were 6 to 10 weeks old and were of C57BL/6 × 129/Sv background.
Generation of MGRKO mice
Mice with conditional deletion of GR in macrophages (MGRKO) were generated using LysM promoter–driven, Cre recombinase–mediated excision of exons 1C and 2 of the GR gene.36,37 Southern-blot analysis was performed as previously described.36 After germ line transmission, we mated heterozygous mice harboring the floxed exon 1C and 2 allele to LysM-Cre transgenic mice. LysM-Cre–negative homozygous floxed GR littermates were used as controls. Experiments were done with sex-matched littermates as controls.
Isolation and culture of mouse peritoneal macrophages
Thioglycollate-elicited macrophages were isolated by peritoneal lavage 3 days following intraperitoneal injection of 2.0 mL of sterile 4% thioglycollate (Becton Dickinson, Sparks, MD). Cells were plated in complete medium containing DMEM (Cambrex BioSciences, Walkersville, MD) with 10% fetal bovine serum (Invitrogen, Grand Island, NY) and penicillin/streptomycin (Invitrogen). After 3 hours of attachment, medium was aspirated and the monolayer was rinsed twice with serum-free DMEM. Fresh complete medium was then added to each well, and the cells were cultured under a humidified atmosphere of 5% CO2 in air at 37°C.
Cytokine measurements
For in vivo cytokine studies, mice were injected intraperitoneally with PBS (Cambrex BioSciences) or LPS (Escherichia coli 0111:B4; Sigma, St Louis, MO) at a dose of 10 mg/kg body weight. Plasma samples were collected after the indicated periods of time and stored at −80°C until assay. Serum proinflammatory cytokine concentrations were measured by enzyme-linked immunosorbent assay (ELISA) for TNF-α and IL-6 according to the manufacturer's instructions (BD Biosciences PharMingen, San Diego, CA).
For in vivo experiments with the p38 MAPK inhibitor, MGRKO mice were injected intraperitoneally with vehicle or SB 203580 hydrochloride (559395; Calbiochem, EMD Biosciences, La Jolla, CA) at a concentration of 2 mg/kg body weight, and 1 hour later they were injected intraperitoneally with LPS at a concentration of 10 mg/kg body weight. Plasma samples were collected after 6 hours of LPS injection and accessed for TNF-α and IL-6 as described in the preceding paragraph.
For in vitro experiments, cells were plated at 106 cells/mL in a 24-well plate (Becton Dickinson Labware, Franklin Lakes, NJ) with 1.0 mL of complete medium. Cells were treated with dexamethasone (Dex; Amtech, St Joseph, MO) or corticosterone (Cort; Sigma) for 3 hours at a concentration of 100 nM (except as indicated) followed by LPS (100 ng/mL) for 18 hours or as indicated. Experiments with MKP-1 inhibitor triptolide (TP; Calbiochem, EMD Biosciences) were performed by treating cells at 0.5- to 2-μM concentrations 3 hours before LPS exposure, with cell harvest 30 minutes after LPS treatment. For the study with p38 MAPK inhibitors, cells were treated with 20 μM of SB 203580 (559389) or PD 169316 (513030; Calbiochem, EMD Biosciences) for 1 hour followed by LPS stimulation. Supernatants were collected and accessed for TNF-α and IL-6 as described earlier.
Survival curves
Control and MGRKO mice were injected intraperitoneally with LPS at a dose of 10 mg/kg body weight and observed for 5 days. To study the effect of p38 MAPK inhibitor, MGRKO mice were injected intraperitoneally with vehicle or SB 203580 hydrochloride at a concentration of 2 mg/kg body weight, and 1 hour later they were injected intraperitoneally with LPS at a dose of 10 mg/kg body weight and observed for 5 days. Experiments were done with age- and sex-matched littermate mice.
Western-blot analysis
For Western-blot analysis, whole-cell extracts were resolved through sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 4% to 12% separating gel (Invitrogen). Proteins were transferred to Hybond enhanced chemiluminescence (ECL) nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ) using a semidry transfer system (BIO-RAD, Hercules, CA) and blocked with 5% dried milk in PBS and 0.1% Tween-20 (Sigma). Blots were probed with anti–phospho-HSP27 (07-646; Upstate, Lake Placid, NY); anti-GR (sc-1004), anti-ERK2 (sc-154), anti–MKP-1 (sc-1199; Santa Cruz Biotechnology, Santa Cruz, CA); anti–COX-2 (160126; Cayman, Ann Arbor, MI); antiactin (A 5060; Sigma); anti–phospho-SAPK/JNK (9251), anti-SAPK/JNK (9252), anti–phospho-p38 MAPK (9211), anti–p38 MAPK (9212), anti–phospho-p44/42 MAPK (9101), anti–phospho-Akt (9271), anti-Akt (9272), and anti-HSP27 (2402) (Cell Signaling Technology, Beverly, MA) antibodies. Binding of horseradish peroxidase (HRP)–labeled goat antirabbit antibody (sc-2004) or goat antimouse antibody (sc-2005; Santa Cruz Biotechnology) was determined using SuperSignal West Chemiluminescent Substrate (Pierce, Rockford, IL). Blots were stripped with Restore Western Blot Stripping Buffer (Pierce) and reprobed with different antibodies.
Statistical analysis
Data are expressed as the mean ± SEM. Statistical significance was tested by unpaired 2-tailed Student t test. The differences were considered to be significant for P values less than .05.
Results
GR in macrophages is essential for survival after TLR4 activation
To define the role of GR in the regulation of innate immune responses, we developed mice with conditional disruption of GR in macrophages. Southern-blot analysis showed efficient recombination of the loxP-flanked GR allele in thioglycollate-elicited macrophages isolated from MGRKO mice and evidence for disruption in spleen but not kidney, liver, or thymus (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article; and data not shown). Western-blot analysis with an anti-GR antibody demonstrated no GR immunoreactivity in thioglycollate-elicited peritoneal macrophages isolated from MGRKO mice (Figure S1). MGRKO mice where grossly indistinguishable from control littermates. However, after TLR4 activation by LPS injection, 88% of the MGRKO mice died as opposed to only 13% of control mice (Figure 1A). Thus, GR action in macrophages is required to limit LPS-induced lethality.
Figure 1.

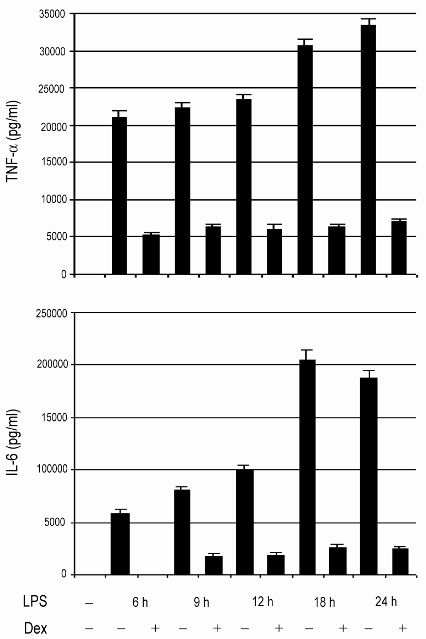
Deletion of GR in macrophages augments LPS-induced lethality and proinflammatory responses. (A) Kaplan-Meier plot of control (n = 8) and MGRKO (n = 8) mice treated with LPS (10 mg/kg). P < .01 for MGRKO versus control. (B) Plasma concentrations of TNF-α and IL-6 in control (n = 7-10) and MGRKO (n = 6-10) mice treated with LPS for the indicated periods of time. Error bars indicate SEM.
Deficiency of GR in macrophages alters systemic secretion of proinflammatory cytokines
To evaluate the involvement of GR in regulating inflammatory responses in vivo, we measured TNF-α and IL-6 after LPS treatment. We found no difference in cytokine secretion between genotypes 1 hour after LPS injection (Figure 1B). After 3 or 6 hours of LPS treatment, we found 2- to 2.5-fold increases in TNF-α and IL-6 secretion in MGRKO compared with the control mice (Figure 1B). Further, we measured a 5-fold increase in TNF-α in MGRKO mice in comparison to control mice 12 hours after LPS treatment (Figure 1B). A large and equivalent rise in serum Cort occurred in both genotypes at 3 and 24 hours after LPS administration (data not shown), implicating that the differential cytokine secretion between genotypes resulted from impaired glucocorticoid-mediated inhibition of cytokine production in the MGRKO mice.
Dex suppresses LPS-mediated inflammatory responses
To analyze direct actions of glucocorticoids on GR in macrophages, we exploited thioglycollate-elicited peritoneal macrophages. To examine the effect of GR deficiency on the secretion of inflammatory cytokines, macrophages from both genotypes were treated with LPS with or without Dex. Treatment with Dex (100 and 1000 nM) suppressed TNF-α and IL-6 by 80% and 90%, respectively, in control macrophages (Figure 2A; Figure S2 for time course). GR-deficient macrophages demonstrated no significant change in cytokine secretion with Dex (Figure 2A), consistent with their loss of GR immunoreactivity.
Figure 2.

Dex suppresses LPS-mediated inflammatory gene expression. (A) Effect of Dex on LPS-mediated induction of TNF-α and IL-6 in control (□) and MGRKO (■) macrophages. Data are shown as mean ± SEM; n = 5; *P < .01 compared with treatment group without Dex in control macrophages. (B) Effect of Dex on LPS-mediated induction of COX-2. Representative of 3 independent experiments.
Induction of COX-2 accounts for elevated levels of prostanoids in chronic inflammatory disorders and has been found to be glucocorticoid regulated. Indeed, augmented prostanoid production following TLR4 engagement results in many of the systematic manifestations associated with endotoxemia.38,39 Similar to cytokine production, pretreatment with Dex abrogated LPS-mediated induction of COX-2 in control macrophages (Figure 2B). Macrophages from MGRKO mice showed complete resistance to Dex (Figure 2B).
GR selectively impairs LPS-mediated activation of p38 MAPK
Members of the MAPK family play crucial roles in multiple aspects of inflammatory responses. Kinetic analysis revealed that LPS-mediated p38 MAPK phosphorylation follows a biphasic pattern. Induction of MAPK was found as early as 15 minutes after LPS stimulation and persisted for 30 minutes (Figure 3A). Phospho–p38 MAPK was undetectable between 1 hour and 3 hours after LPS treatment; however, the abundance of total p38 MAPK was comparable throughout the time course. We observed a second phase of phospho–p38 MAPK induction after 6 hours of LPS treatment, which persisted until the 12-hour time point. To study the effect of Dex on p38 MAPK phosphorylation, we chose the early (15 and 30 min) and late phase (6 h and 12 h) of kinase induction. We observed that pretreatment with Dex suppressed p38 MAPK phosphorylation in the control macrophages during both phases (Figure 3B). Densitometric analysis revealed that Dex-mediated inhibition of p38 MAPK phosphorylation after 15 and 30 minutes of LPS stimulation was 2.2- and 4.8-fold, respectively. At the late phase of induction, phosphorylation of p38 MAPK was completely inhibited in Dex-pretreated control macrophages (Figure 3B). No detectable change in p38 MAPK phosphorylation was observed in MGRKO macrophages by Dex pretreatment (Figure 3B). Similar experiments with Cort, the endogenous murine glucocorticoid, showed marked suppression of p38 MAPK phosphorylation in control macrophages and no detectable change in GR-deficient macrophages (Figure S3A). Temporal analysis showed a similar biphasic activation of JNK (Figure S3B). In contrast to p38 MAPK, phosphorylation of JNK was not altered with Dex treatment (Figure 3C). A similar study with ERK1/2 showed that Dex does not alter ERK phosphorylation (Figure 3D). Additionally, Dex had no effect on Akt phosphorylation in the control macrophages (Figure 3E).
Figure 3.

Dex selectively suppresses LPS-mediated activation of p38 MAPK. (A) Time-dependent effect of LPS (100 ng/mL) on p38 MAPK phosphorylation. Representative of 2 independent experiments. Effect of Dex (100 nM) on LPS-mediated phosphorylation of (B) p38 MAPK in control and MGRKO macrophages and (C) JNK (p46/p54), (D) ERK1/2, or (E) Akt in the control macrophages. Representatives of 3 to 4 independent experiments.
Effect of p38 MAPK inhibitors on proinflammatory gene expression
To address the importance of p38 MAPK in proinflammatory gene expression, we used the p38 MAPK–specific inhibitors SB 203580 or PD 169316. Treatment with these inhibitors significantly inhibited LPS-induced secretion of TNF-α and IL-6 in control and GR-deficient macrophages by approximately 60% and 50%, respectively (Figure 4A; Figure S4).
Figure 4.

p38 MAPK inhibitors impair proinflammatory gene expression and effect of Dex on MKP-1. (A) Effect of SB (20 μM) and PD (20 μM) on LPS-mediated induction of TNF-α and IL-6 in control macrophages; n = 3; **P < .01 and *P < .02 compared with treatment group with LPS. Error bars indicate SEM. Effect of SB (20 μM) on LPS-mediated (B) phosphorylation of HSP27 and (C) induction of COX-2 for the indicated period of time in control macrophages. Representatives of 2 to 3 independent experiments.
HSP27 is a downstream target of p38 MAPK. To examine the efficacy of p38 MAPK inhibitors, the phosphorylation of HSP27 at Ser82 was examined. Treatment with SB 203580 caused 90% reduction in stimulated phospho-HSP27 (Figure 4B), suggesting that the differential attenuation of cytokine production between Dex and the p38 inhibitors was not due to inadequate p38 inhibition. Furthermore, treatment with SB 203580 markedly inhibited LPS-mediated COX-2 induction in control and GR-deficient macrophages, consistent with previous studies that have found both transcriptional and posttranscriptional stimulatory actions of p38 activity for COX-2 induction.40–42 We observed 12-fold suppression of COX-2 6 hours after LPS treatment by SB 203580 (Figure 4C). A similar experiment with GR-deficient macrophages showed a comparable inhibition of COX-2 induction by SB 203580 pretreatment (Figure S5).
Dex induces MKP-1 in control macrophages
MKP-1, a phosphatase induced by ligand-activated GR, suppresses MAPK activation.24,25,43,44 Without Dex treatment, weak induction of MKP-1 was observed at 1 to 3 hours of LPS treatment and thereafter declined to the basal levels in both genotypes (Figure 5A). Treatment with Dex induced MKP-1 18-fold in control macrophages and resulted in prolonged expression (Figure 5A). Dex did not increase expression of MKP-1 in GR-deficient macrophages (Figure 5A).
Figure 5.

Dex induction of MKP-1 expression is the major GR target for suppression of p38 MAPK phosphorylation. (A) Effect of Dex on MKP-1 induction in control and MGRKO macrophages treated with LPS for the indicated periods of time. (B) Effect of triptolide on p38 MAPK phosphorylation in the presence and absence of Dex. Representative of 2 to 3 independent experiments.
To assess the contribution of GR-mediated induction of MKP-1 to attenuation of p38 MAPK phosphorylation, we used triptolide, a pharmacologic inhibitor, to block MKP-1 induction.45–47 Dex in the presence of triptolide suppressed LPS-induced p38 MAPK phosphorylation by only 10% compared with triptolide treatment alone, and p38 MAPK phosphorylation in the presence of Dex plus triptolide remained elevated above LPS-alone levels (Figure 5B). Without triptolide cotreatment, Dex again potently suppressed p38 phosphorylation (Figure 5B).
p38 MAPK inhibition rescues MGRKO mice from LPS-mediated lethality
To assess the relative importance of p38 MAPK regulation by GR in vivo, we treated MGRKO mice with LPS and SB 203580 or vehicle. We observed 88% mortality in the vehicle-treated MGRKO mice, whereas 89% of SB 203580–treated MGRKO mice survived (Figure 6A). In accord with this improved survival, SB 203580 treatment decreased plasma TNF-α and IL-6 concentrations by 2- to 2.5-fold (Figure 6B), resulting in concentrations comparable to control wild-type mice after LPS treatment.
Figure 6.

SB suppresses LPS-induced systemic secretion of proinflammatory cytokines and lethality in MGRKO mice. (A) Effect of SB (●) or vehicle (■) on LPS-mediated mortality in MGRKO mice. For the SB-treated group, mice were injected intraperitoneally with SB (2 mg/kg) 1 hour before LPS (10 mg/kg) treatment. P < .01 for SB-treated mice (n = 9) compared with vehicle-treated mice (n = 8). (B) Effect of SB (□) or vehicle (■) on LPS-mediated induction of TNF-α and IL-6 in MGRKO mice. Mice were injected intraperitoneally with SB or vehicle followed by treatment with LPS for 6 hours. *P < .05 for SB-treated group (n = 5) compared with vehicle-treated group (n = 11). Error bars indicate SEM.
Discussion
These studies use in vivo and ex vivo methods to evaluate the importance of GR modulation of signaling downstream of activated TLR4 and, further, to elucidate the mechanisms by which GR attenuates proinflammatory activity in macrophages. We find that MGRKO mice are highly susceptible to LPS-induced mortality. This finding is consistent with the increased susceptibility of glucocorticoid-deficient, adrenalectomized animals to LPS administration and highlights the macrophage as a key site of action for glucocorticoid enhancement of survival.48
Recent efforts in understanding signaling downstream of TLR4 have revealed an important role of disruption of IRF-3–NFκB interaction by GR in inhibiting proinflammatory cytokine production.6 Activation of TLR4 in macrophages not only results in induction of NFκB activity but also stimulates several kinases that increase proinflammatory gene expression.20–23 Here we demonstrate robust phosphorylation of p38 MAPK, JNK, ERK1/2, and Akt after TLR4 engagement by LPS. Both p38 MAPK and JNK display biphasic patterns of induction, with early (up to 30 min) and late (after 6 h) increases in phosphorylation. This biphasic time course corresponds to the transient induction of MKP-1 that occurs in LPS-stimulated macrophages in the absence of glucocorticoid exposure. Only p38 MAPK among this group of kinases, however, appears to have sensitivity to dexamethasone-mediated suppression in control macrophages (Figure 7). This sensitivity is evident at both early and late phases of p38 induction and occurs in the context of MKP-1 induction that is induced within 30 minutes of LPS stimulation. Based upon the inability of Dex to suppress LPS-induced p38 MAPK phosphorylation in the presence of triptolide, an MKP-1 synthesis inhibitor,45–47 MKP-1 appears to be the primary target for GR-associated inhibition of p38 MAPK.45 It is somewhat surprising that dexamethasone-induced MKP-1 does not down-regulate JNK phosphorylation in control macrophages despite up-regulation of JNK phosphorylation in MKP-1–deficient macrophages.33,34 This finding suggests subcellular compartmentalization of MKP-1 action with steroid treatment or different affinity of MKP-1 for phosphorylated forms of p38 and JNK.
Figure 7.

Figure 7. GR attenuates proinflammatory responses in macrophages by impairing p38 MAPK. While TLR4 engagement activates PI3 kinase (PI3K) and multiple MAP kinase kinases (MKKs) and their downstream kinases, p38 MAPK is selectively glucocorticoid (GC) sensitive by transcriptional activation of MKP-1 through GR.
Given the wealth of data regarding GR-mediated down-regulation of the NFκB pathway, how important are actions of GR in modulating p38 MAPK? p38 was originally identified as a tyrosine-phosphorylated protein in macrophages after stimulation with LPS49 and, more recently, after a more diverse repertoire of stimuli.50 Studies in humans have suggested a key role for p38 signaling after LPS exposure, as healthy volunteers treated with p38 inhibitors displayed reduced ex vivo cytokine production with LPS stimulation.51 In addition, clinical symptoms and inflammatory markers in humans after LPS exposure are improved with p38 inhibitor treatment in vivo as well.52,53 Our ex vivo studies on primary macrophages demonstrate approximately 50% to 60% suppression of cytokine production by p38 inhibitors, whereas dexamethasone inhibits cytokine production 70% to 80%. These findings indicate that p38 inactivation accounts for a large part, but not the entirety of, GR effects and is in accord with an additional role for NFκB. The relative importance of p38-GR interaction for mediating glucocorticoid immunosuppression is highlighted further by the survival of p38-inhibitor–treated MGRKO mice exposed to LPS described in this report and the impaired glucocorticoid-mediated cytokine suppression in MKP-1–deficient macrophages reported by others.
The relative importance of transactivation properties of GR (eg, induction of gene expression at the transcriptional level by DNA binding versus its transrepression of gene expression by protein-protein interaction) has been an area of active debate.5 Previous studies have shown that induction of MKP-1 by the glucocorticoid-GR complex occurs at the transcriptional level, with GR transactivation mutants lacking the ability to activate the MKP-1 promoter in reporter assays.24 Studies in GRdim mice suggested that DNA-binding, transactivation-dependent effects may have less importance than the GR protein-protein interactions.12 Our evidence that MGRKO macrophages fail to induce MKP-1 with dexamethasone treatment while GRdim macrophages are capable of MKP-1 induction35 together implicate significant residual transactivation properties with the GRdim mutation, as suggested in transfection studies.32
The notion of pharmacologic inhibition of p38 MAPK activity as a possible substitute for glucocorticoid-mediated immunosuppression has been an active area for drug discovery.52,54–57 Our data suggest that glucocorticoids normally impact upon p38 activity for a substantial portion of their therapeutic anti-inflammatory actions, and thus targeting p38 directly is a logical goal. Recent efforts to bring p38 inhibitors into clinical practice have had limited success. While efficacious, these compounds lead to heptocellular toxicity, which has impeded their use. The induction of MKP-1 by glucocorticoids in LPS-stimulated macrophages provides an important target for GR action and may serve as a new target for drug development. MKP-1 is a threonine/tyrosine dual-specificity phosphatase that has been shown to limit cytokine hyper-responsiveness in rodents in vivo and macrophages ex vivo through the analysis of MKP-1–deficient mice.33,34 We find that without glucocorticoid stimulation, MKP-1 is only transiently induced with TLR4 activation, but GR activation induces higher levels of MKP-1 expression for longer durations ex vivo. This effect would be anticipated in vivo with activation of the hypothalamic-pituitary-adrenal axis and production of glucocorticoids with sustained inflammation. One attractive mechanism, then, by which anti-inflammatory glucocorticoid effects could be accomplished without detrimental glucocorticoid sequelae or p38 inhibitor toxicity is through pharmacologic augmentation of MKP-1 action.
Supplementary Material
Acknowledgments
We thank Drs Steven Teitelbaum and Wojciech Swat for manuscript review and helpful suggestions.
This work was supported by a grant from the National Institutes of Health (NIH; AI50655; L.J.M.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.B. performed research, analyzed data, and contributed to writing the paper; D.E.B. performed research and analyzed data; J.A.B. contributed a new reagent and performed research; S.K.V. performed research; and L.J.M. designed experiments, performed experiments, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Louis J. Muglia, Washington University School of Medicine, Department of Pediatrics, 660 S. Euclid Ave, Box 8208, St Louis, MO 63110; e-mail: muglia_l@kids.wustl.edu.
References
- 1.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 2.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 3.Wells CA, Ravasi T, Hume DA. Inflammation suppressor genes: please switch out all the lights. J Leukoc Biol. 2005;78:9–13. doi: 10.1189/jlb.1204710. [DOI] [PubMed] [Google Scholar]
- 4.Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its P55 receptor. J Clin Invest. 1997;100:1557–1565. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids: new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 6.Ogawa S, Lozach J, Benner C, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol. 2004;20:455–480. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- 8.Ricote M, Valledor AF, Glass CK. Decoding transcriptional programs regulated by PPARs and LXRs in the macrophage: effects on lipid homeostasis, inflammation, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:230–239. doi: 10.1161/01.ATV.0000103951.67680.B1. [DOI] [PubMed] [Google Scholar]
- 9.Valledor AF, Ricote M. Nuclear receptor signaling in macrophages. Biochem Pharmacol. 2004;67:201–212. doi: 10.1016/j.bcp.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 10.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 11.De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-kappaB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci U S A. 2000;97:3919–3924. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reichardt HM, Tuckermann JP, Gottlicher M, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. doi: 10.1093/emboj/20.24.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 14.Joseph SB, Bradley MN, Castrillo A, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. 2004;119:299–309. doi: 10.1016/j.cell.2004.09.032. [DOI] [PubMed] [Google Scholar]
- 15.Valledor AF, Hsu LC, Ogawa S, Sawka-Verhelle D, Karin M, Glass CK. Activation of liver X receptors and retinoid X receptors prevents bacterial-induced macrophage apoptosis. Proc Natl Acad Sci U S A. 2004;101:17813–17818. doi: 10.1073/pnas.0407749101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi F, Smith KD, Ozinsky A, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 17.Medzhitov R, Janeway CA., Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 18.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 19.Underhill DM, Ozinsky A, Hajjar AM, et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 20.Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 21.O'Neill LA. How Toll-like receptors signal: what we know and what we don't know. Curr Opin Immunol. 2006;18:3–9. doi: 10.1016/j.coi.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 22.Okugawa S, Ota Y, Kitazawa T, et al. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am J Physiol Cell Physiol. 2003;285:C399–C408. doi: 10.1152/ajpcell.00026.2003. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Yamazaki S, Uematsu S, et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature. 2004;430:218–222. doi: 10.1038/nature02738. [DOI] [PubMed] [Google Scholar]
- 24.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–7116. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasa M, Abraham SM, Boucheron C, Saklatvala J, Clark AR. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22:7802–7811. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang LL, Ou CC, Chan JY. Receptor-independent activation of GABAergic neurotransmission and receptor-dependent nontranscriptional activation of phosphatidylinositol 3-kinase/protein kinase Akt pathway in short-term cardiovascular actions of dexamethasone at the nucleus tractus solitarii of the rat. Mol Pharmacol. 2005;67:489–498. doi: 10.1124/mol.104.005595. [DOI] [PubMed] [Google Scholar]
- 27.Cato AC, Nestl A, Mink S. Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE. 2002;2002:RE9. doi: 10.1126/stke.2002.138.re9. [DOI] [PubMed] [Google Scholar]
- 28.Hafezi-Moghadam A, Simoncini T, Yang Z, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 30.Nagaich AK, Rayasam GV, Martinez ED, et al. Subnuclear trafficking and gene targeting by steroid receptors. Ann N Y Acad Sci. 2004;1024:213–220. doi: 10.1196/annals.1321.002. [DOI] [PubMed] [Google Scholar]
- 31.Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schutz G, Angel P. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol. 1999;147:1365–1370. doi: 10.1083/jcb.147.7.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams M, Meijer OC, Wang J, Bhargava A, Pearce D. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol. 2003;17:2583–2592. doi: 10.1210/me.2002-0305. [DOI] [PubMed] [Google Scholar]
- 33.Chi H, Barry SP, Roth RJ, et al. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Q, Wang X, Nelin LD, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–140. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abraham SM, Lawrence T, Kleiman A, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med. 2006;203:1883–1889. doi: 10.1084/jem.20060336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brewer JA, Khor B, Vogt SK, et al. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. 2003;9:1318–1322. doi: 10.1038/nm895. [DOI] [PubMed] [Google Scholar]
- 37.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 38.Masferrer JL, Zweifel BS, Manning PT, et al. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci U S A. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/s0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 40.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 41.Guan Z, Buckman SY, Pentland AP, Templeton DJ, Morrison AR. Induction of cyclooxygenase-2 by the activated MEKK1—SEK1/MKK4—p38 mitogen-activated protein kinase pathway. J Biol Chem. 1998;273:12901–12908. doi: 10.1074/jbc.273.21.12901. [DOI] [PubMed] [Google Scholar]
- 42.Hundley TR, Prasad AR, Beaven MA. Elevated levels of cyclooxygenase-2 in antigen-stimulated mast cells is associated with minimal activation of p38 mitogen-activated protein kinase. J Immunol. 2001;167:1629–1636. doi: 10.4049/jimmunol.167.3.1629. [DOI] [PubMed] [Google Scholar]
- 43.Engelbrecht Y, de Wet H, Horsch K, Langeveldt CR, Hough FS, Hulley PA. Glucocorticoids induce rapid up-regulation of mitogen-activated protein kinase phosphatase-1 and dephosphorylation of extracellular signal-regulated kinase and impair proliferation in human and mouse osteoblast cell lines. Endocrinology. 2003;144:412–422. doi: 10.1210/en.2002-220769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toh ML, Yang Y, Leech M, Santos L, Morand EF. Expression of mitogen-activated protein kinase phosphatase 1, a negative regulator of the mitogen-activated protein kinases, in rheumatoid arthritis: up-regulation by interleukin-1beta and glucocorticoids. Arthritis Rheum. 2004;50:3118–3128. doi: 10.1002/art.20580. [DOI] [PubMed] [Google Scholar]
- 45.Chen P, Li J, Barnes J, Kokkonen GC, Lee JC, Liu Y. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phosphatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol. 2002;169:6408–6416. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- 46.Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem. 2004;279:54023–54031. doi: 10.1074/jbc.M408444200. [DOI] [PubMed] [Google Scholar]
- 47.Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. J Biol Chem. 2005;280:8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]
- 48.Bertini R, Bianchi M, Ghezzi P. Adrenalectomy sensitizes mice to the lethal effects of interleukin 1 and tumor necrosis factor. J Exp Med. 1988;167:1708–1712. doi: 10.1084/jem.167.5.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 50.Ashwell JD. The many paths to p38 mitogen-activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6:532–540. doi: 10.1038/nri1865. [DOI] [PubMed] [Google Scholar]
- 51.Parasrampuria DA, de Boer P, Desai-Krieger D, Chow AT, Jones CR. Single-dose pharmacokinetics and pharmacodynamics of RWJ 67657, a specific p38 mitogen-activated protein kinase inhibitor: a first-in-human study. J Clin Pharmacol. 2003;43:406–413. doi: 10.1177/0091270002250615. [DOI] [PubMed] [Google Scholar]
- 52.Branger J, van den Blink B, Weijer S, et al. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J Immunol. 2002;168:4070–4077. doi: 10.4049/jimmunol.168.8.4070. [DOI] [PubMed] [Google Scholar]
- 53.Fijen JW, Tulleken JE, Kobold AC, et al. Inhibition of p38 mitogen-activated protein kinase: dose-dependent suppression of leukocyte and endothelial response after endotoxin challenge in humans. Crit Care Med. 2002;30:841–845. doi: 10.1097/00003246-200204000-00021. [DOI] [PubMed] [Google Scholar]
- 54.Jackson JR, Bolognese B, Hillegass L, et al. Pharmacological effects of SB 220025, a selective inhibitor of P38 mitogen-activated protein kinase, in angiogenesis and chronic inflammatory disease models. J Pharmacol Exp Ther. 1998;284:687–692. [PubMed] [Google Scholar]
- 55.Miwatashi S, Arikawa Y, Kotani E, et al. Novel inhibitor of p38 MAP kinase as an anti-TNF-alpha drug: discovery of N-[4-[2-ethyl-4-(3-methylphenyl)-1,3-thiazol-5-yl]-2-pyridyl]benzamide (TAK-715) as a potent and orally active anti-rheumatoid arthritis agent. J Med Chem. 2005;48:5966–5979. doi: 10.1021/jm050165o. [DOI] [PubMed] [Google Scholar]
- 56.Nash SP, Heuertz RM. Blockade of p38 map kinase inhibits complement-induced acute lung injury in a murine model. Int Immunopharmacol. 2005;5:1870–1880. doi: 10.1016/j.intimp.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 57.Song GY, Chung CS, Chaudry IH, Ayala A. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. Am J Physiol Cell Physiol. 2001;281:C662–C669. doi: 10.1152/ajpcell.2001.281.2.C662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}