

Abstract
Erythrocytes infected with mature forms of Plasmodium falciparum do not circulate but are withdrawn from the peripheral circulation; they are bound to the endothelial lining and to uninfected erythrocytes in the microvasculature. Blockage of the blood flow, hampered oxygen delivery, and severe malaria may follow if binding is excessive. The NH2-terminal head structure (Duffy binding–like domain 1 [DBL1α]–cysteine-rich interdomain region [CIDR1α]) of a single species of P. falciparum erythrocyte membrane protein 1 (PfEMP1) is here shown to mediate adherence to multiple host receptors including platelet-endothelial cell adhesion molecule 1 (PECAM-1)/CD31, the blood group A antigen, normal nonimmune immunoglobulin M, three virulence-associated receptor proteins, a heparan sulfate–like glucosaminoglycan, and CD36. DBL2δ was found to mediate additional binding to PECAM-1/CD31. The exceptional binding activity of the PfEMP1 head structure and its relatively conserved nature argues that it holds an important role in erythrocyte sequestration and therefore in the virulence of the malaria parasite.
Keywords: malaria, sequestration, cytoadherence, rosetting, ligand
Introduction
Severe malaria is brought about at least in part by the sequestration of Plasmodium falciparum–infected erythrocytes (parasited RBCs [pRBCs]) in postcapillary venules. A local excessive accumulation of both pRBCs and uninfected erythrocytes (RBCs) leading to cessation of the local blood supply is thought to be one explanation for the occurrence of cerebral and most other forms of severe malaria 1. Parasite sequestration is due to the adherence of pRBCs to the vascular endothelium (cytoadherence) and to erythrocytes (rosetting) through multiple endothelial and erythrocyte receptors 2 3 4 5 6 7 8 9 10 11 12 13.
P. falciparum–infected erythrocytes that both adhere to the vascular endothelium and form rosettes are more frequently found in individuals with severe than in those with mild malaria 14 15 16 17 18. It was recently established that patients with severe malaria carry pRBCs that bind to multiple endothelial and erythrocyte receptors (our unpublished data). Such parasites were therefore generated in vitro by phenotypic selection using micromanipulation of the parasite FCR3S1 19. A good example is clone FCR3S1.2, where the pRBCs not only form rosettes and autoagglutinates but also readily bind to platelet-endothelial cell adhesion molecule 1 (PECAM-1)/CD31, CD36, IgM, the blood group antigen A, and to a heparan sulfate (HS)-like glycosaminoglycan (GAG) 7 11 12 19. Stable in vitro–cloned parasites that so mimic the polyadhesive phenotype of pRBCs of patients with severe malaria can thus be explored.
There is compelling evidence that P. falciparum erythrocyte membrane protein 1 (PfEMP1) is an adhesin 10 12 20 21 22 23 24, but whether it is the only adhesin and how it is involved in binding to different receptors remain to be explored. Here, we provide evidence that PfEMP1 is a multiadhesive parasite ligand and that most of the activity is localized to the semiconserved head structure composed of the Duffy binding–like domain 1 (DBL1α) and the cysteine-rich interdomain region (CIDR1α) mediating the binding to several independent host receptors.
Materials and Methods
The Parasite.
FCR3S1.2 was obtained by micromanipulation cloning from FCR3S1 19, a parasite previously cloned by limiting dilution 25. The parasites were cultured according to standard methods.
The Adherence of Soluble Receptors to pRBCs of FCR3S1.2.
The infected erythrocytes of FCR3S1.2 were studied for their capacity to adhere to soluble fluorescence-labeled receptor proteins as follows. A 200 μl aliquot of the resuspended parasite culture of an ∼8% parasitemia and a 5% hematocrit was washed three times with 100 mM Nacitrate in PBS and once in PBS before adding different receptors as specified below. The binding was examined under incident UV light using a Nikon Optiphot-2 after a room temperature 60-min incubation on a rotator, three washes with PBS, and counterstaining with ethidium bromide (0.001% in PBS). The estimation of IgM binding was performed as described previously 11.
Blood group A antigen (GalNAc-1-3Gal-2-1-Fuc) bound to biotinylated BSA via a spacer (-O-p-trifluoroacetamidophenylethyl) was purchased from IsoSep. The trisaccharide-biotin-BSA conjugates or control biotin-BSA were diluted in double dilutions from 200 to 50 μg/ml in PBS and mixed with a 200 μl aliquot of the culture as above. An FITC-avidin conjugate (1:100 dilution; Sigma-Aldrich) was added after a 60-min room temperature incubation and three washes in PBS. The binding was visualized as outlined above.
Soluble PECAM-1/CD31 purified from Chinese hamster ovary (CHO) cells (cat. no. ADP6) was purchased from R&D Systems. Four distinct CD36–glutathione S-transferase (GST) fusion proteins covering amino acids (aa) 67–298 were expressed and purified from Escherichia coli and held in PBS with 1% Triton X-100 26. 500 μg of a mixture of the four CD36 fusion proteins was labeled with the fluorescent dye Alexa 488 according to the protocols of the producer (Molecular Probes). Intracellular adhesion molecule 1 (ICAM-1) and PECAM-1/CD31 were similarly directly labeled with Alexa 488. The fluorescence-labeled receptors (CD36, CD31, and ICAM-1) were added at double dilutions ranging from 200 to 50 μg/ml to the parasite culture as above after three washes in PBS. The binding was visualized as outlined above.
Adherence of pRBCs to Receptors Expressed on Transfected CHO or L Cells.
The methods used were as described 7 with some minor modifications. In brief, the binding of pRBCs of FCR3S1.2 was assessed with the cells adherent to coverslips. CHO cells (K1/CCL61), transfected CHO cells expressing CD36 at the cell surface (CHO-CD36), L cells, or transfected L cells expressing PECAM-1/CD31 (L cell–PECAM-1/CD31) were seeded at a density of ∼25,000 cells/coverslip (Thermonox; Nunc) and cultured in RPMI 1640 with 0.6% Hepes, 0.2% NaHCO3, 10% FCS, 0.5 mg/ml gentamicin, and 1% penicillin-streptomycin for 2 d before use (37°C, 2% CO2). The pRBCs to be assayed were fractionated on a Percoll gradient 19 to yield ∼95% late stage–infected RBCs, which were resuspended in binding medium (RPMI 1640, 25 mM Hepes, 25 μg/ml, pH 6.8). 1 ml of a 2% hematocrit suspension of the pRBCs was overlaid on the transfected cells and incubated at 37°C for 60 min with gentle rocking every now and then. The cells were washed three times with binding medium and stained with Giemsa. The number of pRBCs bound per 100 CHO or L cells was estimated counting a minimum of 500 cells for the determination of the binding capacity of the pRBCs.
Cloning and Expression of DBL1α, CIDR1α, and DBL2δ of FCR3S1.2var1 in E. coli.
The cloning and expression of DBL1α and the acidic terminal segment (ATS) were conducted as described 12. Gene fragments encoding CIDR1α (aa 516–822) and DBL2δ (aa 905–1307) were PCR amplified with primers (C1 5′-TCC AAC ATA AAG GTG GTA ATC AA-3′ and C2 5′-TGT CTT ACC ATC ACT TAT ACA A-3′ for CIDR1α; D4.1 5′-TCA CCG GAG TAC GAC CCA-3′ and D4.2 5′-ATT TTC TAC TTT ACA ATC CAC TTT-3′ for DBL2δ), cloned in the pGEX-4T plasmid (Amersham Pharmacia Biotech), and expressed in E. coli (BL21). The GST fusion proteins were expressed and purified according to the instructions of the manufacturer 12 27. The purity was determined by using SDS-PAGE and Western blot as described 12.
Binding of Recombinant DBL1α, CIDR1α, and DBL2δ to Receptors on Solid Phase.
The interaction of recombinant DBL1α-GST, CIDR1α-GST, and DBL2δ-GST with different receptors was first studied using a solid phase assay system. 100 μl of CD36, PECAM-1/CD31, IgM, or E-selectin was coated in ELISA plates (cat. no. 3455; Immulon) at a concentration of 5 μg/ml in NaHCO3 buffer (pH 9.5) overnight at 4°C. The plates were subsequently blocked for 1 h at room temperature with 3% BSA in PBS. 100 μl of serially double-diluted DBL1α-GST, CIDR1α-GST, DBL2δ-GST, or GST (100 to 0.75 μg/ml) was added to each well and incubated at room temperature for 60 min. Biotin-labeled anti-GST mAb (G-1160, IgG2b, 1:600 dilution; Sigma-Aldrich) and streptavidin–alkaline phosphatase (ALP) (1:1,000 dilution; Sigma-Aldrich) were then added to detect the fusion proteins after three washes in PBS-Tween. A final 100 μl of substrate buffer was added to each well after three washes in PBS-Tween, and each reaction was determined with an automatic ELISA reading system at OD 405 nm. Each experiment was repeated three times, and the final results shown are the means ± 2 SD. The background binding generated by GST alone was always negligible but deduced before the calculations. In the case of blood group antigen binding, the fusion proteins (DBL1α-GST, CIDR1α-GST, DBL2δ-GST, and GST; 10 μg/ml) were coated on ELISA plates as above. Biotinylated BSA coupled with the blood group A antigen or biotin-BSA were serially double diluted from 100 to 0.75 μg/ml and incubated with each fusion protein for 60 min at room temperature followed by three washes with PBS-Tween. 100 μl of substrate buffer was added to each well after a further incubation with streptavidin-ALP (1:1,000 dilution; Sigma-Aldrich) and three washes in PBS-Tween. The results were assessed as above. For testing heparin binding activity of the three domains, 100 μl of serially double-diluted DBL1α-GST, CIDR1α-GST, DBL2δ-GST, or GST (100 to 0.75 μg/ml) was incubated with 20 μl of heparin-sepharose (Amersham Pharmacia Biotech) for 30 min followed by three washes with PBS-Tween. The beads were transferred to an ELISA plate after further incubation with biotin-labeled anti-GST antibodies and streptavidin-ALP. The binding was assessed as above.
Binding of Recombinant DBL1α, CIDR2α, and DBL2δ to Cells Expressing CD36 or PECAM-1/CD31.
CHO cells, CHO-CD36, L cells, or L cell–PECAM-1/CD31 were cultured in normal medium for 2 d before the assays. The cells were detached with a rubber policeman and suspended in PBS. Serially double-diluted fusion proteins (DBL1α-GST, CIDR1α-GST, DBL2δ-GST, and GST) from 200 to 50 μg/ml were added to the cell suspensions (200 μl) and incubated at room temperature for 60 min. The cells were washed three times with PBS and subsequently stained with an Alexa 488 (Molecular Probes) labeled anti-GST mAb (G-1160, 1:100 dilution; Sigma-Aldrich) for 60 min. Surface fluorescence was studied under incident UV light as above after three washes in PBS.
The importance of glycosylation for the binding of the recombinant fusion proteins to CHO cells was also studied. Normal CHO cells and CHO-CD36 were cultured for 2 d with or without the addition of tunicamycin (1 μg/ml; see also reference 28) in the culture medium. The cells were detached with a rubber policeman and suspended in PBS. Some of the cells not treated with tunicamycin were further incubated with heparinase (8.5 × 10−3 IU/ml) for 3 h at 25°C. The cells were washed three times in PBS, and the binding assay was performed as above.
Cloning DBL1α, CIDR1α, and DBL2δ of FCR3S1.2var1 in pRE4 Vector and Transfection of COS-7 Cells.
For transient expression of FCR3S1.2var1 gene fragments encoding DBL-1, CIDR1α, and DBL2δ on the surface of COS-7 cells, the three domains were PCR amplified with the following three pairs of primers: DBL1α with DL-1.1 5′-ATC GAT CAG CTG CCG TGC AAA AAA GAT GGA A-3′ and DL-1.2 5′-ATC GAT GGG CCC TGA TAT TTC TTT TGT GTA TTT-3′; CIDR1α with CR-1 5′-ATC GAT CAG CTG ACT AGT GGT GCT AGT GGT A-3′ and CR-2 5′-ATC GAT GGG CCC GTC TTT TAT TGG CTT CCA TTC-3′; and DBL2δ with DL-4.1 5′-ATC GAT CAG CTG ACA CCA AGT GGT GAA CC-3′ and DL-4.2 5′-ATC GAT GGG CCC CCG ACA GAA ACT CTC TCC-3′. The PCR product of each amplification was precipitated with ethanol. Both the PCR products and the pRE4 vector were digested with ApaI and PuvII. The fragments were further purified after digestions from agarose using standard methods. 1 μl of the digested vector and each PCR fragment were mixed for ligation at room temperature for 4 h. 3 μl of each ligation solution was used to transform competent bacterial cells (Top10F′; Invitrogen). The bacterial clones with correct recombinant plasmids were determined by PCR amplification with two primers (Sig-1, 5′-GTG ATT TTG TTT GCT GTC ATA-3 and Tm-1 5′-CAG CAG GGT GCT CGT GTA-3′). The sequences of the three constructs were further determined by sequencing. The plasmids were isolated with a Plasmid Isolation kit (Bio-Rad Laboratories). 1 mg of each recombinant plasmid (DBL1α-pRE4, CIDR1α-pRE4, DBL2δ-pRE4, or the vector pRE4) was purified with the Plasmid Midiprep kit (Invitrogen). The plasmids were dissolved in 1 ml of sterile dH2O (1 μg/μl) and kept at −20°C for later use.
COS-7 cells from the American Tissue Culture Collection were routinely cultured in a normal DMEM (Life Technologies, Inc.) containing 10% heat-inactivated FCS. The cells were seeded on coverslips in 12-well culture plates overnight or until 50–60% confluence. 4 μg of the plasmid was mixed with 24 μg of liposome (Invitrogen) in 2 ml DMEM without serum and incubated for 5 min at room temperature. The COS cells were washed once with PBS, and the transfection solutions were applied to the cells immediately. After 4 h of transfection, the transfection solutions were removed and replaced with normal COS cell medium. The cells were cultured at 37°C in a CO2 incubator for 48 h. The expression of DBL1α, CIDR1α, and DBL2δ on the COS cell surface was confirmed through the surface immunofluorescence by using mAbs ID3 or DL6, which recognize a short sequence of herpes simplex virus glycoprotein D that is expressed by the vector on either side of the insert as described 29 30.
Binding of Soluble Receptors to Transfected COS-7 Cells.
Human myeloma IgM (Jackson ImmunoResearch Laboratories), recombinant CD36, and PECAM-1/CD31 were directly labeled with Alexa G-448 as above and kept at 4°C before use. 1 ml of different dilutions of the receptors (from 200 to 50 μg/ml in PBS) was incubated with COS cells transfected with the construct (DBL1α-pRE4, CIDR1α-pRE4, DBL2δ-pRE4, or pRE4) for 60 min. After the incubation, the cells were washed three times with either RPMI 1640 or PBS. The fluorescence was estimated with cells either on coverslips or in suspension.
Erythrocyte Binding to Transfected COS-7 Cells.
Aliquots of blood group A+ and O Rh+ erythrocytes were treated with heparinase as described 12. Both untreated and heparinase-treated RBCs were subsequently washed three times with RPMI 1640 and resuspended in malaria culture medium. 1 ml of a 2% erythrocyte suspension was added to the COS-7 cells transfected with DBL1α, CIDR1α, or DBL2δ or to untransfected COS cells and incubated at 37°C for 60 min as described 10 30. The cells were washed with binding buffer, and the rosetting rate was assessed as described 10.
Results
FCR3S1.2-infected Erythrocytes Bind Soluble Receptors.
To ascertain the adhesive profile of the pRBCs of FCR3S1.2, we studied the binding of various soluble fluorescence-labeled receptors to the live pRBC surface including the blood group A antigen, CD36, PECAM-1/CD31, heparin, and human IgM. Prominent binding of all of the receptor conjugates except soluble ICAM-1–fluorescein was seen to the pRBC surface (Fig. 1; heparin-FITC, not shown). The labeling varied in intensity between the different conjugates: the PECAM-1/CD31 and soluble CD36 fluorescence was the strongest, showing an even distribution all around the pRBC surface (Fig. 1). The adherence of all of the soluble receptors to the pRBCs of a sister clone (FCR3S1.6) 19 that lacks adhesive properties was also studied, but no or very weak binding was seen. These findings confirm the specificity of the assays and the panadhesive profile of this parasite clone, which is also summarized in Table (see also references 7, 11, 13, and 19).
Figure 1.
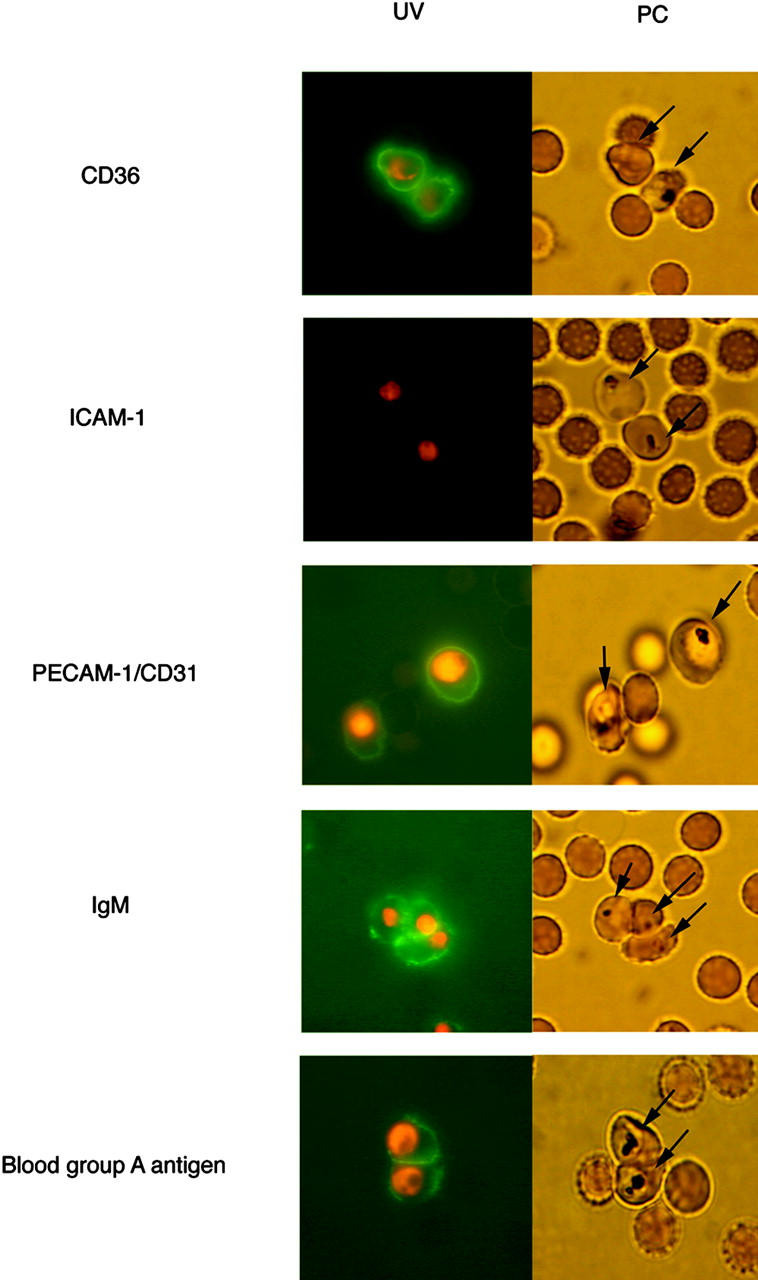
The binding of fluorescence-labeled soluble receptors to the surface of erythrocytes infected with FCR3S1.2. Fluorescence-labeled recombinant human CD36, ICAM-1, or PECAM-1/CD31 was directly incubated with the infected cells. The binding of human nonimmune IgM was detected by incubating the pRBCs with FITC-labeled rabbit anti–human Ig, whereas that of the blood group antigen A was by incubating the infected cells with an A trisaccharide coupled to biotinylated BSA and FITC-avidin. Infected (arrows) and uninfected RBCs are shown with phase–contrast (PC) and in parallel with UV light (UV). The binding shown was at a receptor concentration of 100 μg/ml, and the parasites were counterstained with ethidium bromide. The fluorescence rates were 65–70% (CD36), 0–5% (ICAM-1), 70–80% (PECAM-1/CD31), 75–85% (IgM), and 60–70% (blood group A). For further details, see Materials and Methods.
Table 1.
Adhesive Phenotypes of the Infected Erythrocytes of P. falciparum Clone FCR3S1.2
| CHO-CD36 binding | 400 ± 50 |
|---|---|
| CHO–ICAM-1 binding | 40 ± 12 |
| CHO binding | 6 ± 3 |
| L cell–PECAM-1/CD31 binding | 800 ± 80 |
| L cell binding | ≈5 |
| Thrombospondin | 0 |
| IgM binding | ≈90% |
| IgG binding | ≈20% |
| Blood group A antigen binding | ≈90% |
| Heparin-like GAG binding | ≈90% |
| Rosetting | 90 ± 5% |
| Autoagglutination | >40% |
Binding of Recombinant PfEMP1 to Multiple Receptors.
Domain-like recombinant fragments (DBLIα-GST, CIDR1α-GST, DBL2δ-GST, and GST as control) of the PfEMP1 of FCR3S1.2 were expressed and purified from E. coli and used to dissect the binding to different receptors or receptor proteins. The various fusion proteins were tested in ELISA assays for their capacity to adhere to the blood group A antigen, PECAM-1/CD31, CD36, E-selectin, heparin, and IgM (Fig. 2). We also studied the binding activities of recombinant DBL1α-GST, CIDR1α-GST, DBL2δ-GST, or GST to normal CHO cells (K1/CCL61), transfected CHO cells expressing CD36, normal L cells, or transfected L cells expressing PECAM-1/CD31 (Fig. 3). The binding of DBL1α, CIDR1α, and DBL2δ to the panel of receptors was further examined using a COS cell expression system in which the PfEMP1 domains cloned into the pRE4 vector were expressed at the surface of the COS cells (Fig. 4).
Figure 2.
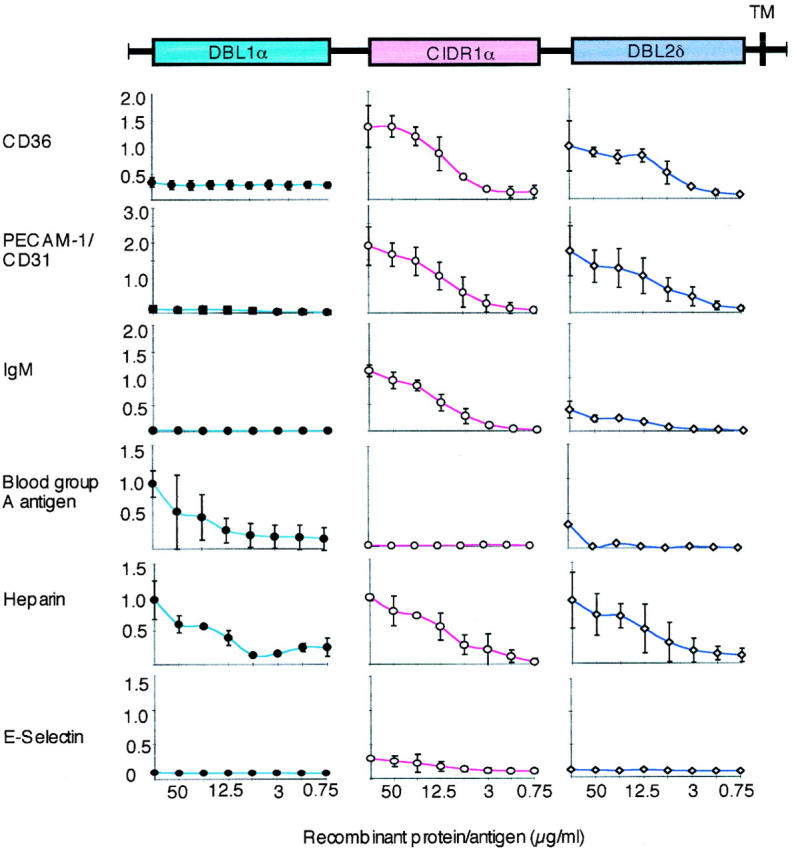
The interaction of the recombinant PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) as GST fusion proteins with different receptors (CD36, PECAM-1/CD31, blood group antigen A, heparin, and E-selectin) or receptor proteins (IgM) bound to solid phase as measured by ELISA. The domain-like primary structure of FCR3S1.2var1-PfEMP1 is shown at the top of the figure (colored boxes). Each receptor studied (left) for interaction with a GST fusion protein is shown as a separate curve (DBL1α, blue •; CIDR1α, pink ○; DBL2δ, violet ⋄). The ranges of absorption values are shown on the y-axis and the concentrations (in μg/ml) on the x-axis. The results are the mean of at least three separate experiments ± 2 SD. For further details, see Materials and Methods.
Figure 3.

The interaction of the recombinant PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) as GST fusion proteins with receptors expressed on normal or transfected cells (some of the cells were treated with heparinase to remove HS from the cell surface) visualized by a fluorescence-labeled anti-GST antibody. The results are those of at least three separate experiments. 95% untreated and 5% treated CHO cells (CHO and CHO-CD36) bind DBL1α, respectively. More than 80% untreated CHO-CD36 cells bind CIDR1α, whereas <30% CHO-CD36 bind DBL2δ. For further details, see Materials and Methods.
Figure 4.

The binding of fluorescence-labeled receptors, receptor proteins, or erythrocytes to COS-7 cells transiently transfected with the different PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) of FCR3S1.2var1. The transfected cell type used is shown at the top of the figure. The fluorescence-labeled protein or erythrocyte used is shown in the right hand corner of each photograph. The results are those of at least three separate experiments. For further details, see Materials and Methods.
CIDR1α Mediates Binding to CD36.
Both the CIDR1α and the DBL2δ domains bound in an ELISA assay in which human CD36 was coated on the plates whereas the other fusion proteins (DBL1α-GST and GST) were not (Fig. 2). The CIDR1α-GST also bound to CHO cells expressing CD36, whereas binding to DBL2δ-GST was weak (Fig. 3) and neither DBL1α-GST nor GST bound to these cells when heparinase treated (Fig. 3, and data not shown). The binding of CD36 to the PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) was further studied using the COS cell expression system. COS cells expressing CIDR1α specifically bound CD36 (Fig. 4 and Table ), whereas neither COS-DBL1α, COS-DBL2δ, nor the untransfected COS cells showed any binding to CD36. Thus, it seems that CIDR1α is the critical PfEMP1 domain involved in CD36 binding but that DBL2δ could also play a part.
Table 2.
Binding of Different Receptors and RBCs to var Fragment–transfected COS-7 Cells
| COS-DBL1α | COS-CIDR1α | COS-DBL2δ | |
|---|---|---|---|
| Transfection efficiency | 1–3% | 6–8% | 3–6% |
| IgM | − | + | − |
| CD36 | − | + | − |
| PECAM-1/CD31 | − | + | + |
| O Rh+ RBCs | 50 ± 15 | 0 | 0 |
| O Rh+ RBCs (heparinase treated) | 0 | ND | ND |
| A+ RBCs | 45 ± 20 | 0 | 0 |
| A+ RBCs (heparinase treated) | 40 ± 15 | ND | ND |
CIDR1α and DBL2δ Bind to PECAM-1/CD31.
Both the CIDR1α and the DBL2δ domains bound in an ELISA assay in which human PECAM-1/CD31 was coated on the plates, whereas the other fusion proteins (DBLIα-GST and GST) were not (Fig. 2). The CIDR1α-GST and DBL2δ-GST both also bound to L cells (>60% fluorescence rate) expressing PECAM-1/CD31, whereas neither DBL1α-GST nor GST bound to these cells (Fig. 3, and data not shown). Again, the binding of PECAM-1/CD31 to the PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) was further studied using the COS cell expression system. COS cells expressing CIDR1α or DBL2δ did specifically bind PECAM-1/CD31 (Fig. 4, and Table ) whereas neither COS-DBL1α nor the untransfected COS cells showed any binding to PECAM-1/CD31.
To delineate the relative importance of either CIDR1α or DBL2δ in adherence of PECAM-1/CD31 to the pRBCs, we investigated the capacity of the recombinants (DBL1α-GST, CIDR1α-GST, DBL2δ-GST, and GST) to abrogate the binding of fluorescence-labeled sPECAM-1/CD31. Inhibition was obtained with both the CIDR1α and the DBL2δ domains (40% inhibition at 200 μg/ml for CIDR1α; 60% inhibition at 200 μg/ml when CIDR1α and DBL2δ were mixed at a ratio of 1:1, 100 μg/ml each), whereas no inhibition was seen with either DBL1α-GST or GST. The CIDR1α domain was somewhat more efficient in binding inhibition than DBL2δ (data not shown). Thus, it seems that both CIDR1α and DBL2δ are involved in PECAM-1/CD31 binding.
CIDR1α Binds IgM.
The binding of the three fusion proteins (DBL1α-GST, CIDR1α-GST, and DBL2δ-GST) and GST to ELISA plates precoated with human IgM clearly showed that the CIDR1α region bound to this serum protein (Fig. 2). Neither DBL1α, DBL2δ, nor GST had the capacity to bind IgM (Fig. 2, and data not shown). Further, in complementary experiments we similarly found that CIDR1α-transfected COS cells did specifically bind IgM whereas neither COS-DBL1α, COS-DBL2δ, nor the untransfected COS cells were able to bind immunoglobulins (Fig. 4, and Table ). Taken together, the data suggest that CIDR1α is the PfEMP1 domain that binds normal nonimmune IgM.
DBL1α Mediates Rosetting through an HS-like GAG and the Blood Group A Antigen.
Potential GAG-binding motifs 31 were identified in all three PfEMP1 domains of FCR3S1.2var1 12 and, as expected, all three GST fusion proteins (DBL1α-GST, CIDR1α-GST, and DBL2δ-GST) bound dose-dependently to heparin-sepharose (Fig. 2) whereas no binding was seen with GST alone (data not shown). To confirm these findings, we studied the interaction of recombinant DBL1α-GST, CIDR1α-GST, DBL2δ-GST, or GST alone to normal CHO cells, transfected CHO cells expressing CD36 at the cell surface, or transfected L cells expressing PECAM-1/CD31. Surprisingly, DBL1α-GST, but not the other recombinant proteins (CIDR1α-GST, DBL2δ-GST, or GST), bound to all CHO cells including the nontransfected ones. This suggested that DBL1α-GST might adhere to HS-like GAGs. Further proof of this specificity was generated, as it was found that the binding activity was abrogated when the CHO cells were cultured in the presence of tunicamycin (1 μg/ml) for 2 d or when the cells were treated with heparinase (8.5 × 10−3 IU/ml) before the binding assays (Fig. 3). The binding of DBL1α to the CHO cells is therefore most likely to be mediated via HS-like molecules on the CHO cell surface. The involvement of DBL1α, CIDR1α, or DBL2δ in HS-mediated binding at the cellular level was further studied using a COS cell expression system. The COS-DBL1α did bind blood group O RBCs, whereas the other transfectants did not (COS-CIDR1α, COS-DBL2δ, and COS; Table and Fig. 4). The binding of blood group O RBCs to COS-DBL1α could again be abrogated by the pretreatment of the erythrocytes with heparinase (8.5 × 10−3 IU/ml). Taken together, the data confirm our findings that DBL1α has high affinity for an HS-like GAG expressed on the erythrocyte surface and suggest that the other two domains, CIDR1α and DBL2δ, are less involved in this interaction. Recombinant DBL1α-GST also bound dose-dependently to the trisaccharide GalNAc-Gal-Fuc (blood group A antigen linked to BSA), whereas the other two PfEMP1 domains (CIDR1α-GST and DBL2δ-GST; Fig. 2) and GST alone did not (data not shown). COS cells transiently transfected with DBL1α bound both blood group O and A erythrocytes, whereas neither COS-CIDR1α– nor COS-DBL2δ–expressing cells or the untransfected COS cells did so (Table and Fig. 4). Importantly, while the binding of blood group O RBCs to COS-DBL1α was abrogated when the erythrocytes were pretreated with heparinase (8.5 × 10−3 IU/ml), the binding of blood group A RBCs to COS-DBL1α still remained after heparinase treatment even though the number of RBCs bound to the COS-DBL1α–expressing cells was somewhat reduced (Table ). These data show that DBL1α has affinity for the blood group A antigen.
Discussion
The data presented in this report demonstrate that P. falciparum–infected erythrocytes may bind to a large number of diverse receptors through a single species of PfEMP1. The expression of PfEMP1 by FCR3S1.2 at the infected RBC surface was previously established by both surface iodination and immunoprecipitation using specific anti-PfEMP1 antibodies 12 19 27. A PfEMP1 polypeptide with a molecular mass of ∼280 kD was identified. Immunoprecipitation and transcript analysis of single cells further indicated that the parasite expresses only one species of PfEMP1 at the infected erythrocyte surface 27, a fact that has been repeatedly confirmed throughout this study. Antibodies raised to the FCR3S1.2var1 DBL1α domain have been found to stain live infected erythrocytes, confirming that it is var1 that encodes the PfEMP1 of FCR3S1.2 (data not shown). Mild trypsinization of infected RBCs of FCR3S1.2 readily deleted the PfEMP1 from the cell surface, at least the iodinated portion, and the adhesive events incurred by this parasite 32. We have also found, in recent experiments not presented here, that immunoglobulins, heparin, or blood group A can selectively precipitate radioiodinated PfEMP1 from FCR3S1.2. This suggests that it is PfEMP1 that mediates the major binding events of this parasite.
The binding of DBL1α to GAGs is structurally specific for heparin or HS/HS-like GAGs and dependent on a 2-N-sulfated glucosamine, as is the rosetting binding of intact cells 13. All three domains (DBL1α, CIDR1α, and DBL2δ), as expected, were found to bind to heparin-sepharose, but it is not likely that the CIDR1α and the DBL2δ domains avidly participate in the cellular interactions leading to rosette formation. Arguing for this are the findings that only the DBL1α domain and not CIDR1α or DBL2δ bound to HS-like GAGs on normal CHO cells. Further, only the DBL1α domain and not CIDR1α or DBL2δ supported binding of erythrocytes when the domains were expressed at the surface of COS cells. Taken together, the data suggest a prominent role for DBL1α and an HS-like GAG in the formation of stable rosettes.
Individuals of the blood group A antigen type have been found to come down with severe disease more frequently than those of other ABO blood groups 33. Importantly, the blood group A antigen has also been discovered to be a receptor mediating rosetting of both fresh isolates and laboratory-adapted strains 8 34. Blood group antigen–dependent rosetting has been found to be as sensitive to trypsin as is PfEMP1 19 and, further, PfEMP1 encoded by FCR3S1.2var1 can be precipitated from the parasite using a polymerized blood group A antigen. Here, with the help of four additional experimental systems, we provide evidence that it is the DBL1α domain of PfEMP1 that mediates binding to the blood group antigens.
The infected erythrocytes of FCR3S1.2 also display adhesive features other than rosetting. This motivated us to check all three PfEMP1 domains for binding to serum proteins and endothelial receptors such as IgM, CD36, PECAM-1/CD31, and E-selectin. Surprisingly, DBL1α did not adhere to any of these receptors. On the other hand, dramatic binding was seen with CIDR1α to CD36, PECAM-1/CD31, and IgM, whereas DBL2δ had PECAM-1/CD31 as the main specificity. Not one of the fusion proteins bound to E-selectin (Fig. 2). These findings are the condensation of results of a large number of in vitro assays. Still, some uncertainty remains on the importance for adherence of DBL2δ to CD36, as weak binding was seen in both the solid phase and in the CHO-CD36 assay, whereas no binding of CD36 was detected to DBL2δ-transfected COS cells. Baruch et al. 24 have shown that CIDR1α binds to CD36 via a peptide–peptide interaction involving a degenerate CIDR1α sequence containing multiple conserved cysteine residues, a sequence which is also present in the CIDR1α of FCR3S1.2var1 PfEMP1. Thus, it seems likely that the CIDR1α domain plays a major role in endothelial binding to CD36. When we tested the binding of the three domains (DBL1α, CIDR1α, and DBL2δ) to ICAM-1, some affinity for CIDR1α was also observed but it was weak (data not shown). Yet it is tempting to suggest that CIDR1α has an affinity for a structure present in the members of the immunoglobulin superfamily studied (IgM and PECAM-1/CD31) but a separate binding site for CD36. The details of the adhesive interactions of CIDR1α are complex, and the relative importance of the interactions characterized could very well vary in different situations and at different time points depending on the availability of each receptor at each vascular location in the same host.
In conclusion, the data here show that three PfEMP1 domains (DBL1α, CIDR1α, and DBL2δ) of one PfEMP1 species mediate multiple independent interactions with a diverse set of host receptors, as summarized in Fig. 5. The findings provide a molecular explanation of the multiadhesive phenotype of P. falciparum and suggest its importance in the development of severe malaria. The DBL1α-CIDR1α head structure may also prove to be an important vaccine candidate, particularly if antibodies directed to conserved determinants in DBL1α-CIDR1α are common in those protected against severe malaria.
Figure 5.

Schematic summary of the different binding activities of the DBL1α, CIDR1α, and DBL2δ domains of PfEMP1 encoded by FCR3S1.2var1. P. falciparum expresses PfEMP1 molecules (here shown corkscrew-like) on a knob-like structure at the infected erythrocyte surface. The DBL1α domain participates in rosetting through binding to an HS-like GAG and to the blood group A antigen. The CIDR1α domain binds to CD36 and to members of the immunoglobulin superfamily, including IgM and PECAM-1/CD31, whereas the DBL2δ domain binds mainly to PECAM-1/CD31.
Acknowledgments
We are very grateful to Drs. Gary Cohen and Roselyn Eisenberg, University of Pennsylvania, Philadelphia, PA, for providing the plasmid pRE4 and the mAbs, ID3 and DL6, which were essential for this work. We thank Dr. William A. Muller for the gift of L cells transfected with PECAM-1/CD31. We thank Fredrik Pettersson for help with the thrombospondin assays, and are very grateful to Dr. A. Rowe for sharing her COS transformation protocols.
The project was funded by the Swedish Cancer Society (Cancerfonden), the United Nations Development Programme/World Bank World Health Organization Special Programme for Research and Training in Tropical Diseases (TDR), an INCO-DC grant from the European Commission, and the Swedish Medical Research Council.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); ALP, alkaline phosphatase; CHO, Chinese hamster ovary; CIDR, cysteine-rich interdomain region; DBL, Duffy binding–like; GAG, glycosaminoglycan; GST, glutathione S-transferase; HS, heparan sulfate; ICAM-1, intracellular adhesion molecule 1; PECAM-1, platelet-endothelial cell adhesion molecule 1; PfEMP1, P. falciparum erythrocyte membrane protein 1; pRBC, parasited RBC; RT, reverse transcriptase.
References
- Miller L.H., Good M.F., Milon G. Malaria pathogenesis. Science. 1994;264:1878–1883. doi: 10.1126/science.8009217. [DOI] [PubMed] [Google Scholar]
- Barnwell J.W., Asch A.S., Nachman R.L., Yamaya M., Aikawa M., Ingravallo P. A human 88-kD membrane glucoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum–infected erythrocytes. J. Clin. Invest. 1989;84:77–87. doi: 10.1172/JCI114234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendt A.R., Simmons D.L., Tansey J., Newbold C.I., Marsh K. Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum . Nature. 1989;341:57–59. doi: 10.1038/341057a0. [DOI] [PubMed] [Google Scholar]
- Robert C., Pouvelle B., Meyer P., Muanza K., Fujioka H., Aikawa M., Scherf A., Gysin J. Chondroitin-4-sulfate (proteoglycan), a receptor for Plasmodium falciparum-erythrocyte adhesion. Res. Immunol. 1995;146:383–393. doi: 10.1016/0923-2494(96)81042-x. [DOI] [PubMed] [Google Scholar]
- Rogerson S.J., Chaiyaroj C.S., Ng K., Reeder J.C., Brown G.V. Chondroitin sulfate A is a cell surface receptor for Plasmodium falciparum–infected erythrocytes. J. Exp. Med. 1995;182:15–20. doi: 10.1084/jem.182.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold I.C., Warn P., Black G., Berendt A., Craig A., Snow B., Msobo M., Peshu N., Marsh K. Receptor-specific adhesion and clinical disease in Plasmodium falciparum . Am. J. Trop. Med. Hyg. 1997;57:389–398. doi: 10.4269/ajtmh.1997.57.389. [DOI] [PubMed] [Google Scholar]
- Treutiger C.J., Heddini A., Fernandez V., Muller W.A., Wahlgren M. PECAM-1/CD31, an endothelial receptor for binding Plasmodium falciparum-infected erythrocytes. Nat. Med. 1997;3:1405–1408. doi: 10.1038/nm1297-1405. [DOI] [PubMed] [Google Scholar]
- Carlson J., Wahlgren M. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. J. Exp. Med. 1992;176:1311–1317. doi: 10.1084/jem.176.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handunnetti S.M., Schravendijk M.R., Hasler T., Barnwell J.W., Greenwalt D.E., Howard R.J. Involvement of CD36 on erythrocytes as a rosetting receptor for Plasmodium falciparum-infected erythrocytes. Blood. 1992;80:2097–2104. [PubMed] [Google Scholar]
- Rowe A.J., Moulds J.M., Newbold C.I., Miller L.H. P. falciparum rosetting is mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature. 1997;388:292–295. doi: 10.1038/40888. [DOI] [PubMed] [Google Scholar]
- Scholander C., Treutiger C.J., Hultenby K., Wahlgren M. Novel fibrillar structure confers adhesive property to malaria-infected erythrocytes. Nat. Med. 1996;2:204–208. doi: 10.1038/nm0296-204. [DOI] [PubMed] [Google Scholar]
- Chen Q., Barragan A., Fernandez V., Sundström A., Schlichtherle M., Sahlén A., Carlson J., Datta S., Wahlgren M. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum . J. Exp. Med. 1998;187:15–23. doi: 10.1084/jem.187.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragan A., Spillmann D., Wahlgren M., Carlson J. Erythrocyte glycans as Plasmodium falciparum rosetting receptorsmolecular background of strain specific rosette disruption by glycosaminoglycans and sulfated glycoconjugates. Exp. Parasitol. 1999;91:133–143. doi: 10.1006/expr.1998.4349. [DOI] [PubMed] [Google Scholar]
- Carlson J., Helmby H., Hill A.V.S., Brewster D., Greenwood B.M., Wahlgren M. Human cerebral malariaassociation with erythrocyte rosetting and lack of anti-rosetting antibodies. Lancet. 1990;336:1457–1460. doi: 10.1016/0140-6736(90)93174-n. [DOI] [PubMed] [Google Scholar]
- Treutiger C.J., Hedlund I., Helmby H., Carlson J., Jepson A., Twumasi P., Kwiatkowski D., Greenwood B.M., Wahlgren M. Rosette formation in Plasmodium falciparum isolates and anti-rosette activity of sera from Gambians with cerebral or uncomplicated malaria. Am. J. Trop. Med. Hyg. 1992;46:503–510. doi: 10.4269/ajtmh.1992.46.503. [DOI] [PubMed] [Google Scholar]
- Ringwald P., Lepers J.P., Le Bras J., Rakotomalala C., Rabodonirina M., Vuillez J.P., Razanamparany M., Peyron F., Roux J. Parasite virulence factors during falciparum malariarosetting, cytoadherence, and modulation of cytoadherence by cytokines. Infect. Immun. 1992;61:5198–5204. doi: 10.1128/iai.61.12.5198-5204.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe A.J., Obeiro J., Newbold C.I., Marsh K. Plasmodium falciparum rosetting is associated with malaria severity in Kenya. Infect. Immun. 1995;63:2323–2326. doi: 10.1128/iai.63.6.2323-2326.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udomsangpetch R., Taylor B.J., Looareesuwan S., White N.J., Elliott J.F., Ho M. Receptor specificity of clinical Plasmodium falciparum isolatesnon-adherence to cell-bound E-selectin and vascular cell adhesion molecule-1. Blood. 1996;88:2754–2760. [PubMed] [Google Scholar]
- Fernandez V., Treutiger C.J., Nash G.B., Wahlgren M. Multiple adhesive phenotypes linked to rosetting-binding of erythrocytes in Plasmodium falciparum malaria. Infect. Immun. 1998;66:2969–2975. doi: 10.1128/iai.66.6.2969-2975.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R.J. Malarial proteins at the membrane of Plasmodium falciparum-infected erythrocytes and their involvement in cytoadherence to endothelial cells. Prog. Allergy. 1988;41:98–147. doi: 10.1159/000415221. [DOI] [PubMed] [Google Scholar]
- Su X.Z., Heatwole V.M., Wertheimer S.P., Guinet F., Herrfeldt J.A., Peterson D.S., Ravetch J.A., Wellems T.E. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995;82:89–99. doi: 10.1016/0092-8674(95)90055-1. [DOI] [PubMed] [Google Scholar]
- Baruch D.I., Gormley J.A., Ma C., Howard R.J., Pasloske B.L. Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intracellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA. 1996;93:3497–3502. doi: 10.1073/pnas.93.8.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch D.I., Pasloske B.L., Singh H.B., Bi X., Ma C., Feldman M., Taraschi T.F., Howard R.J. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995;82:77–87. doi: 10.1016/0092-8674(95)90054-3. [DOI] [PubMed] [Google Scholar]
- Baruch D.I., Ma C., Singh H.B., Bi X., Pasloske B.L., Howard R.J. Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36conserved function with variant sequence. Blood. 1997;90:3766–3775. [PubMed] [Google Scholar]
- Udomsangpetch R., Wåhlin B., Carlson J., Berzins K., Torii M., Aikawa M., Perlmann P., Wahlgren M. Plasmodium falciparum–infected erythrocytes form spontaneous erythrocyte rosettes. J. Exp. Med. 1989;169:1835–1840. doi: 10.1084/jem.169.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce S.F., Roy P., Nicholson A.C., Hajjar D.P., Febbraio M., Silverstein P.L. Recombinant glutathione S-transferase/CD36 fusion proteins define an oxidized low density lipoprotein-binding domain. J. Biol. Chem. 1998;273:34875–34881. doi: 10.1074/jbc.273.52.34875. [DOI] [PubMed] [Google Scholar]
- Chen Q., Fernandez V., Sundström A., Schlichtherle M., Datta S., Hagblom P., Wahlgren M. Developmental selection of var gene expression in Plasmodium falciparum . Nature. 1998;394:392–395. doi: 10.1038/28660. [DOI] [PubMed] [Google Scholar]
- Duksin D., Mahoney W.C. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J. Biol. Chem. 1982;257:3105–3109. [PubMed] [Google Scholar]
- Cohen G.H., Wilcox W.C., Sodora D.L., Long D., Levin J.Z., Eisenberg R.J. Expression of herpes simplex virus type 1 glycoprotein D deletion mutants in mammalian cells. J. Virol. 1988;62:1932–1940. doi: 10.1128/jvi.62.6.1932-1940.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitnis C.E., Miller L.H. Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 1994;180:497–506. doi: 10.1084/jem.180.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin A.D., Weintraub H.J.R. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Fernandez V., Hommel M., Chen Q., Hagblom P., Wahlgren M. Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum–infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J. Exp. Med. 1999;190:1393–1404. doi: 10.1084/jem.190.10.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher P.R., Boone P. Severe malaria associated with blood group. Am. J. Trop. Med. Hyg. 1993;58:122–123. doi: 10.4269/ajtmh.1998.58.122. [DOI] [PubMed] [Google Scholar]
- Udomsangpetch R., Todd J., Carlson J., Greenwood B.M. The effects of haemoglobin genotype and of ABO blood group on the formation of rosettes by Plasmodium falciparum infected red blood cells. Am. J. Trop. Med. Hyg. 1993;48:149–153. doi: 10.4269/ajtmh.1993.48.149. [DOI] [PubMed] [Google Scholar]