

Abstract
Misregulation of the innate immune response and other immune-related processes have been suggested to play a critical role in the pathogenesis of a number of different neurodegenerative diseases, including age related macular degeneration. In an animal model for photoreceptor degeneration, several genes of the innate and acquired immune system were found to be differentially regulated in the retina during the degenerative process. In addition to this differential regulation of individual genes, we found that in the rd1 retina a significantly higher number of genes involved in immune-related responses were expressed at any given time during the degenerative period. The peak of immune-related gene expression was at postnatal day 14, coinciding with the peak of photoreceptor apoptosis in the rd1 mouse. We directly tested the potential involvement of acquired and innate immune responses in initiation and progression of photoreceptor degeneration by analyzing double mutant animals. Retinal morphology and photoreceptor apoptosis of rd1 mice on a SCID genetic background (no mature T- and B-cells) or in combination with a RAG-1 (no functional B- and T-cells) or a C1qα (no functional classical complement activation pathway) knockout was followed during the degenerative process using light microscopy or TUNEL staining, respectively. Although complement factor C1qα was highly up-regulated in the rd1 retina concomitantly with the degenerative process, lack of this protein did not protect the rd1 retina. Similarly, retinal degeneration and photoreceptor apoptosis appeared to proceed normally in the rd1 mouse lacking functional B- and T-cells. Our results suggest that both, the classical complement system of innate immunity and a functional acquired immune response are not essential for the degenerative process in the rd1 mouse retina.
Keywords: retinal degeneration, immune response, rd1, SCID, Rag1−/ −, complement system
INTRODUCTION
Recent evidence supports an involvement of immune-related factors in the development and progression of retinal degenerative diseases in human patients. Local inflammation may not only be involved in the formation of drusen, a hallmark of AMD (Hageman et al. 2001; Johnson et al. 2002) but inflammation triggered by the debris generated by dying RPE cells may also lead to photoreceptor cell death and loss of vision (Johnson et al. 2002). Furthermore, mutations in complement factor H and factor B were shown to significantly increase the risk to develop age-related macular degeneration (AMD) (Edwards et al. 2005; Hageman et al. 2005; Haines et al. 2005; Klein et al. 2005; Umeda et al. 2005; Gold et al. 2006). Topographical analysis of retinas affected by AMD revealed that rod cell death precedes cone cell death (Curcio 2001). The investigation of the mechanisms by which rod cells die in human patients may therefore be a valuable approach to address early changes in the pathology of AMD (Adler et al. 1999; Reme et al. 2003). Many induced and inherited animal models have been described for the study of photoreceptor degeneration (Hafezi et al. 2000; Wenzel et al. 2005). Among these, the rd1 mouse constitutes the best characterized model in which a recessive mutation in the β-subunit of the phosphodiesterase (β-PDE) gene (Farber 1995)) causes an early onset of rapid retinal degeneration. The mutation results in high levels of cGMP (Lolley et al. 1974), which leaves an increased number of the cGMP-gated channels in the open state, allowing intracellular calcium to rise to toxic levels and rapid rod degeneration to follow (Fox et al. 1999). The degenerative process also correlates with defects in energy metabolism (Acosta et al. 2005), although a causal effect has not yet been demonstrated. Based on observed changes in gene expression, we have previously suggested that photoreceptor degeneration in the rd1 mouse is a process starting with Ca2+-toxicity followed by secondary insults involving multi-destructive pathways such as apoptosis and potentially an immune response (Rohrer et al. 2004). This was based on the observation that the gene ontology terms (GO terms) ‘defense response’, ‘complement activity’ and ‘stress response’ were significantly over-represented in the differentially-expressed genes during the degenerative process in the retina of the rd1 mouse. The increased expression of inflammatory chemokines (MCP-1, MCP-3, MIP-1α , MIP-1β, and RANTES) along with tumor necrosis factor (TNF-a) is additional evidence supporting a potential inflammatory response in these retinas (Zeng et al. 2005). In addition, Andrieu-Soler and coworkers (Andrieu-Soler et al. 2005) have documented the presence of F4/80 positive infiltrating inflammatory cells (macrophages and/or activated microglial cells) in the degenerating rd1 mouse retina. Finally, the presence of antigenic changes such as GFAP expression in Muller cells (Sheedlo et al. 1995) has been reported in these retinas.
Recently, the pathway of classical complement activation has been implicated in the development of glaucoma and an up-regulation of C1q in glaucomatous eyes has been documented mainly in Muller cells and in the region of the inner limiting membrane (Stasi et al. 2006). In a model of ocular hypertension, C1q expression was similarly localized to the nerve fiber layer and the ganglion cell layer (Kuehn et al. 2006). Thus, the retina may respond to different types of retinal injury by an up-regulation of the classical complement activation cascade. As for glaucoma and the model of ocular hypertension (Kuehn et al. 2006; Stasi et al. 2006), the classical complement pathway of the innate immune response was found to be up-regulated in the rd1 mouse model of photoreceptor apoptosis and retinal degeneration at the mRNA level. In addition, genes of the acquired immune system were induced as well. To test the involvement of these two immune pathways in the pathogenesis of the rd1 mouse retina, and to analyze the role of individual immune-related genes, we combined the rd1 mutation with one mutation targeting the classical complement pathway (C1qα −/ −), and two mutations targeting the innate immune response (severe combined immune deficiency (SCID) mice and recombination activating protein RAG1 knockout mice (Rag-1−/ −)), respectively. Photoreceptor degeneration as well as removal of cellular debris in the three double-knockout mice was found to proceed at the same rate as in the single knockout (rd1) mice. Our results suggest that both the classical complement system of innate immunity and acquired immune responses appear not to be essential for the degenerative process in the rd1 mouse retina, but leave open the possibility that photoreceptor degeneration in this model is dependent on the alternative pathway of the complement system.
MATERIALS AND METHODS
Mice
All procedures concerning animals were in accordance with the regulations of the Veterinary Authority of Zurich and the Medical University of South Carolina Animal Care and Use Committee, and with the statement of The Association for Research in Vision and Ophthalmology for the use of animals in research. All mice were housed under standard conditions in a 12h:12h light:dark cycle. C3H/SCID mice carrying the rd1 mutation on the SCID genetic background were purchased from Jackson labs (Bar Harbor, Maine, USA). Rag1−/ − mice are described elsewhere (Mombaerts et al. 1992) and were genotyped by PCR using the following four primers: 1) 5’-TAC CCT GAG CTT CAG TTC-3’ (Rag1 forward wt); 2) 5’-CAA CAT CTG CCT TCA CGT C-3’ (Rag1 reverse wt); 3) 5’-CTT GGG TGG AGA GGC TAT TC-3’ (Rag1 forward ko); 4) 5’-AGG TGA GAT GAC AGG AGA TC-3’ (Rag1 reverse ko). Amplification products were 500bp for the wt and 280bp for the knockout allele. C1qα −/ − mice on a pure C57BL/6 background were generated by Botto and colleagues (Botto et al. 1998). Genotyping of C1qα −/ − mice was done by PCR using the following three primers: 1) 5’-GGG GCC TGT GAT CCA GAC AG-3’ (C1qα forward); 2) 5’-TAA CCA TTG CCT CCA GGA TGG-3’ (C1qα reverse); 3) 5’-GGG GAT CGG CAA TAA AAA GAC-3’ (Neo reverse). Wildtype sequence gave an amplification product of 360bp, knockout sequence of 160bp. To determine the successful generation of double knockouts, mice were genotyped for the rd1 mutation using upstream primer 5’-CAT CCC ACC TGA GCT CAC AGA AAG-3’ and downstream primer 5’-GCC TAC AAC AGA GGA GCT TCT AGC-3’. Amplification products were digested with DdeI (Promega, Madison, WI, USA) to detect rd1 specific restriction fragments. The amplification product of wild type DNA is not cut by DdeI.
Gene expression profiling
The data used here has previously been published by our group (Rohrer et al. 2004). In this previous publication, only highly expressed genes (log expression level of >5.5; median gene expression level is ~5.2), with low standard deviation and high correlation between repeats were analyzed, eliminating many genes of median and low expression levels that were never-the-less consistent between repeats; here we included all genes that passed the requirement for low standard deviation and high correlation between repeats.
The platforms used to analyze this data were Affymetrix, Matlab (Version 7.0), including its Statistics Toolbox and BioConductor. CEL and CHP files for each time point (P6 through P21) were imported and normalized using Robust Multi-chip Average (RMA). Three sequential steps were taken to identify differentially expressed genes. First, the data was filtered based on absence and presence calls obtained from Affymetrix. The criterion of at least 3 presence values throughout the time-series replicates was adopted. Second, at each time point and for each genotype, the correlation between the repeats was determined, accepting a correlation of >0.8 between repeats as described previously (Rohrer et al., 2004). And third, a one-way ANOVA was performed per gene per time point to identify genes that are differentially expressed between rd1 and wt, requiring a P value of <0.05.
Gene ontology analysis
Gene ontology terms (biological process) and the relation scheme between the terms were obtained at the Gene Ontology Consortium (http://www.geneontology.org) for all genes present on the array. In determining the GO terms to be used for further analysis of these sets of genes, the following keywords were used: immune, lymphocyte, antigen, antibody, MHC, complement, T cell, B cell, defense. These keywords were then used to determine the genes that were associated with each keyword and the GO term and ID to which they belonged.
Cluster analysis
Hierarchical Clustering was performed on the resulting 58 genes (see Figure 2) using Cluster and TreeView (Eisen et al., 1998). The clustering method was average linkage, using correlation as a similarity measure. The most specific GO Biological Process Term (the one with the highest index) was included in the cluster analysis to attempt to determine whether any GO terms were overexpressed in the gene set. GO IDs and additional information can be found in supplemental materials.
Figure 2.

Gene expression profiling. Hierarchical clustering of the gene expression profiles of the control (C57BL/6) and experimental (rd1) mouse retinas (down-regulated (green); up-regulated (red)). Genes were selected based on standard deviation between repeats of the RMA transformed data and absent present calls. The gene symbol for each of the 58 genes is included. Also included are the GO Biological Process terms for each gene. The color scheme (yellow-blue) represents the similarity of the GO terms as identified by the relation scheme between the terms. The different GO term are distributed fairly evenly throughout the map both in the up- and down-regulated genes in both sets of arrays.
Semi-quantitative RT-PCR
Retinas of rd1/rd1 and of wildtype mice (B6;129) were isolated at post-natal days 11, 14, 21 and 37. Total RNA was prepared using the RNeasy mini kit (Qiagen, Hilden, Germany) including a DNase treatment to digest residual genomic DNA. Total RNA (500 ng) from each of three retinas from three animals were pooled and used for reverse transcription. Real-time PCR was performed on a LightCycler (Roche Diagnostics, Rotkreuz, Switzerland) using an equivalent of 10 ng of total retinal RNA per reaction and the SYBR Green JumpStart Taq ReadyMix (Sigma, Saint Louis, Missouri, USA). Primers used were: β-actin, forward: 5’-CAA CGG CTC CGG CAT GTG C-3’ and reverse: 5’-CTC TTG CTC TGG GCC TCG-3’; opsin, forward: 5’-TTT TAT GTG CCC TTC TCC AAC G-3’ and reverse: 5’-ATG ATA GCG TGA TTC TCC CCG-3’; C1qα , forward: 5’-AAG ATG TCT GCC GAG CAC CCA AC-3’ and reverse: 5’-TTC ACG CCC TTC AGT CCT TG-3’. cDNA was amplified in triplicates and relative quantification was done using the LCS4.0 software (Roche Diagnostics, Rotkreuz, Switzerland).
Morphology and TUNEL staining
For light microscopy, eyes were enucleated and fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.3. For each eye, the superior and the inferior retina was prepared, washed in cacodylate buffer, incubated in osmium tetroxide for 1 h, dehydrated in increasing ethanol concentrations, and embedded in Epon 812. Sections (5 μm) were prepared from the lower central retina, counterstained with methylene blue, and analyzed using an Axiophot microscope (Zeiss, Oberkochen, Germany).
Terminal transferase-mediated dUTP nick end labeling was performed as described (Wenzel et al. 2000). Briefly, retinal tissue was fixed in 2% paraformaldehyde for 2 h at 4°C and embedded in paraffin. The ‘in situ cell death detection kit’ (Roche diagnostics, Rotkreuz, Switzerland) was used with minor modifications. Staining was on 5 μm sections prepared from the lower central retina.
RESULTS
Our main goal was to characterize differential expression of immune-related genes in the rd1 mouse, and to test whether any of the genes that were isolated were critically involved in the degenerative process. We therefore chose to assay gene expression in the retina of rd1 mice covering the time course of photoreceptor degeneration using DNA microarrays (Rohrer et al., 2004). We directly tested a potential essential involvement of the acquired immune response and of the classical activation pathway of the innate immune response in the degenerative process, using double mutant mice lacking essential genes required for those two pathways.
Immune and defense genes in the rd1 mouse retina
In order to analyze genes associated with immune response, gene ontology (GO) terms were identified based on keywords as described above, which resulted in 157 GO terms and 577 corresponding genes. Thus, out of the ~6000 genes and ~6000 ESTs present on the U74Av2 array, approximately 5 % of the probe sets (577) were identified in this way. Please note that a given gene may have been identified by more then one GO term.
To determine whether the number of immune genes expressed varied with genotype, the data were analyzed with a 1 between (genotype) and 1 within (% immune genes expressed) analysis of variance (ANOVA). As expected, a higher percentage of the immune genes were expressed in the rd1 retina during the time course of photoreceptor degeneration when compared with the C57BL/6 control based on the criteria of absent present calls established by the Affymetrix software (genotype effect: F(1,18) = 6.9; P = 0.02) (Figure 1).
Figure 1.
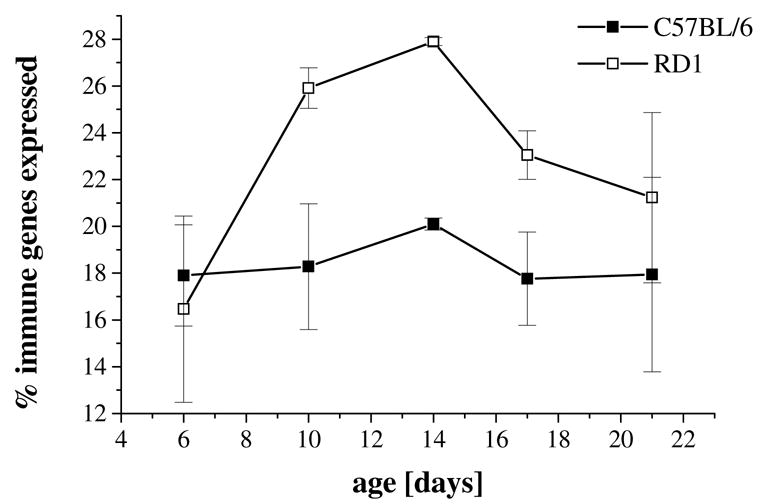
Percent immune/defense genes used in the rd1 mouse retina. Analysis of variance revealed that a higher percentage of the immune genes were expressed in the rd1 retina (open squares) across the five time points covering the time course of photoreceptor degeneration when compared with the C57BL/6 control retina (filled squares) (genotype effect: F(1,18) = 6.9; P = 0.02). Data are expressed as mean between repeats ± SEM.
The identification of differentially expressed genes is based on correlation between replicates, the requirement of the genes to be present (present call according to Affymetrix) in at least 3 out of the 10 time-series replicates and significant P values between age-matched genotypes (e.g., P6 C57BL/6 versus P6 rd1), which resulted in a list of 58 genes (Figure 2; see also supplemental material, Table S1-3). Thus, ~10% (57/577) of the potential genes involved in the immune response were found to be differentially expressed in the juvenile rd1, when compared to the wild type retina.
Genes were subsequently clustered based on expression profiles (Figure 2, left hand side of the figure), and the most specific GO terms were used to classify these 58 genes (Figure 2, right hand side of the figure; see also supplemental material, Table S4). The color-coding demonstrated that components of all GOs are differentially regulated, but do not fall exclusively into either the up- or down-regulated gene clusters. A prominent cluster with three genes involved in the classical pathway of complement activation with high expression at P14, the peak of photoreceptor degeneration in the rd1 mouse (Portera-Cailliau et al., 1994) was identified. In addition a number of genes involved in T-cell activation (GO terms: antigen processing, endogenous antigen via MHC class I; positive regulation of T-cell activation) are found to be up-regulated in the rd1 mouse retinas. T-Cells are responsible for coordinating all Acquired Immune Responses. Based on these findings, the involvement of the classical complement pathway and the acquired immune system were tested in the progression of cell death in the rd1 mouse.
Inactivation of the classical complement activation pathway
Several members of the classical complement activation pathway (C1qα ; C1qγ , C1qβ and C1r) were identified as being differentially regulated during the phase of retinal degeneration in the rd1 mouse. C1q binding to C1r-receptors is involved in the activation of the classical complement pathway. We have previously confirmed the up-regulation of C1qβ in the rd1 mouse (Rohrer et al. 2004) by quantitative real time PCR (QRT-PCR), and extended this analysis to the expression of C1qα , using β-actin and rhodopsin as controls (Table 1). Similar to the expression of C1qβ (Lohr et al. 2006), C1qα expression levels peaked in the rd1 mouse retina at P14 and decreased to almost basal levels towards the end of the period of photoreceptor apoptosis (P21). To test the potential role of complement activation in the degenerative process, we combined the rd1 mutation with a C1qα null background. A potential influence of the lack of C1qα on the rd1-induced degeneration of photoreceptor cells was assessed morphologically at different postnatal days in rd1/rd1; C1qα +/− and rd1/rd1; C1qα −/ − littermates (Figure 3). Lack of C1qα did not influence initiation or progression of retinal degeneration in the rd1 mouse retina. After P11, photoreceptors start to degenerate independently of the presence or absence of a functional C1qα gene. At P17, one to two rows of photoreceptor nuclei are remaining. At P21 and P35, almost all photoreceptors have been lost and the ONL almost completely disappeared. Importantly, clearance of dead photoreceptor cells was not delayed in C1qα deficient mice although C1q has been implicated in the opsonization of dying cells (Fishelson et al. 2001; Botto and Walport 2002).
Table 1.
| Genotype/age | opsin rel to actin | C1qα rel to actin | C1qα rel to opsin |
|---|---|---|---|
| wt P11 | 1.00 | 1.00 | 1.00 |
| wt P14 | 3.27 | 0.71 | 0.22 |
| wt P17 | 5.70 | 0.65 | 0.11 |
| wt P21 | 8.62 | 0.61 | 0.07 |
| rd1/rd1 P11 | 1.10 | 2.56 | 2.30 |
| rd1/rd1 P14 | 0.58 | 13.10 | 22.48 |
| rd1/rd1 P17 | 0.01 | 4.70 | 361.00 |
| rd1/rd1 P21 | 0.000436 | 1.88 | 4320.00 |
Figure 3.
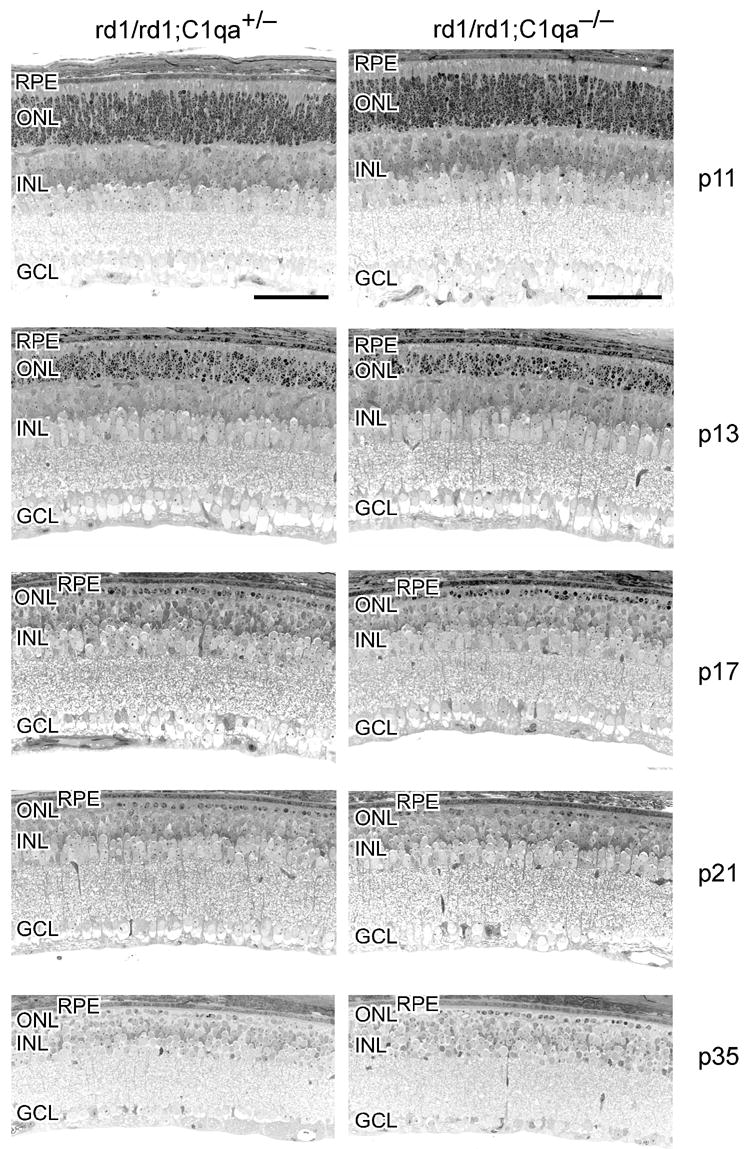
Lack of C1qα does not influence retinal degeneration in the rd1 mouse. Retinal morphology of rd1/rd1;C1qα +/− (left column) and of rd1/rd1;C1qα −/ − (right column) littermates was analyzed at postnatal days 11, 13, 17, 21 and 35, as indicated. Photoreceptor degeneration proceeded similarly in both types of mice. Abbreviations: RPE, retinal pigment epithelium; ONL, outer nuclear layer; INL, inner nuclear layer; GC, ganglion cell-layer. Scale bar: 50μm.
Lack of functional B- and T-cells
The acquired immune response relies on functional B- and T-cells. A number of genes required for T-cell activation are found to be up-regulated in the rd1 mouse retinas (Ap3d1, Cd1d1, H2-D1, H2-K1, H2-D1, Prkcq, B2m and Egr1). The SCID/SCID mutation affects the VDJ recombinase system, which is required to arrange the V, D, and J gene segments. Therefore, SCID mice do not express surface Ig’s on precursor T- and B-cells and hence they do not develop mature T- and B-cells (Nonoyama et al. 1993; Nally et al. 2005). Retinal degeneration in the rd1/SCID (C3H/SCID) mouse was followed morphologically (Figure 4) and by TUNEL staining of apoptotic cells (Figure 5). The presence of the SCID mutation did not influence initiation and progression of photoreceptor apoptosis induced by the rd1 mutation. After P10, the ONL progressively thinned with one row of photoreceptor nuclei remaining at P21. The appearance of TUNEL-positive cells with a peak at P14 also suggests that the SCID mutation did not influence the mode of degeneration. At P21, when almost all photoreceptors have been cleared from the ONL, TUNEL-positive staining was rare. Cells of the INL and GCL were hardly affected by the rd1 mutation, as expected. The scattered TUNEL-positive nuclei in the INL at P10 were due to regular programmed cell death during postnatal development. Several lines of SCID mice show a ‘leakiness’ with respect to serum levels of Ig’s. The strain used here, however, appears to be one of the ‘non-leaky’ strains as shown by the very low serum Ig levels (Nonoyama et al. 1993; Nally et al. 2005). Nevertheless, we additionally generated rd1/Rag-1−/ − double-mutant mice and analyzed retinal development and degeneration by morphological evaluation of retinal tissue at different postnatal days. Mice deficient in RAG1 activity are unable to initiate V(D)J recombination in Ig and TCR genes, and therefore lack functional T- and B-lymphocytes (Mombaerts et al. 1992). Similar to rd1/SCID mice, lack of RAG1 activity did not notably influence induction and/or progression of photoreceptor apoptosis in the rd1 mice (Figure 4). These findings confirmed our results from the rd1/SCID double-deficient mice.
Figure 4.
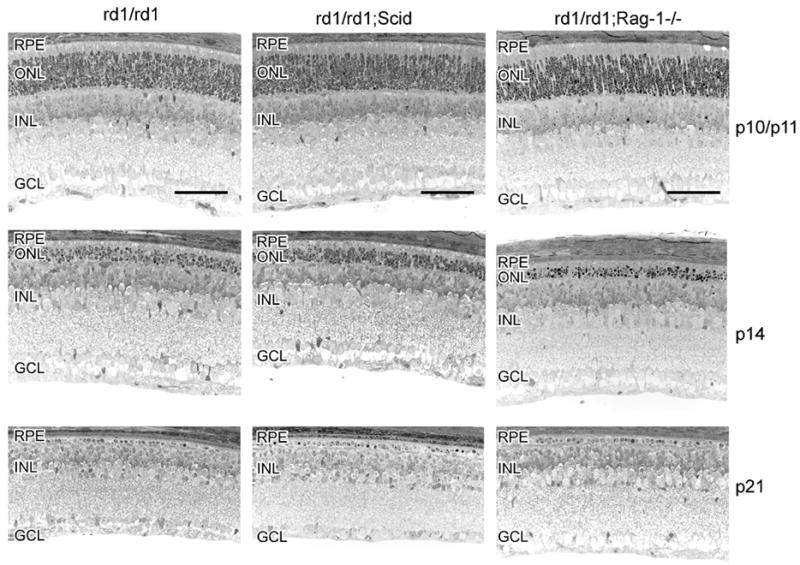
SCID mutation or lack of RAG1 does not influence retinal degeneration in the rd1 mouse. Retinal morphology of rd1/rd1 (left column) and of rd1/rd1;SCID (middle column) was analyzed at postnatal days 10 (rd1/rd1 and rd1/rd1;SCID), 11 (rd1/rd1/Rag-1−/ −), 14, 17 and 21, as indicated. Photoreceptor degeneration proceeded similarly in all types of mice. Scale bar: 50μm. Abbreviations as in Figure 3.
Figure 5.
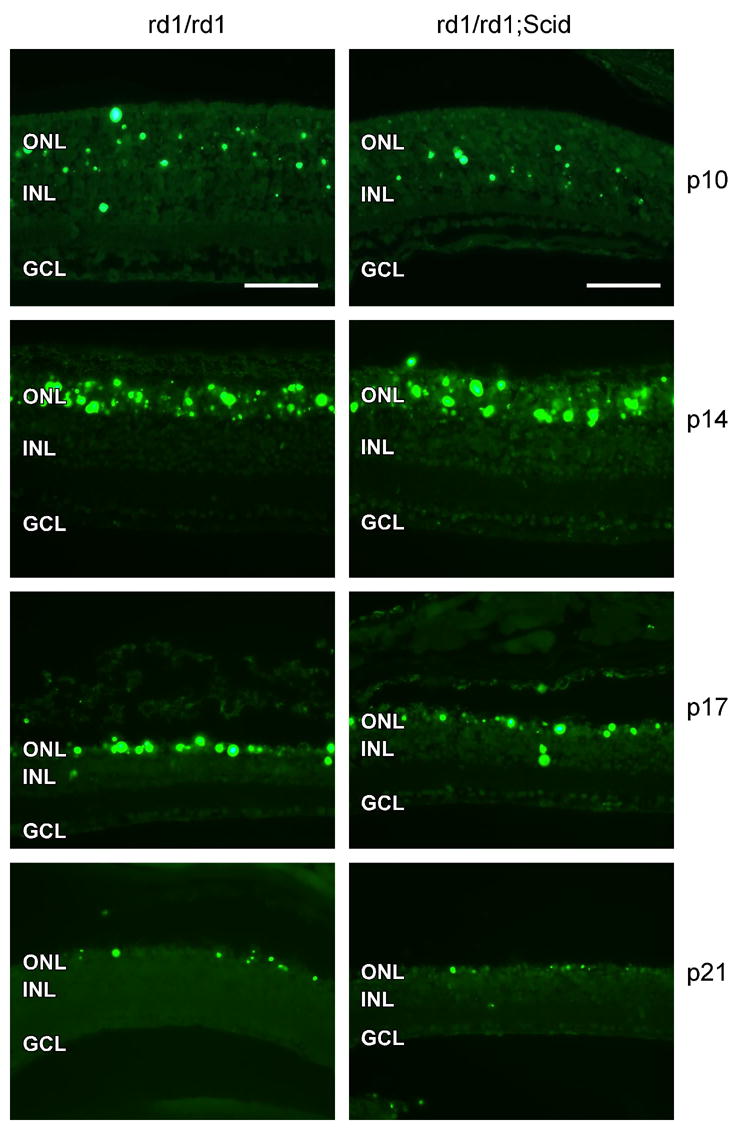
SCID mutation does not influence retinal degeneration in the rd1 mouse. TUNEL staining of apoptotic cells in rd1/rd1 (left column) and in rd1/rd1;SCID (right column) was performed at postnatal 10, 14, 17 and 21, as indicated. Amount and distribution of apoptotic cells were similar in both types of mice. Scale bar: 50μm. Abbreviations as in Figure 3.
DISCUSSION
Recent genetic evidence has strongly implicated polymorphisms in the complement control protein factor H (Edwards et al. 2005; Hageman et al. 2005; Haines et al. 2005; Klein et al. 2005; Umeda et al. 2005) and factor B (Gold et al. 2006) as major risk (or, depending on the polymorphism, protective) factors for AMD. Based on these results, it has been proposed that inadequate control of complement-driven immune-response may be an important factor in the pathogenesis of AMD.
Although the rd1 mouse is not investigated as a model system for AMD, the analysis of the degenerative processes in the rd1 retina may nevertheless significantly advance our understanding of the pathology in age related macular degeneration. As Curcio and coworkers (Curcio et al. 1996) very convincingly showed, rod photoreceptors are the first cells to die in AMD. Since rods are known to secrete cone-protecting factors (Leveillard et al. 2004), saving the rods from cell death may increase the survival time of the cones (Curcio et al. 2000; Sahel 2005). Furthermore, recent data shows that immune-related genes, including genes expressing factors of the complement system are up-regulated in the rd1 retina (Rohrer et al. 2004; Zeng et al. 2005) as they are in the retina of AMD patients (Hageman et al. 2001), suggesting that the degeneration in the rd1 mouse may share some features with molecular processes during the course of AMD in human patients.
Although genes of both, the innate and the acquired immune response were induced in the retina of rd1 mice, neither lack of the classical complement system nor the acquired immune response did notably influence the degeneration or the clearing/phagocytosis of apoptotic cells. Although the retina clearly reacts to photoreceptor degeneration by an up-regulation of genes involved in the tissue defense response against intruders and/or debris, this mechanism does not appear to be essential for the degeneration and/or the maintenance of the integrity of retinal cells not directly affected by the gene mutation per se. When using knockout mice to analyse specific features during postnatal development, the problem of the potential lack of specificity has to be kept in mind (e.g., (Crusio 1996; Gerlai 1996; Lathe 1996; Crawley et al. 1997; Gass et al. 1998)). Two issues are of particular concern, indirect effects due to deficits in organs not directly targeted; and the possible effects of hitchhiking genes, which are genes surrounding the targeted gene in the embryonic stem cell strain. To circumvent these problems, Gerlai (Gerlai 1996) recommended to characterize more than one independently created animal line. Thus, the identical (although negative) finding in the rd1/ RAG1 and the rd1/SCID mice, which both lack functional T- and B-lymphocytes, strengthens our conclusion.
Two distinct populations of macrophages have been demonstrated in the retina: perivascular cells and microglia (Provis et al. 1995; Provis et al. 1996). Microglia not only are phagocytes, clearing away cell debris (Egensperger et al. 1996) but they are also the primary immunocompetent cells in the central nervous system (CNS) and may be involved in surveillance of tissue integrity and the protection of neuronal tissue during injury and disease (Streit et al. 2005). On the other hand, microglia produce a multitude of factors which may be toxic to neurons thereby contributing to cell death in the CNS upon activation (Block and Hong 2005). In addition, glial cells may express most if not all of the activation and regulatory proteins of the complement system (Barnum 1995; Barnum 2002) and microglia up-regulate complement factor C1q in response to transient ischemia in the brain (Schafer et al. 2000).
It has been shown recently that microglial migration and accumulation is enhanced in the degenerating retina such as in that of the RCS rat (Roque et al. 1996), the rd1 (Zeiss and Johnson 2004; Andrieu-Soler et al. 2005; Zeng et al. 2005) or the rd2 mouse (Hughes et al. 2003). Zeng and coworkers (Zeng et al. 2005) have additionally shown in the rd1 retina that activated microglia express a multitude of signaling molecules (chemokines MCP-1, MCP-3, MIP-1α , MIP-1β, and RANTES) and microglia-derived toxic factor (TNF-α ) which may contribute to three different processes: 1st, increased photoreceptor death due to the release of cytotoxic substances; 2nd, recruitment of monocytes/macrophages to the site of injury through the release of chemokines and 3rd, removal of cellular debris. On the other hand, Hughes and colleagues (Hughes et al. 2004) have questioned the role of microglia in rd2 photoreceptor degeneration by showing that minocycline, a potent anti-inflammatory semi-synthetic tetracycline antibiotic, does not exert its neuroprotective effect(s) via the suppression of microglial activation.
Here, we have focused on the possible involvement of immune-related factors in the degenerative process of rd1 mouse photoreceptors. Although the individual factors analyzed here are not essential for the degenerative process in the rd1 retina, it is possible, that they contribute to the process in a synergistic way. Inactivation of one of these factors or even pathways may not be sufficient to induce a distinct phenotype due to redundancy. The complement system for example can be activated by each of three distinct pathways (classical, alternative, and lectin), and complement factor H is the major fluid phase inhibitor of the alternative pathway (Holers 2000). Involvement of the alternative pathway in the pathology of the rd1 retina will be tested in the future once the required mouse mutants become freely available.
Supplementary Material
Gene expression analysis of defense and immune genes in the rd1/rd1 mouse retina. Based on correlation between repeats (Rohrer et al. 2004), p- values between age-matched samples and the requirement of the genes to be expressed in at least 3 out of the 10 time-series replicates, a list of 58 genes was produced. For each gene, the gene symbol, gene description and gene expression data (average of two replicates, RMA data), is provided. This data set is used for Figure 2, and the determination of up- and down-regulation (Table S2) and fold change calculations (Table S3). Please note that the order of the genes in Tables S1–3) is the same as plotted in Figure 2.
P-values based on n-way ANOVA using pairwise comparison at each time point to determine significance of expression. This calculation represents the significance of the difference between experiment (rd1) and control (C57BL/6) age-matched samples. At least one time point in the series was required to have a significant p-value. Many of these genes have significant p-values at more than one time point, and in some cases, all time points, indicating their importance through the process of retinal degeneration.
Differences in Expression (fold change) between age-matched samples for rd1 and C57BL/6 for each gene at each time point. This data was calculated using the differences in the RMA averages and transforming these log differences into fold change using a power function. As seen in this table, many of the genes have the highest fold changes around P10 and P14, the critical time points in the retinal degeneration mechanism.
The GO Biological Process terms used to classify the 58 genes are listed. Please note that each gene is characterized by more then one GO term, however only the most significant GO term is listed here and was used to characterize the genes in figure 2. Some of the top GO terms in this list include immune response, complement activation, defense response and humoral immune response, all of which were expected in this data set.
Acknowledgments
We would like to thank Coni Imsand, Gaby Hoegger, Hedi Wariwoda, Heather Lohr and Kathryne Hulse for excellent technical assistance, and Luanna Bartholomew for critical review of the manuscript. This work was supported by the Swiss National Science Foundation (3100A0-105793); a grant from a research priority program of the university of Zurich ‘Integrative Human Physiology’; grants from the European Community (EVI-GenoRet; LSHG-CT-512036) and the National Institutes of Health (EY13520 and EY014793); and an unrestricted grant to MUSC from Research to Prevent Blindness, Inc., New York, NY. The authors are especially thankful for the generous support by the H. Messerli Fond. BR is a Research to Prevent Blindness Olga Keith Weiss Scholar.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta ML, Fletcher EL, Azizoglu S, Foster LE, Farber DB, Kalloniatis M. Early markers of retinal degeneration in rd/rd mice. Mol Vis. 2005;11:717–728. [PubMed] [Google Scholar]
- Adler R, Curcio C, Hicks D, Price D, Wong F. Cell death in age-related macular degeneration. Mol Vis. 1999;5:31. [PubMed] [Google Scholar]
- Andrieu-Soler C, Aubert-Pouessel A, Doat M, Picaud S, Halhal M, Simonutti M, Venier-Julienne MC, Benoit JP, Behar-Cohen F. Intravitreous injection of PLGA microspheres encapsulating GDNF promotes the survival of photoreceptors in the rd1/rd1 mouse. Mol Vis. 2005;11:1002–1011. [PubMed] [Google Scholar]
- Barnum SR. Complement biosynthesis in the central nervous system. Crit Rev Oral Biol Med. 1995;6:132–146. doi: 10.1177/10454411950060020301. [DOI] [PubMed] [Google Scholar]
- Barnum SR. Complement in central nervous system inflammation. Immunol Res. 2002;26:7–13. doi: 10.1385/IR:26:1-3:007. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell'Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology. 2002;205:395–406. doi: 10.1078/0171-2985-00141. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Belknap JK, Collins A, Crabbe JC, Frankel W, Henderson N, Hitzemann RJ, Maxson SC, Miner LL, Silva AJ, Wehner JM, Wynshaw-Boris A, Paylor R. Behavioral phenotypes of inbred mouse strains: implications and recommendations for molecular studies. Psychopharmacology (Berl) 1997;132:107–124. doi: 10.1007/s002130050327. [DOI] [PubMed] [Google Scholar]
- Crusio WE. Gene-targeting studies: new methods, old problems. Trends Neurosci. 1996;19:186–187. doi: 10.1016/s0166-2236(96)20023-2. discussion 188–189. [DOI] [PubMed] [Google Scholar]
- Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye. 2001;15:376–383. doi: 10.1038/eye.2001.140. [DOI] [PubMed] [Google Scholar]
- Curcio CA, Medeiros NE, Millican CL. Photoreceptor loss in age-related macular degeneration. Invest Ophthalmol Vis Sci. 1996;37:1236–1249. [PubMed] [Google Scholar]
- Curcio CA, Owsley C, Jackson GR. Spare the rods, save the cones in aging and age-related maculopathy. Invest Ophthalmol Vis Sci. 2000;41:2015–2018. [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Egensperger R, Maslim J, Bisti S, Hollander H, Stone J. Fate of DNA from retinal cells dying during development: uptake by microglia and macroglia (Muller cells) Brain Res Dev Brain Res. 1996;97:1–8. doi: 10.1016/s0165-3806(96)00119-8. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber DB. From mice to men: the cyclic GMP phosphodiesterase gene in vision and disease. The Proctor Lecture. Invest Ophthalmol Vis Sci. 1995;36:263–275. [PubMed] [Google Scholar]
- Fishelson Z, Attali G, Mevorach D. Complement and apoptosis. Mol Immunol. 2001;38:207–219. doi: 10.1016/s0161-5890(01)00055-4. [DOI] [PubMed] [Google Scholar]
- Fox DA, Poblenz AT, He L. Calcium overload triggers rod photoreceptor apoptotic cell death in chemical-induced and inherited retinal degenerations. Ann N Y Acad Sci. 1999;893:282–285. doi: 10.1111/j.1749-6632.1999.tb07837.x. [DOI] [PubMed] [Google Scholar]
- Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp HP, Schutz G. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem. 1998;5:274–288. [PMC free article] [PubMed] [Google Scholar]
- Gerlai R. Gene-targeting studies of mammalian behavior: is it the mutation or the background genotype? Trends Neurosci. 1996;19:177–181. doi: 10.1016/s0166-2236(96)20020-7. [DOI] [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R, Chang S, Yannuzzi LA, Merriam JC, Barbazetto I, Lerner LE, Russell S, Hoballah J, Hageman J, Stockman H. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006 doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezi F, Grimm C, Simmen BC, Wenzel A, Reme CE. Molecular ophthalmology: an update on animal models for retinal degenerations and dystrophies. Br J Ophthalmol. 2000;84:922–927. doi: 10.1136/bjo.84.8.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Holers VM. Phenotypes of complement knockouts. Immunopharmacology. 2000;49:125–131. doi: 10.1016/s0162-3109(00)80298-2. [DOI] [PubMed] [Google Scholar]
- Hughes EH, Schlichtenbrede FC, Murphy CC, Broderick C, van Rooijen N, Ali RR, Dick AD. Minocycline delays photoreceptor death in the rds mouse through a microglia-independent mechanism. Exp Eye Res. 2004;78:1077–1084. doi: 10.1016/j.exer.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Hughes EH, Schlichtenbrede FC, Murphy CC, Sarra GM, Luthert PJ, Ali RR, Dick AD. Generation of activated sialoadhesin-positive microglia during retinal degeneration. Invest Ophthalmol Vis Sci. 2003;44:2229–2234. doi: 10.1167/iovs.02-0824. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer's A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:11830–11835. doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn MH, Kim CY, Ostojic J, Bellin M, Alward WL, Stone EM, Sakaguchi DS, Grozdanic SD, Kwon YH. Retinal synthesis and deposition of complement components induced by ocular hypertension. Exp Eye Res. 2006;83:620–628. doi: 10.1016/j.exer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Lathe R. Mice, gene targeting and behaviour: more than just genetic background. Trends Neurosci. 1996;19:183–186. doi: 10.1016/s0166-2236(96)20022-0. discussion 188–189. [DOI] [PubMed] [Google Scholar]
- Leveillard T, Mohand-Said S, Lorentz O, Hicks D, Fintz AC, Clerin E, Simonutti M, Forster V, Cavusoglu N, Chalmel F, Dolle P, Poch O, Lambrou G, Sahel JA. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–759. doi: 10.1038/ng1386. [DOI] [PubMed] [Google Scholar]
- Lohr HR, Kuntchithapautham K, Sharma AK, Rohrer B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp Eye Res. 2006 doi: 10.1016/j.exer.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Lolley RN, Schmidt SY, Farber DB. Alterations in cyclic AMP metabolism associated with photoreceptor cell degeneration in the C3H mouse. J Neurochem. 1974;22:701–707. doi: 10.1111/j.1471-4159.1974.tb04283.x. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Nally JE, Fishbein MC, Blanco DR, Lovett MA. Lethal infection of C3H/HeJ and C3H/SCID mice with an isolate of Leptospira interrogans serovar copenhageni. Infect Immun. 2005;73:7014–7017. doi: 10.1128/IAI.73.10.7014-7017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonoyama S, Smith FO, Bernstein ID, Ochs HD. Strain-dependent leakiness of mice with severe combined immune deficiency. J Immunol. 1993;150:3817–3824. [PubMed] [Google Scholar]
- Portera-Cailliau C, Sung CH, Nathans J, Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci U S A. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provis JM, Diaz CM, Penfold PL. Microglia in human retina: a heterogeneous population with distinct ontogenies. Perspect Dev Neurobiol. 1996;3:213–222. [PubMed] [Google Scholar]
- Provis JM, Penfold PL, Edwards AJ, van Driel D. Human retinal microglia: expression of immune markers and relationship to the glia limitans. Glia. 1995;14:243–256. doi: 10.1002/glia.440140402. [DOI] [PubMed] [Google Scholar]
- Reme CE, Grimm C, Hafezi F, Iseli HP, Wenzel A. Why study rod cell death in retinal degenerations and how? Doc Ophthalmol. 2003;106:25–29. doi: 10.1023/a:1022423724376. [DOI] [PubMed] [Google Scholar]
- Rohrer B, Pinto FR, Hulse KE, Lohr HR, Zhang L, Almeida JS. Multidestructive pathways triggered in photoreceptor cell death of the rd mouse as determined through gene expression profiling. J Biol Chem. 2004;279:41903–41910. doi: 10.1074/jbc.M405085200. [DOI] [PubMed] [Google Scholar]
- Roque RS, Imperial CJ, Caldwell RB. Microglial cells invade the outer retina as photoreceptors degenerate in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 1996;37:196–203. [PubMed] [Google Scholar]
- Sahel JA. Saving cone cells in hereditary rod diseases: a possible role for rod-derived cone viability factor (RdCVF) therapy. Retina. 2005;25:S38–S39. doi: 10.1097/00006982-200512001-00015. [DOI] [PubMed] [Google Scholar]
- Schafer MK, Schwaeble WJ, Post C, Salvati P, Calabresi M, Sim RB, Petry F, Loos M, Weihe E. Complement C1q is dramatically up-regulated in brain microglia in response to transient global cerebral ischemia. J Immunol. 2000;164:5446–5452. doi: 10.4049/jimmunol.164.10.5446. [DOI] [PubMed] [Google Scholar]
- Sheedlo HJ, Jaynes D, Bolan AL, Turner JE. Mullerian glia in dystrophic rodent retinas: an immunocytochemical analysis. Brain Res Dev Brain Res. 1995;85:171–180. doi: 10.1016/0165-3806(94)00203-c. [DOI] [PubMed] [Google Scholar]
- Stasi K, Nagel D, Yang X, Wang RF, Ren L, Podos SM, Mittag T, Danias J. Complement component 1Q (C1Q) upregulation in retina of murine, primate, and human glaucomatous eyes. Invest Ophthalmol Vis Sci. 2006;47:1024–1029. doi: 10.1167/iovs.05-0830. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Conde JR, Fendrick SE, Flanary BE, Mariani CL. Role of microglia in the central nervous system's immune response. Neurol Res. 2005;27:685–691. doi: 10.1179/016164105X49463a. [DOI] [PubMed] [Google Scholar]
- Umeda S, Suzuki MT, Okamoto H, Ono F, Mizota A, Terao K, Yoshikawa Y, Tanaka Y, Iwata T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis) Faseb J. 2005;19:1683–1685. doi: 10.1096/fj.04-3525fje. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Marti A, Kueng-Hitz N, Hafezi F, Niemeyer G, Remé CE. c-fos controls the 'private pathway' of light-induced apoptosis of retinal photoreceptors. J Neurosci. 2000;20:81–88. doi: 10.1523/JNEUROSCI.20-01-00081.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Zeiss CJ, Johnson EA. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Invest Ophthalmol Vis Sci. 2004;45:971–976. doi: 10.1167/iovs.03-0301. [DOI] [PubMed] [Google Scholar]
- Zeng HY, Zhu XA, Zhang C, Yang LP, Wu LM, Tso MO. Identification of sequential events and factors associated with microglial activation, migration, and cytotoxicity in retinal degeneration in rd mice. Invest Ophthalmol Vis Sci. 2005;46:2992–2999. doi: 10.1167/iovs.05-0118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene expression analysis of defense and immune genes in the rd1/rd1 mouse retina. Based on correlation between repeats (Rohrer et al. 2004), p- values between age-matched samples and the requirement of the genes to be expressed in at least 3 out of the 10 time-series replicates, a list of 58 genes was produced. For each gene, the gene symbol, gene description and gene expression data (average of two replicates, RMA data), is provided. This data set is used for Figure 2, and the determination of up- and down-regulation (Table S2) and fold change calculations (Table S3). Please note that the order of the genes in Tables S1–3) is the same as plotted in Figure 2.
P-values based on n-way ANOVA using pairwise comparison at each time point to determine significance of expression. This calculation represents the significance of the difference between experiment (rd1) and control (C57BL/6) age-matched samples. At least one time point in the series was required to have a significant p-value. Many of these genes have significant p-values at more than one time point, and in some cases, all time points, indicating their importance through the process of retinal degeneration.
Differences in Expression (fold change) between age-matched samples for rd1 and C57BL/6 for each gene at each time point. This data was calculated using the differences in the RMA averages and transforming these log differences into fold change using a power function. As seen in this table, many of the genes have the highest fold changes around P10 and P14, the critical time points in the retinal degeneration mechanism.
The GO Biological Process terms used to classify the 58 genes are listed. Please note that each gene is characterized by more then one GO term, however only the most significant GO term is listed here and was used to characterize the genes in figure 2. Some of the top GO terms in this list include immune response, complement activation, defense response and humoral immune response, all of which were expected in this data set.