

Abstract
Hypoxia is a common cause of cell death and is implicated in many disease processes including stroke and chronic degenerative disorders. In response to hypoxia, cells express a variety of genes, which allow adaptation to altered metabolic demands, decreased oxygen demands, and the removal of irreversibly damaged cells. Using polymerase chain reaction–based suppression subtractive hybridization to find genes that are differentially expressed in hypoxia, we identified the BH3-only Bcl-2 family protein Noxa. Noxa is a candidate molecule mediating p53-induced apoptosis. We show that Noxa promoter responds directly to hypoxia via hypoxia-inducible factor (HIF)-1α. Suppression of Noxa expression by antisense oligonucleotides rescued cells from hypoxia-induced cell death and decreased infarction volumes in an animal model of ischemia. Further, we show that reactive oxygen species and resultant cytochrome c release participate in Noxa-mediated hypoxic cell death. Altogether, our results show that Noxa is induced by HIF-1α and mediates hypoxic cell death.
Keywords: Noxa, HIF-1α, hypoxia, apoptosis, ROS
Introduction
Hypoxia is a well-known cause of cell injury with implications in many pathologic conditions including cerebral ischemia, myocardial infarction, and chronic degenerative disorders (1). In response to hypoxia, cells express a variety of gene products such as erythropoietin, vascular endothelial growth factor, glycolytic enzymes, and p27 to satisfy altered metabolic demands or diminish proliferative rates (2–4). However, under the extreme hypoxic conditions beyond the cellular adaptive capability, a number of biochemical mechanisms involved in necrotic or apoptotic cell death pathways are activated to remove irreversibly injured cells (2).
To date, transcriptional factor hypoxia-inducible factor 1 α (HIF-1α), tumor suppressor p53, and bcl-2 family proteins are known to play fundamental roles in adaptive or death process in response to hypoxia (5–7). HIF-1α is induced, stabilized, and translocated to the nucleus to regulate transcription of a variety of genes involved in the adaptive response such as increased O2 delivery and angiogenesis under situation of oxygen deficiency (8, 9). Although HIF-1α participates largely in adaptive process to hypoxia, paradoxically it also mediates hypoxic cell death via the interaction with p53 or modulation of effector expression (10, 11). Tumor suppressor gene p53, stabilized by HIF-1α, facilitates growth arrest or cell death under hypoxic condition via (a) its downstream genes such as p21/Waf1/Cip1 and bax or (b) direct translocation to mitochondria to induce mitochondrial permeability transition (12–14). Recently, it has been reported that Bcl-2 family proteins are involved in cellular hypoxic injury. For example, BNip3, a proapoptotic BH3-only member of Bcl-2 family proteins, is under the regulation of HIF-1α and induces apoptotic cell death (11, 15, 16). However, despite central roles of these molecules, molecular mechanisms of cell death after hypoxic injury and their downstream effector molecules remain poorly understood.
We have been attempting to identify novel proapoptotic genes mediating hypoxic cell death using PCR-based suppression subtractive hybridization. Among the differentially expressed genes in response to hypoxia we found Noxa. Noxa is a proapoptotic member of BH3-only Bcl-2 family proteins and its expression is dependent on p53 through p53 consensus binding sequence (p53-CBS) on Noxa promoter (17). However, little is known about functions of Noxa in physiologic status or roles in the process of cell injury. Here we report that Noxa is transcriptionally activated in hypoxic condition and mediates p53-independent hypoxic cell death. Its up-regulation of expression in hypoxia depends on HIF-1α via hypoxia-responsive element (HRE) on Noxa promoter. In addition, we show that Noxa mediates hypoxic cell death by generation of reactive oxygen species (ROS). Inhibition of endogenous Noxa expression by antisense (AS) oligonucleotide suppressed cell death and reduced infarction volumes induced by hypoxia in ischemic animal model. Based on our results, we propose that Noxa is another member of BH3-only Bcl-2 family proteins that responds to hypoxia and mediates hypoxic cellular injury.
Materials and Methods
Cell Culture, Hypoxia Condition, Caspase-3 Assay, and Cell Death Analysis.
SK-N-MC, Saos-2, and H719 cells were cultured in RPMI 1640 medium under 5% CO2. For hypoxic condition, cells in degassed serum- and glucose-deficient medium were transferred to a hypoxic chamber with 0.5% O2/5% CO2/94.5% N2. Caspase-3 activity was measured using the Colorimetric Caspase-3 Assay Kit (Calbiochem) according to the manufacturer's instructions. Apoptotic cells, exhibiting altered nuclear morphology after DAPI staining, were enumerated manually as previously described (18) or counted using trypan blue exclusion method.
Subtractive Hybridization.
Subtractive hybridization was performed with a PCR-Select cDNA Subtraction Kit (CLONTECH Laboratories, Inc.) according to the manufacturer's protocol. In brief, subtractive cDNA library was prepared with poly(A)+ RNAs purified from SK-N-MC cells incubated under normoxic (20% O2; driver) and hypoxic (0.5% O2; tester) conditions for 3 h. The tester cDNAs were digested with RsaI and linked with adaptor oligonucleotides. The tester cDNA was hybridized with an excess of driver cDNA digested with RsaI (the first hybridization) and subtracted single-stranded cDNA was converted to double-stranded DNA by the second hybridization with another adaptor-linked first hybridization. The subtracted cDNA was amplified by PCR with the primer set located in adapters and inserted into TA vector (CLONTECH Laboratories, Inc.).
Plasmid Construction and Transient Transfection.
DNA encoding human Noxa, p53, or HIF-1α was amplified by PCR from human kidney cDNA library (CLONTECH Laboratories, Inc.) and subcloned into the pcDNA3.1 or TA vector tagged with myc/V5 (Invitrogen). Cells were transfected with expression plasmid using Transfast Reagent (Promega).
Luciferase Assay.
The luciferase plasmid used was pGL2 (Promega). Four fragments of the Noxa promoter sequence were amplified by PCR from human kidney genomic DNA using the KpnI site–containing upstream primer, F1 (5′-GGTACCTTGA GACAGAGTCTTATT-3′), F2 (5′-GGTACCGACGGGGTTTCACCATATT-3′), F3 (5′-GGTACCTTTCTGCACCCGTAAAGTAG-3′), and F4 (5′-GGTACCCCACTCCCAGCTCCCGC-3′) primers, respectively, together with a single downstream primer R1 (5′-AAGCTTTCAGCTACAGAGCCCGGG-3′) containing HindIII site. Each Noxa promoter fragment was digested with both KpnI and HindIII and it was in-frame cloned into pGL2 luciferase reporter vector to generate pGL2-Noxa-1, pGL2-Noxa-2, pGL2-Noxa-3, and pGL2-Noxa-4, in order. For pGL2-Noxa-5 construct, a 256-bp fragment ranging from −1,120 to −1,375 was amplified by PCR using sense (SE; 5′-GGTACCGACGGGGTTTCACCATATT-3′) and AS (5′-ACTGAGGTTTGTAGAAAATT-3′) primers and introduced into KpnI and HindIII site of pGL2Basic. Mutant form of pGL2-Noxa-5 was constructed by changing core sequence of HRE (CGTG) to GCAC using splicing overlap extension method (19). Expression vector carrying HIF-1α was cotransfected with luciferase reporter constructs into p53-deleted Saos-2 cells by calcium phosphate precipitation method. Luciferase activity was measured on samples containing equivalent amounts of protein using luminometer and luciferase assay reagents (Promega).
Electrophoretic Mobility Shift Assay (EMSA) and Supershift Analysis.
EMSA was performed on nuclear extracts from Saos-2 cells. The extract preparation and binding reaction were performed as previously described (20). The double-stranded oligonucleotides used included (a) commercially available consensus HIF-1α gel shift oligonucleotide 5′-TCTGTACGTGACCACACTCACCTC-3′ (Santa Cruz Biotechnology, Inc.) for positive control, (b) putative HIF-1α binding site of Noxa promoter 5′-ATTACAGGCGTGAGCCACCGCGT-3′, and (c) mutant Noxa oligonucleotide 5′-ATTACAGGGCACAGCCACCGCGT-3′. For supershift analysis, 1 μg monoclonal antibody against HIF-1α (Santa Cruz Biotechnology, Inc.) or isotype control antibody (Sigma-Aldrich) was added to the reaction mixture before the addition of labeled oligonucleotides. For the binding competition experiments, unlabeled oligonucleotides were added to the reaction mixture in 100-fold excess.
Semiquantitative RT-PCR Analysis.
1 μg total RNA extracted from normoxia- or hypoxia-stimulated cells was converted to cDNA. The RT-PCR exponential phase was determined on 20–32 cycles to allow semiquantitative comparisons. PCR regimen for Noxa involved 94°C, 5 min initial denaturation step, followed by 32 cycles at 94°C for 30 s, 58°C for 45 s, and 72°C for 45 s on a Gene Amp PCR system 9700 (PerkinElmer). The resulting PCR products were separated in 2% agarose gels and visualized by ethidium bromide staining. For semiquantitative evaluation, densitometric analysis (Bio-Rad Laboratories) was performed.
AS Oligonucleotide.
To inhibit expression of endogenous Noxa, SE (5′-GCCGCCGTGGCCGCCTCTAC-3′) and AS (5′-CATCTCCGCCGGTGCCGCCG-3′) oligonucleotides were delivered by streptolysin-O (SL-O) technique as previously described (21). In brief, after washing in serum-free RPMI medium, 106 cells were exposed to dithiothreitol-activated SL-O at 20 μmol/liter SE or AS oligonucleotide in serum-free RPMI. After incubation of 10 min at 37°C, complete medium was used to reseal the cells.
Immunocytochemistry.
Saos-2 cells were fixed with 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100. After blocking with 3% BSA in PBS, the cells were incubated with anti-Noxa (Zymed Laboratories) or anti–cytochrome c (BD Biosciences) antibody for 2 h at room temperature. These antibodies were stained with secondary antibody conjugated with FITC or Texas Red and viewed using a Nikon E800 microscope equipped with a cooled CCD camera.
Cell Fractionation and Immunoblot Analysis.
The harvested cells were washed and suspended in hypotonic solution (10 mM HBSS, 10 mM MgCl2, 42 mM KCl) for 5 min on ice, passed through a 30-gauge needle, and centrifuged at 800 g for 10 min at 4°C. The supernatants were centrifuged at 150,000 g for 1 h. The mitochondrial fraction (pellet) and cytosolic fraction (supernatant) were collected separately. The proteins were separated using 15% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. The blots were incubated with anti-Noxa or anti–cytochrome c antibody followed by enhanced chemiluminescence–based detection (Amersham Biosciences). Densitometric analysis (Bio-Rad Laboratories) was performed for quantitative evaluation.
Focal Cerebral Ischemia in Rat.
Reversible focal cerebral ischemia was performed in male Wistar rats weighing 300–350 g by occluding middle cerebral artery for 2 h as previously described (22). After reperfusion, rats were killed at indicated time points by halothane overdose. Brains were removed, cut into eight coronal slices, and stained with triphenyl tetrazolium chloride. Infarct volume was calculated after measuring the infarct areas on coronal brain sections as previously described (23). Noxa AS (5′-CATGTTGTTATCCTCCAG-3′) and SE (5′-GACCTCCTATTGTTGTAC-3′) oligonucleotides (2 μg in 1 μl) were injected into the right lateral ventricle 4 h before the onset of ischemia.
In Situ Hybridization.
cRNA probes (SE or AS) were prepared by in vitro transcription using T7 or SP6 polymerase from the coding region of rat Noxa cDNA (sequence data are available from GenBank/EMBL/DDBJ under accession no. XM_225860) subcloned into pGEM-T vector (Promega). The probes were labeled with FITC and in situ hybridization reactions were performed according to the manufacturer's instructions (InnoGenex) using Fast Red as chromogens.
Terminal Deoxynucleotidyl Transferase-mediated dUTP Digoxigenin Nick End Labeling (TUNEL) Assay and Immunohistochemistry.
Coronal slices of rat brain were fixed in 10% buffered neutral formalin. 5-μm thick tissue sections were analyzed immunohistochemically using anti-Noxa antibody and ABC kit (Vector Laboratories). TUNEL staining was performed on paraffin-embedded tissue sections using In Situ Cell Death Detection Kit (Roche) according to the manufacturer's protocol.
Statistical Analysis.
All data were presented as mean ± SD from three or more independent experiments. Statistical comparison between different treatments was performed by Student's t test. Differences with P value <0.05 were considered statistically significant.
Results
Hypoxia Induces Noxa mRNA and Protein Expressions.
Subtractive hybridization was performed to identify proapoptotic genes induced by hypoxia in SK-N-MC neuroblastoma cells. Among the putative 25 hypoxia-regulated genes detected by subtractive hybridization, we have focused on the Noxa gene. Noxa, initially cloned as a PMA-responsive immediate early gene from acute T cell leukemia (24), is a proapoptotic member of BH3-only Bcl-2 family proteins and known to be a candidate mediator of p53-induced apoptosis (17). To confirm the results of subtractive hybridization, RT-PCR analysis was performed using mRNA extracted from hypoxia-exposed SK-N-MC neuroblastoma cells. An increase of Noxa transcripts was initially found at 1 h of hypoxia with pronounced induction up to sixfold around 6 h of stimulation (Fig. 1 A). A statistically significant increase of Noxa transcripts (Fig. 1 A, *, P < 0.05) was seen from 4 h of hypoxia (Fig. 1 A). A representative RT-PCR result was shown in Fig. 1 B. At protein levels, a significant increase (Fig. 1 B, *, P < 0.05) was seen from 2 h of hypoxia with maximal induction around 12 h after hypoxic insult (Fig. 1 C). A representative blot was presented in Fig. 1 D. Up-regulation of Noxa transcription in hypoxia was seen in a variety of human and mouse cells including endothelial cells, neurons, kidney cells, neuroblastomas, leukemias, and breast cancers (unpublished data), indicating that our findings were not cell type specific.
Figure 1.

Hypoxia induces transcriptional and translational up-regulation of Noxa. SK-N-MC neuroblastoma cells in serum- and glucose-deficient medium were subjected to hypoxic condition (0.5% O2) for the indicated times. (A) cDNA was synthesized from the total RNAs extracted from SK-N-MC cells exposed to hypoxia and subjected to RT-PCR analysis. 32 and 26 cycles of amplification were performed for Noxa and GAPDH, respectively. Fold increases of Noxa expression (mean ± SD) were presented using densitometry. Noxa expression level at 0 h was arbitrarily defined as 1. *, a statistically significant difference in Noxa expression levels (P < 0.05 in A and C). (B) A representative RT-PCR result. (C) 30 μg cell lysates extracted from hypoxia-exposed cells were subjected to Western blot analysis using polyclonal anti-Noxa and β-tubulin antibodies. Fold increases of Noxa protein expression was presented. Noxa expression level at 0 h was arbitrarily defined as 1. (D) Representative Western blot.
Noxa Up-regulation in Response to Hypoxia Is Independent of p53.
A previous report showed that genotoxic stress such as DNA damage or infrared exposure induced p53-dependent Noxa transcription via p53-CBS on Noxa promoter (17). Therefore, we wished to address whether hypoxia-induced Noxa up-regulation was mediated by p53. p53-deleted Saos-2 cells or H719 cells with wild-type p53 were exposed to hypoxia or DNA-damaging agent etoposide for each indicated time point and expression levels of Noxa and p21 were determined. p21 is known to be consistently up-regulated by p53 in response to DNA damage. Consistent with previous reports, genotoxic stress transcriptionally up-regulated p53, p21, and Noxa mRNA in H719 cells with wild-type p53 (Fig. 2, A and B) . In p53-deleted Saos-2 cells, etoposide failed to induce both Noxa and p21 expression (Fig. 2, A and B), suggesting that Noxa and p21 transcriptional up-regulations depend on p53 status in response to genotoxic stress. In contrast, Noxa mRNA, but not p21, was up-regulated in both H719 and Saos-2 cells by hypoxic stress (Fig. 2 A, right). In protein levels, Noxa expression by hypoxia was in line with mRNA (Fig. 2, C and D). Together, these results suggest that Noxa transcription and translation are regulated regardless of p53 status in response to hypoxia. To reinforce our results on the roles of hypoxia and p53 in Noxa transactivation machinery, we introduced wild-type p53 into Saos-2 cells. After 24 h of transfection, cells were exposed to hypoxia or treated with etoposide for 8 h and then Noxa expression levels were determined. Significant up-regulation of Noxa expression was seen in untreated or etoposide-treated Saos-2 cells with wild-type p53 as compared with mock-transfected cells (Fig. 2, E and F). Hypoxic exposure of Saos-2 cells transfected with wild-type p53 did not increase Noxa expression level significantly as compared with mock-transfected cells (Fig. 2, E and F). These results further support that Noxa transactivation machinery responds to hypoxia in a p53-independent manner.
Figure 2.

Noxa is transcriptionally up-regulated by hypoxia in a p53-independent manner. H719 cells with wild-type p53 and p53-deleted Saos-2 cells were incubated with 10 μM etoposide or cultured under 0.5% O2 in serum- and glucose-deficient medium for the indicated times. (A) RT-PCR analysis. cDNA synthesized from total RNAs extracted from etoposide-treated or hypoxia-exposed cells were subjected to RT-PCR analysis for p53, p21, and Noxa. Quantitations of expression levels were achieved after adjustment for the expression levels of the housekeeping gene, GAPDH. The cycle numbers for amplification were 30 for p53, 32 for p21 and Noxa, and 26 for GAPDH. (B) Determination of relative Noxa mRNA expression was achieved by densitometric analysis (*, P < 0.05). Noxa expression level of normoxic cells was defined as 1. C, untreated normoxic; E, etoposide; H, hypoxia. (C) 30 μg cell lysates extracted from etoposide-treated or hypoxia-exposed cells were subjected to Western blot analysis using anti-p53, anti-p21, anti-Noxa, and β-tubulin antibodies. (D) Determination of relative Noxa protein expression was achieved by densitometric analysis (*, P < 0.05). Noxa expression level of normoxic cells was defined as 1. (E) Saos-2 cells were transiently transfected with 1 μg pcDNA-p53. After 24 h, cells were untreated or treated with etoposide or hypoxia for 8 h. Cell lysates were subjected to Western blot analysis using anti-p53 or anti-Noxa antibody. p53 TF, p53 transfection. (F) Determination of relative Noxa protein expression was achieved by densitometric analysis (*, P < 0.05). Noxa expression level of untreated normoxic cells with mock transfection was defined arbitrarily as 1.
Noxa Promoter Responds to Hypoxia via HIF-1α.
Previous results led us to determine the promoter region of Noxa that responds to hypoxia. Promoter assay was performed using luciferase reporter gene linked to upstream region from initiation codon ATG of Noxa, as shown in Fig. 3 A. Luciferase activities were increased to 4.8-fold and 5.2-fold in pGL2-Noxa-1 and pGL2-Noxa-2 as compared with pGL2Basic, respectively. No significant increase of luciferase activity was observed when transfected with pGL2-Noxa-3 and pGL2-Noxa-4 (Fig. 3 B). This result suggests that the hypoxia-responsive region of Noxa promoter is located at −1,375 to −1,120 bp from the initiation codon ATG. To confirm our results, we inserted this 256-bp fragment into pGL2Basic to construct pGL2-Noxa-5 and performed promoter assay. Luciferase activity of pGL2-Noxa-5 was increased to 5.7-fold, confirming that this 256-bp fragment of Noxa promoter responds to hypoxia (Fig. 3 B). HIF-1α is known to be a major transcription regulator in hypoxic condition. Thus, to determine whether the hypoxic up-regulation of Noxa mRNA was driven by HIF-1α, we cotransfected Saos-2 cells with luciferase reporter plasmid and pcDNA-HIF-1α. As shown in Fig. 4 A, luciferase activities of pGL2-Noxa-1, pGL2-Noxa-2, and pGL2-Noxa-5 were increased to fivefold, indicating that Noxa responded to hypoxia via HIF-1α. Next, we analyzed the HRE consensus motif proposed by Wenger and Gassmann (25): (T, G, C) (A, G) CGTG (C, G, A) (G, T, C) (G, T, C) (G, T, C). In this 256-bp fragment, no more than a single base mismatch outside the CGTG core sequence was allowed. We found a single HRE motif fitting Wenger's and Gassmann's consensus sequence located at −1,275 bp upstream of the translation start codon. We constructed a site-directed mutant plasmid of HRE motif (pGL2-Noxa-M5), changing from 5′-GGCGTG-3′ to 5′-GGGCAC-3′. pGL2-Noxa-M5, an HRE mutant form of pGL2-Noxa-5, lost luciferase activity (Fig. 4 B). Our data demonstrate that Noxa promoter responds to HIF-1α through the HRE motif located at −1,275 from the ATG start codon. To prove direct involvement of HIF-1α in the regulation of Noxa transcription, we performed EMSA with an oligonucleotide containing the extended HRE consensus sequence derived from the human Noxa promoter (nucleotides from −1,290 to −1,268). A known consensus HIF-1α binding oligonucleotide and mutated core HRE sequence were used for positive and negative control, respectively. As evident from Fig. 4 C, the addition of 32P-labeled Noxa promoter oligonucleotide to nuclear protein extracts from Saos-2 cells cultured under hypoxic condition resulted in complex formation (Fig. 4 C, lane 3). The specificity of this complex was confirmed by competitive inhibition with an excess of the same unlabeled cold oligonucleotide (Fig. 4 C, lane 5) and a supershift analysis using anti–HIF-1α. The addition of anti–HIF-1α antibody to binding reaction mixture resulted in the supershift of complex (Fig. 4 C, lane 6), whereas control anti-Flag antibody did not influence the mobility of complex (Fig. 4 C, lane 7). Taken together, our data clearly show that hypoxic regulation of human Noxa is mediated by HIF-1α.
Figure 3.


Activation of Noxa promoter by hypoxia. (A) The Noxa promoter and various luciferase reporter constructs. HRE sites (closed circle) and p53-CBS (closed box) were shown. Numbering refers to the region of the Noxa promoter inserted into the parental pGL2Basic vector relative to the ATG translation initiation site as +1. (B) Saos-2 cells were transiently transfected with 1 μg indicated luciferase reporter plasmid. After 24 h of transfection, the cells were subjected to hypoxia for 6 h and then luciferase activity was determined. For each construct tested, fold increases of luciferase activities with hypoxia versus luciferase activity in normoxia (arbitrarily defined as 1) were presented. The mean ± SD of three independent experiments is shown.
Figure 4.
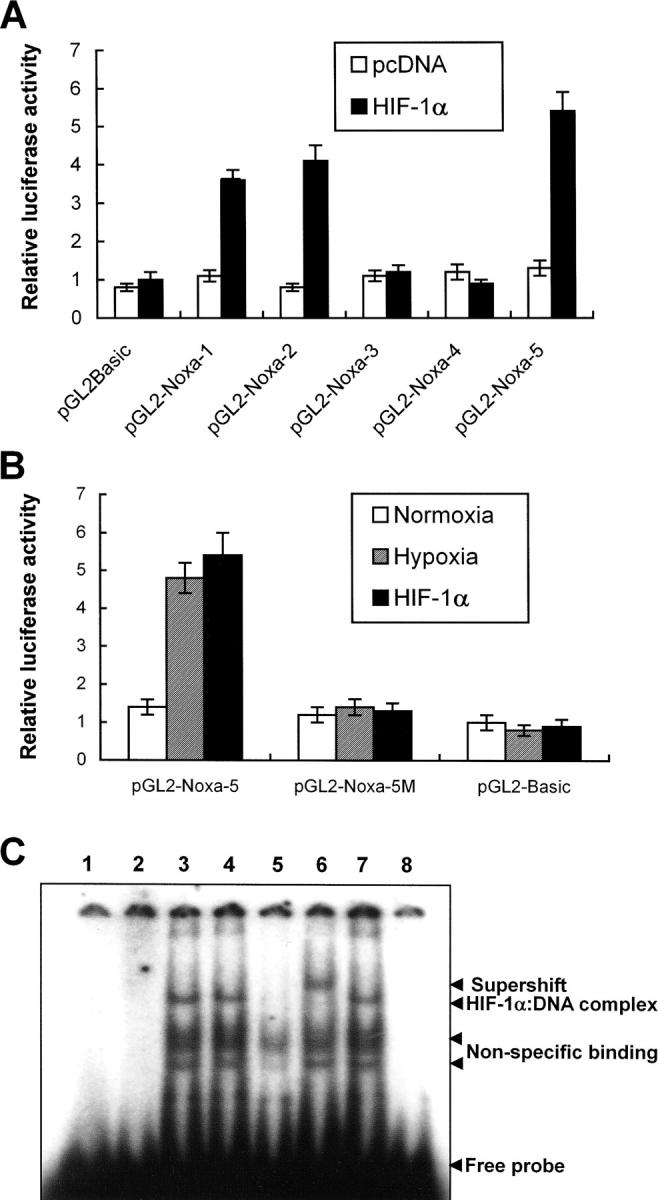
Noxa promoter responds to hypoxia via HIF-1α. (A) Saos-2 cells were transiently cotransfected with 1 μg indicated luciferase report plasmid and pcDNA-HIF-1α or mock vector, and then cultured in normoxia. After 24 h of cotransfection, luciferase activities were determined. The values represent the average luciferase activity of three independent experiments. The bars indicate standard error. (B) Luciferase reporter plasmid containing a 256-bp fragment with one putative HRE of Noxa promoter, starting from −1,120 bp of ATG initiation codon, was constructed (pGL2-Noxa-5). Mutant form of pGL2-Noxa-5 was constructed by changing the core sequence of HRE (CGTG) to GCAC. Saos-2 cells were transiently transfected with the indicated luciferase reporter plasmid and exposed to hypoxia for 6 h, or cotransfected with pcDNA-HIF-1α. Luciferase activities were determined as described above. The mean ± SD of three independent experiments is shown. (C) EMSA and supershift analysis of human Noxa. Lane 1, 32P-Noxa oligonucleotide; lane 2, 32P-Noxa oligonucleotide and nuclear extracts from Saos-2 cells cultured in normoxia (NN); lane 3, 32P-consensus oligonucleotide and nuclear extracts from Saos-2 cells cultured in hypoxia (NH); lane 4, 32P-Noxa oligonucleotide and NH; lane 5, 32P-Noxa oligonucleotide and 100-fold excess of unlabeled cold Noxa oligonucleotide with NH; lane 6, 32P-Noxa oligonucleotide and anti–HIF-1α antibody with NH; lane 7, 32P-Noxa oligonucleotide and isotype control antibody with NH; lane 8, 32P mutant oligonucleotide and NH.
Suppression of Endogenous Noxa Expression Protects against Hypoxic Cell Death.
To assess the role of Noxa in cell death after hypoxic assaults, we performed transfection and suppression assays in Saos-2 cells. Cells were transiently transfected with pcDNA-Noxa or mock vector and after 24 h of transfection cells were subjected to hypoxic or normoxic condition. Cell death rates were determined using trypan blue exclusion method at the indicated time points. As presented in Fig. 5 A, Noxa-transfected cells with hypoxic exposure showed significantly higher cell death rates compared with Noxa-transfected normoxia-cultured or mock-transfected hypoxia-exposed cells (Fig. 5 A, *, P < 0.05), suggesting that Noxa might sensitize the cells to hypoxic injury. To further characterize the roles of Noxa in hypoxic injury, we synthesized SE and AS oligonucleotides corresponding to Noxa coding sequences. Using AS and SE, we examined whether inhibition of endogenous Noxa expression with AS might affect the extent of cell death after hypoxia. Cell death analysis showed that suppression of Noxa expression by AS rescued the cells from hypoxic injury as opposed to SE (Fig. 5 B, *, P < 0.05). Time-dependent attenuations of Noxa expression by SL-O–delivered AS are presented in Fig. 5 C. Taken together, our results indicated that Noxa mediated hypoxia-induced cell death.
Figure 5.
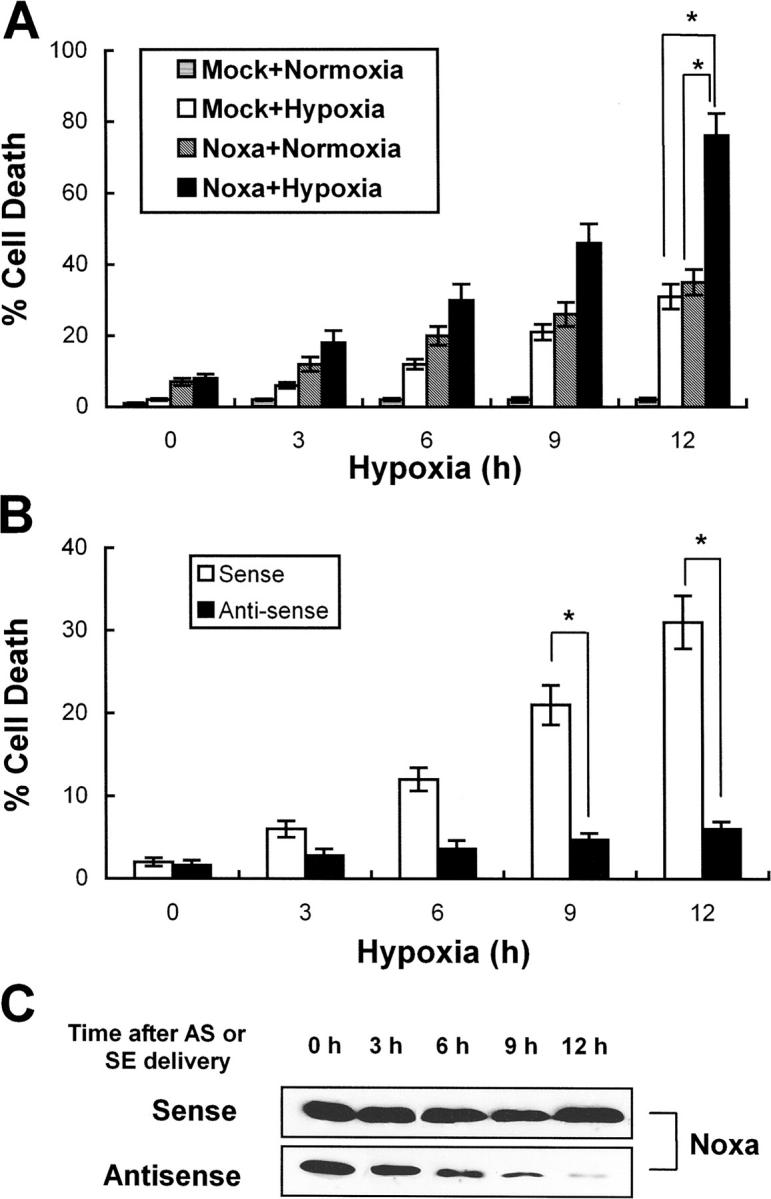
Noxa mediates cell death in response to hypoxia. (A) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa or mock vector. After 18 h of transfection, cells were cultured in normoxic condition or subjected to hypoxia for the indicated time points. Dead cells were counted by trypan blue exclusion method (*, P < 0.05). (B) Saos-2 cells were treated with AS or SE oligonucleotides 6 h before hypoxic assault and then subjected to hypoxia for the indicated times. Dead cells were counted by the trypan blue exclusion test. The mean ± SD of three independent experiments is shown (*, P < 0.05). (C) Representative Western blot for Noxa after treatment with AS or SE.
Noxa Facilitates Cell Death by Generation of ROS.
ROS plays a central role in the arena of cell injuries in response to hypoxia by perturbation of mitochondria, e.g., permeability transition or membrane peroxidation. It has been known that Noxa, targeting to mitochondria, induces loss of mitochondrial membrane potential (Δψm; reference 17). In addition, mitochondria are the major organelle responsible for intracellular ROS generation. Therefore, we hypothesized that Noxa might mediate hypoxic cell death by production of ROS. To examine the possibility that Noxa may perturb mitochondria by ROS generation, we transiently transfected Saos-2 cells with pcDNA-Noxa and determined the changes of ROS level using ROS-sensitive fluorescent dye, 2′, 7′-dichlorofluorescein diacetate (DCF-DA). As presented in Fig. 6 A, an increase in the ROS level was observed in Noxa-transfected cells in comparison with mock-transfected cells. Next, we analyzed the suppression effects of endogenous Noxa on ROS production in hypoxia. Cells whose endogenous Noxa expression was blocked with AS were cultured in hypoxic condition for 3 h followed by 1 h of reoxygenation. Cells were then sequentially stained with DCF-DA and propidium iodide (PI) followed by FACS® analysis. The populations of living cells, as indicated by their ability to exclude PI, were analyzed to discard the possibility that ROS were byproducts of cell death. As shown in Fig. 6 B, AS suppressed ROS generation induced by hypoxia, whereas SE failed to reduce ROS levels. Further, to establish that ROS contributed directly to Noxa-mediated hypoxic cell death, cells were transiently transfected with 1 μg pcDNA-Noxa in the presence or absence of antioxidant N-acetylcysteine (NAC). NAC reduced the ROS level and protected cell death in pcDNA-Noxa–transfected cells in a concentration-dependent manner (Fig. 6 C). Our results showed that ROS generation was crucial in Noxa-triggered cell death.
Figure 6.

ROS mediates Noxa-induced hypoxic cell death. (A) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa or mock vector. After 16 h of transfection, cells were stained with DCF-DA and subjected to flow cytometric analysis. (B) The cells were untreated or treated with 20 μmol/liter SE or AS oligonucleotides before 4 h of hypoxic exposure, subjected to hypoxia for 3 h, and followed by reoxygenation for 1 h. The cells were double stained with PI and DCF-DA and analyzed by flow cytometry using CELLQuest™ software. (C) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa or mock vector. After 16 h of transfection, cells were treated with or without NAC for an additional 12 h and cell death assay was performed using the trypan blue exclusion test.
Noxa Induces ROS-dependent Cytochrome c Release and Caspase Activation in Hypoxia.
We have demonstrated that Noxa mediated hypoxic cell death via generation of ROS. Previous characterization of Noxa indicated that Noxa was a member of BH3-only mitochondrial proteins that dissipated mitochondrial membrane potential with resultant cytochrome c release (17). Thus, we wished to address whether ROS induced by Noxa mediated cytochrome c release. Saos-2 cells were transiently transfected with pcDNA-Noxa or mock vector. ROS increase and cytochrome c release were sequentially determined from 8 h of transfection. As shown in Fig. 7 A, ROS increases were well correlated with cytochrome c releases. In agreement with cytochrome c release, caspase-3 activities were increased up to ninefold after 16 h of transfection (Fig. 7 B). To reinforce our results, Noxa- or mock-transfected cells were incubated with or without 10 mM NAC for an additional 6 h. Cells with cytochrome c release were enumerated manually based on immunocytochemistry staining or were determined by Western blots after cell fractionation (Fig. 7 C). In addition, cytochrome c release was suppressed in NAC-incubated cells (Fig. 7 C), suggesting that generation of ROS was responsible for cytochrome c release in Noxa-mediated cell death. Next, the cells treated with AS were subjected to hypoxic incubation and the number of cells with cytochrome c release was compared with that of SE-treated cells. As shown in Fig. 7 D, cytochrome c release was detected in ∼1, 2, 5, 7, and 9% of AS-treated cells in comparison with 2, 7, 27, 36, and 49% of SE-treated cells at the indicated time points. Attenuation of cytochrome c release by Noxa AS is shown in Fig. 7 D, right. Taken together, our data indicate that Noxa induced ROS-dependent cytochrome c release and caspase-3 activation.
Figure 7.

Noxa induces ROS-dependent cytochrome c release in response to hypoxia. (A) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa and change of ROS levels was sequentially determined at the indicated time point by flow cytometric analysis (left). Cytochrome c releases at the corresponding time points were determined by Western blot after cell fractionation (right). Mt., mitochondria; Cyt., cytoplasm. (B) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa or mock and then caspase-3 activity was determined at the indicated time point. Caspase-3 activity of mock-transfected cells after 8 h of transfection was arbitrarily defined as 1. (C) Saos-2 cells were transiently transfected with 1 μg pcDNA-Noxa or mock vector. After 16 h of transfection, cells were treated with or without 10 mM NAC for an additional 8 h and cytochrome c release was determined by immunocytochemistry using anti–cytochrome c antibody. Percentage of cell numbers of three independent experiments with standard errors is presented (left). Representative Western blot for cytochrome c release after cell fractionation was presented (right). (D) Cells were treated with SE or AS oligonucleotides for 4 h, exposed to hypoxia for 3 h, and reoxygenated for an additional hour. Numbers of cells with released cytochrome c were counted at indicated time points (time was counted after 1 h of reoxygenation) and average percentages with standard errors are presented (left). Representative Western blot for cytochrome c after treatment with AS or SE is shown (right).
Noxa Level Increases after Transient Ischemia in Rat Brain Middle Cerebral Artery Occlusion (MCAO) Model.
To address whether Noxa mRNA and protein expressions were enhanced in in vivo brain ischemia/hypoxia, we performed in situ hybridization using cRNA probe and immunohistochemistry in serial coronal sections derived from the rat transient MCAO model. After transient MCAO for 2 h, rats were killed from 6 h of MCA occlusion with a 1-h interval and their brains were removed. Enhanced Noxa mRNA and protein expressions were detected in peri-ischemic areas (penumbra) of the ipsilateral (infarcted) hemisphere from 4 h of infarction, peaking at 12 h after ischemia (Fig. 8, A–D) . TUNEL assay performed in brain slices adjacent to those used for analysis of Noxa expression revealed apoptotic nuclei in the ipsilateral hemisphere from 14 h of ischemia as compared with the contralateral hemisphere (Fig. 8, E and F).
Figure 8.
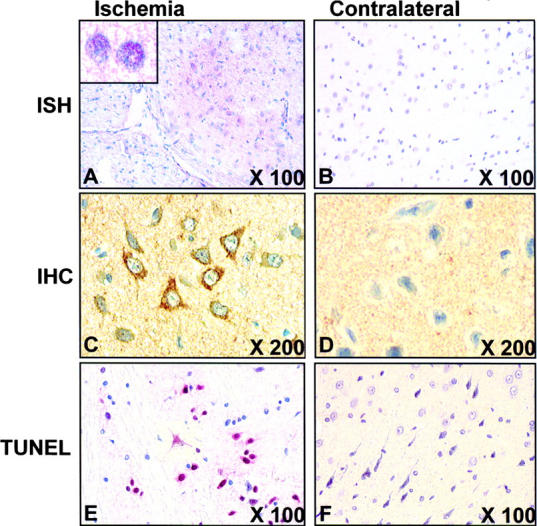
Expression of Noxa in ischemic rat brain from MCAO model. Brain was infarcted with MCA occlusion for 2 h, reperfused for 14 h, and then removed. Sections from infarcted or contralateral brain were (A) in situ hybridized with FITC-labeled cRNA probe (inset at high magnification) and (B) immunostained with anti-Noxa antibody or TUNEL stained. Fast Red substrate solution was used for chromagen in in situ hybridization and TUNEL method. DAB was used for immunostaining. Original magnifications are shown in each panel.
Liposomal Delivery of Noxa AS Oligonucleotide Reduces Acute Cerebral Infarction.
To determine whether suppression of Noxa expression may affect the extent of cerebral ischemia in vivo, AS in cationic liposomes was administered into the right lateral ventricle of rats 4 h before MCAO. The rats were killed after 16 h of MCA occlusion and their brains were removed for evaluation of infarction volume. There was a significant decrease in infarct volume in rats given the Noxa AS compared with SE (Fig. 9, A and C) . Attenuations of Noxa expressions according to slice levels were observed in Western blot analysis (Fig. 9 B). The infarction volume of the control animal (SE) was 154.24 ± 28.38 mm3, whereas that of the experimental group (AS) was reduced to 48.86 ± 13.46 mm3 (Fig. 9 C, *, P < 0.05).
Figure 9.
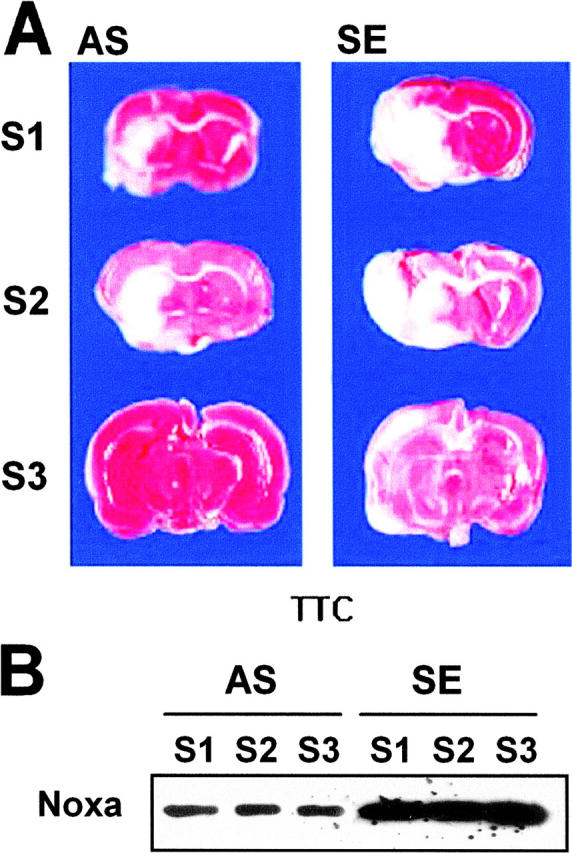

Protection of brain from ischemia by suppression of Noxa expression. SE or AS oligonucleotides (2 μg in 1 μl) were injected into the lateral ventricle 4 h before MCA occlusion for 2 h. Brain was reperfused for 14 h, removed, and then cut into eight coronal slices. (A) Representative section stained with triphenyl tetrazolium chloride (TTC). (B) Western blot analysis of Noxa from the corresponding slices of rat brain. (C) Infarction volume was calculated after measuring the infarct areas on coronal brain sections as described in Materials and Methods. The infarction volume of the control animal (SE) was 154.24 ± 28.38 mm3, whereas that of the experimental group (AS) was reduced to 48.86 ± 13.46 mm3 (*, P < 0.05).
Discussion
Hypoxia/ischemia is important in human pathophysiology because it participates in numerous pathological processes such as ischemic stroke, myocardial infarction, and chronic degenerative disorders. When exposed to hypoxia, cells transactivate a variety of gene products to adapt to altered metabolic status or to remove irreversibly damaged cells (2). Among the transcriptional factors responding to hypoxia, HIF-1α and p53 appear to be key modulators because they transactivate numerous genes responsible for increased oxygen delivery, cell cycle arrest, or apoptosis (25–28). Although survival genes and their molecular pathways that protect against cell death in response to hypoxia are rather well documented, death-promoting genes induced by hypoxia are less clarified. Here we have demonstrated that Noxa, a member of BH3-only Bcl-2 family proteins, is an HIF-1α–dependent hypoxia-inducible gene and mediates hypoxic cell death in an ROS-dependent manner.
Our initial experiments by subtraction hybridization, RT-PCR, and Western analysis strongly suggest that hypoxia up-regulates Noxa expression. Noxa, initially identified as a PMA-responsive gene in leukemia cells (24), is known to be a downstream effector molecule of tumor suppressor p53 because genotoxic stress such as DNA damage or X rays induced Noxa up-regulation through p53-CBS on Noxa promoter (17). Consistent with this finding, p53 and its downstream effectors, p21 and Noxa, were up-regulated by genotoxic agent etoposide in H719 cells with wild-type p53, but not in p53-deleted Saos-2 cells. This result indicates that genotoxic stress up-regulated Noxa expression via p53-dependent pathways. However, we cannot completely discard the possibility that additional molecules or pathways other than p53 might be involved in the transactivation pathway of Noxa expression by genotoxic stress. Interestingly, Noxa, but not p21, was transcriptionally and translationally up-regulated by hypoxic stress in p53-deleted Saos-2 cells. In addition, hypoxic stress on Saos-2 cells with reintroduced p53 did not induce a significant additional increase of Noxa expression. Dissociation of transactivation property of p53 between genotoxic damage and hypoxia may explain the reasons why restoration of p53 had no additive effect on the induction of Noxa in hypoxia. A previous report showed that hypoxia induced p53 accumulation, but in contrast to DNA damage, hypoxia failed to induce endogenous downstream p53 effector mRNA and proteins (29). Whatever mechanisms are involved in p53-dependent transactivation pathways, our data suggest that Noxa expression is regulated regardless of p53 status in response to hypoxia. These results prompted us to perform Noxa promoter assay.
The data obtained from luciferase reporter assay, mutant plasmid, and EMSA clearly showed that Noxa expression was regulated in a p53-independent manner. HIF-1α is a master regulator of oxygen homeostasis (9). As a whole, HIF-1α functions as a survival factor because its target genes participate in increasing oxygen delivery or reducing metabolic demands (30–32). However, HIF-1α serves as a pro-death gene depending on the severity of hypoxic stimulus (1, 27). To date, it seems that proapoptotic activity of HIF-1α is driven from interaction with p53 or modulation of proapoptotic Bcl-2 family protein (33, 34). Our data demonstrated that Noxa is a first member of a proapoptotic gene regulated directly by both HIF-1α and p53, thus linking two major transactivating systems responsible for cellular response to hypoxia (Fig. 10) . Noxa is categorized into BH3-only members of Bcl-2 family proteins (17). Recently, it has been proposed that BNip3, another member of the proapoptotic BH3-only protein, is a target molecule of HIF-1α in response to hypoxia (15). Although both BNip3 and Noxa are BH3-only Bcl-2 proteins that respond to hypoxia, it seems like that Noxa functions differently from BNip3. First, Noxa responds to hypoxia in early stage (Fig. 1), whereas BNip3 accumulates in cells after exposure to prolonged hypoxia (15). This suggests that Noxa may participate in the early stage of ischemic damage. This finding has potential clinical relevance for selection of targets in ischemic disorders at the early stage. Second, we and others reported that the ability of BNip3 to facilitate cell death depends on the transmembrane domain, but not on the BH3 domain (18, 19). However, the BH3 domain is crucial for cell death by Noxa (17), suggesting different cell death pathways. And third, as discussed below, Noxa facilitates apoptosis through an ROS-dependent pathway (Fig. 6), whereas BNip3 does so through an ROS-independent pathway (unpublished data). Based on functional differences between Noxa and BNip3, our data presented here categorize Noxa as a new member of the BH3-only proteins that responds to hypoxia. It also suggests the possibility that BH3-only proteins might be the major downstream effectors of HIF-1α in hypoxic cell death.
Figure 10.

A schematic pathway of HIF-1α, p53, and their downstream effectors in apoptosis induced by hypoxic injury.
It is well known that hypoxia increases cellular ROS generation, probably from mitochondrial electron transport complexes (35). Once generated, ROS induces membrane peroxidation, leading to destabilization of the mitochondrial and/or other cellular membranes. Also, it might be required to create a redox environment that facilitates the functions of other proapoptotic proteins such as Bax and BNip3 (36). Our data clearly show that Noxa induces cell death by production of ROS, leading to cytochrome c release and caspase activation because (a) antioxidant NAC inhibited cytochrome c release in Noxa-transfected cells (Fig. 7 C) and (b) suppression of endogenous Noxa expression by AS significantly reduced cytochrome c release in cells exposed to hypoxia (Fig. 7 D). However, it is not clear whether Noxa-induced ROS initiates a cytochrome c–dependent apoptotic machinery. Nevertheless, our data provide clear evidence that ROS is crucial for Noxa-induced cell death.
Our results from cell death assay and MCAO animal model using AS oligonucleotide strongly suggest that Noxa mediates hypoxic cell death and that Noxa is a candidate target molecule of ischemia therapy in the acute stage. Hypoxia participates in numerous pathological processes such as ischemic stroke, myocardial infarction, and chronic degenerative disorders. Thus, hypoxia-induced cell death genes are important targets for the development of drugs or other treatment modalities. Overall, data presented here led us to conclude that Noxa is an HIF-1α–induced proapoptotic gene that mediates hypoxic cell death by generation of ROS. It will be interesting to study the potential biological significance of Noxa in transgenic and knockout mice.
Acknowledgments
This work was supported by a grant (no. R01-2001-000-00200-0) from the Basic Research Program of the Korea Science & Engineering Foundation. This work was also funded by the Korea Science & Engineering Foundation through the Biomedical Research Center for Reactive Oxygen Species at Kyung Hee University (no. R13-2002-020-01002-0).
Abbreviations used in this paper: AS, antisense; DCF-DA, 2′, 7′-dichlorofluorescein diacetate; EMSA, electrophoretic mobility shift assay; HIF-1α, hypoxia-inducible factor 1 α; HRE, hypoxia-responsive element; MCAO, middle cerebral artery occlusion; NAC, N-acetylcysteine; p53-CBS, p53 consensus binding sequence; PI, propidium iodide; ROS, reactive oxygen species; SE, sense; SL-O, streptolysin-O; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP digoxigenin nick end labeling.
References
- 1.Halterman, M.W., and H.J. Federoff. 1999. HIF-1α and p53 promote hypoxia-induced delayed neuronal death in models of CNS ischemia. Exp. Neurol. 159:65–72. [DOI] [PubMed] [Google Scholar]
- 2.Bunn, H.F., and R.O. Poyton. 1996. Oxygen sensing and molecular adaptation to hypoxia. Physiol. Rev. 76:839–885. [DOI] [PubMed] [Google Scholar]
- 3.Semenza, G.L. 1999. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 15:551–578. [DOI] [PubMed] [Google Scholar]
- 4.Gardner, L.B., Q. Li, M.S. Park, W.M. Flanagan, G.L. Semenza, and C.V. Dang. 2001. Hypoxia inhibits G1/S transition through regulation of p27 expression. J. Biol. Chem. 276:7919–7926. [DOI] [PubMed] [Google Scholar]
- 5.Semenza, G.L., and G.L. Wang. 1992. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 12:5447–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Banasiak, K.J., Y. Xia, and G.G. Haddad. 2000. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. 62:215–249. [DOI] [PubMed] [Google Scholar]
- 7.Saikumar, P., Z. Dong, J.M. Weinberg, and M.A. Venkatachalam. 1998. Mechanisms of cell death in hypoxia/reoxygenation injury. Oncogene. 17:3341–3349. [DOI] [PubMed] [Google Scholar]
- 8.Semenza, G.L. 2000. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 88:1474–1480. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet, P., Y. Dor, J.M. Herbert, D. Fukumura, K. Brusselmans, M. Dewerchin, M. Neeman, F. Bono, R. Abramovitch, P. Maxwell, et al. 1998. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 394:485–490. [DOI] [PubMed] [Google Scholar]
- 10.Halterman, M.W., C.C. Miller, and H.J. Federoff. 1999. Hypoxia-inducible factor-1α mediates hypoxia-induced delayed neuronal death that involves p53. J. Neurosci. 19:6818–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruick, R.K. 2000. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA. 97:9082–9087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomasevic, G., M. Shamloo, D. Israeli, and T. Wieloch. 1999. Activation of p53 and its target genes p21(WAF1/Cip1) and PAG608/Wig-1 in ischemic preconditioning. Brain Res. Mol. Brain Res. 70:304–313. [DOI] [PubMed] [Google Scholar]
- 13.Sansome, C., A. Zaika, N.D. Marchenko, and U.M. Moll. 2001. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett. 501:97–98. [DOI] [PubMed] [Google Scholar]
- 14.Saikumar, P., Z. Dong, Y. Patel, K. Hall, U. Hopfer, J.M. Weinberg, and M.A. Venkatachalam. 1998. Role of hypoxia-induced Bax translocation and cytochrome c release in reoxygenation injury. Oncogene. 17:3401–3415. [DOI] [PubMed] [Google Scholar]
- 15.Sowter, H.M., P.J. Ratcliffe, P. Watson, A.H. Greenberg, and A.L. Harris. 2001. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 61:6669–6673. [PubMed] [Google Scholar]
- 16.Vande Velde, C., J. Cizeau, D. Dubik, J. Alimonti, T. Brown, S. Israels, R. Hakem, and A.H. Greenberg. 2000. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 20:5454–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- 18.Chen, G., R. Ray, D. Dubik, L.F. Shi, J. Cizeau, R.C. Bleackley, S. Saxena, R.D. Gietz, and A.H. Greenberg. 1997. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J. Exp. Med. 186:1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, J.Y., J.J. Cho, J. Ha, and J.H. Park. 2002. The carboxy terminal C-tail of BNip3 is crucial in induction of mitochondrial permeability transition in isolated mitochondria. Arch. Biochem. Biophys. 398:147–152. [DOI] [PubMed] [Google Scholar]
- 20.Shoshani, T., A. Faerman, I. Mett, E. Zelin, T. Tenne, S. Gorodin, Y. Moshel, S. Elbaz, A. Budanov, A. Chajut, et al. 2002. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 22:2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spiller, D.G., R.V. Giles, J. Grzybowski, D.M. Tidd, and R.E. Clark. 1998. Improving the intracellular delivery and molecular efficacy of antisense oligonucleotides in chronic myeloid leukemia cells: a comparison of streptolysin-O permeabilization, electroporation, and lipophilic conjugation. Blood. 91:4738–4746. [PubMed] [Google Scholar]
- 22.Bruce-Keller, A.J., W. Boling, M.S. Kindy, J. Peschon, P.J. Kraemer, M.K. Carpenter, F.W. Holtsberg, and M.P. Mattson. 1996. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF-receptors. Nat. Med. 2:788–794. [DOI] [PubMed] [Google Scholar]
- 23.Culmsee, C., R.K. Stumm, M.K.H. Schafer, E. Weihe, and J. Krieglstein. 1999. Clenbuterol induces growth factor mRNA, activates astrocytes, and protects rat brain tissue against ischemic damage. Eur. J. Pharmacol. 379:33–45. [DOI] [PubMed] [Google Scholar]
- 24.Hijikata, M., N. Kato, T. Sato, Y. Kagami, and K. Shimotohno. 1990. Molecular cloning and characterization of a cDNA for a novel phorbol-12-myristate-13-acetate-responsive gene that is highly expressed in an adult T-cell leukemia cell line. J. Virol. 64:4632–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wenger, R.H., and M. Gassmann. 1997. Oxygen and the hypoxia-inducible factor-1. Biol. Chem. 378:609–616. [PubMed] [Google Scholar]
- 26.Zaman, K., H. Ryu, D. Hall, K. O'Donovan, K.I. Lin, M.P. Miller, J.C. Marquis, J.M. Baraban, G.L. Semenza, and R.R. Ratan. 1999. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J. Neurosci. 19:9821–9830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piret, J.P., D. Mottet, M. Raes, and C. Michiels. 2002. Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem. Pharmacol. 64:889–892. [DOI] [PubMed] [Google Scholar]
- 28.Moll, U.M., and A. Zaika. 2001. Nuclear and mitochondrial apoptotic pathways of p53. FEBS Lett. 493:65–69. [DOI] [PubMed] [Google Scholar]
- 29.Koumenis, C., R. Alarcon, E. Hammond, P. Sutphin, W. Hoffman, M. Murphy, J. Derr, Y. Taya, S.W. Lowe, M. Kastan, et al. 2001. Regulation of p53 by hypoxia: dissociation of transcriptional repression and apoptosis from p53-dependent transactivation. Mol. Cell. Biol. 21:1297–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blagosklonny, M.V. 2001. Hypoxia-inducible factor: Achilles' heel of antiangiogenic cancer therapy. Int. J. Oncol. 19:257–262. [DOI] [PubMed] [Google Scholar]
- 31.Semenza, G.L., P.H. Roth, H.M. Fang, and G.L. Wang. 1994. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269:23757–23763. [PubMed] [Google Scholar]
- 32.Goda, N., H.E. Ryan, B. Khadivi, W. McNulty, R.C. Rickert, and R.S. Johnson. 2003. Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol. Cell. Biol. 23:359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vogelstein, B., D. Lane, and A.J. Levine. 2000. Surfing the p53 network. Nature. 406:307–310. [DOI] [PubMed] [Google Scholar]
- 34.Miyashita, T., and J.C. Reed. 1995. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 80:293–299. [DOI] [PubMed] [Google Scholar]
- 35.Chuanyu, L., and M.J. Robert. 2002. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 282:C227–C241. [DOI] [PubMed] [Google Scholar]
- 36.Gottlieb, E., M.G. Vander Heiden, and C.B. Thompson. 2000. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 20:5680–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]