

Abstract
Phosphorylated histone H2AX (γH2AX) is generated in nucleosomes flanking sites of DNA double-strand breaks, triggering the recruitment of DNA-damage response proteins such as MDC1 and 53BP1. Here, we study shortened telomeres in senescent human cells. We show that most telomeres trigger γH2AX formation, which spreads up to 570 kb into the subtelomeric regions. Furthermore, we reveal that the spreading patterns of 53BP1 and MDC1 are very similar to that of γH2AX, consistent with a structural link between these factors. Moreover, different subsets of telomeres signal in different cell lines, with those that signal tending to equate to the shortest telomeres of the corresponding cell line, thus linking telomere attrition with DNA-damage signalling. Notably, we find that, in some cases, γH2AX spreading is modulated in a manner suggesting that H2AX distribution or its ability to be phosphorylated is not uniform along the chromosome. Finally, we observe weak γH2AX signals at telomeres of proliferating cells, but not in hTERT immortalised cells, suggesting that low telomerase activity leads to telomere uncapping and senescence in proliferating primary cells.
Keywords: ChIP-chip, DNA-damage response, H2AX, senescence, telomere
Introduction
DNA double-strand breaks (DSBs) are highly toxic lesions that, if unrepaired or repaired incorrectly, compromise genome stability and can lead to cancer (Khanna and Jackson, 2001). To deal with DSBs and other lesions in DNA, cells have evolved a network of responses, referred to as the DNA-damage response (DDR). A hallmark of the DDR is the recruitment of proteins to damaged chromatin regions; some of these factors engage in DNA repair, whereas others trigger DNA-damage checkpoints that delay cell-cycle progression and coordinate repair processes (Zhou and Elledge, 2000; Rouse and Jackson, 2002). Some DDR factors, such as MDC1 and 53BP1, accumulate in large nuclear aggregates that appear as IR-induced nuclear foci (IRIF) by fluorescence microscopy (Schultz et al, 2000; Goldberg et al, 2003; Stewart et al, 2003), and accumulating evidence suggests that IRIF are required for accurate and coordinated DSB repair in the context of chromatin (Fernandez-Capetillo et al, 2004).
A key factor of IRIF formation in mammalian cells is the histone H2A variant H2AX, a component of the nucleosome core that comprises 5–20% of total cellular H2A in higher organisms (Rogakou et al, 1998; Fernandez-Capetillo et al, 2003, 2004). H2AX is phosphorylated on serine 139 near its carboxyl terminus (the phosphorylated form is referred to as γH2AX) in extended regions of chromatin flanking DSBs (Rogakou et al, 1998), and this response is mediated by ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs), two members of the phosphoinositide 3-kinase-related protein kinase (PIKK) family (Stiff et al, 2004; Falck et al, 2005). Rapidly after H2AX phosphorylation, the DNA-damage mediator protein, MDC1/NFBD1, is recruited to damage sites by its tandem BRCA1 carboxy-terminal (BRCT) domain, specifically interacting with the γH2AX C-terminal phospho-epitope (Lukas et al, 2004; Stucki et al, 2005). The yeast Saccharomyces cerevisiae lacks a H2AX variant, but instead, PIKK-family proteins phosphorylate the C-terminus of histone H2A in response to DNA damage, and this phosphorylation spreads about 25 kb to each side of a HO-induced DSB and is absent in the immediate vicinity of the break (Shroff et al, 2004). By contrast, γH2AX spreading in mammals has been estimated to occur over regions up to 3 Mb adjacent to DSBs, as measured by two-dimensional gel electrophoresis and immunofluorescence microscopy studies (Rogakou et al, 1998, 1999). The underlying similarities and differences between the mechanisms of chromatin spreading of phospho-H2AX/H2A in yeast and mammals, however, are currently unknown. Through the use of fluorescence microscopy experiments, it has been shown that, in addition to MDC1, other DDR factors such as 53BP1, the Mre11-Rad50-Nbs1 (MRN) complex, ATM and BRCA1 also spread over long distances together with γH2AX (for example, Bekker-Jensen et al, 2006). Mainly due to the lack of a suitable system to induce site-specific DSBs in mammals, however, molecular data about the spreading distances and molecular architecture of DSB-modified nucleosomal fibres in higher eukaryotes are not yet available.
Telomeres are specialised structures at the ends of linear chromosomes, and in most organisms they consist of repetitive short-sequence elements that bind proteins to form a complex termed Shelterin that sequesters the telomeric DNA and prevents it being recognised as DNA damage (D'Adda Di Fagagna et al, 2004; de Lange, 2005). To counteract gradual telomere shortening caused by the inability of the standard DNA replication apparatus to fully replicate chromosomal ends, the ribonucleoprotein complex, telomerase, adds additional telomeric repeats to chromosomal termini in a regulated fashion. However, whereas this occurs in human stem cells and in the germ line, most somatic human cells lack sufficient telomerase to maintain telomere length. Consequently, telomeres gradually shorten upon the continual passage of somatic human cells (Martens et al, 2000), ultimately leading to replicative senescence, the irreversible loss of replicative capacity occurring upon extended culture (Hayflick and Moorhead, 1961). Significantly, age-related diseases and premature ageing syndromes are sometimes characterised by short telomeres (Blasco, 2005).
Notably, the major hallmarks of a DDR are observed in cells experiencing replicative senescence, with γH2AX, MDC1, 53BP1 and other DDR factors accumulating on the critically shortened telomeres in such cells or in cells which have entered senescence as a result of artificial telomere uncapping (d'Adda di Fagagna et al, 2003; Takai et al, 2003; Shay and Wright, 2004; Zou et al, 2004; Hockemeyer et al, 2005). Under these conditions, cell-cycle arrest is triggered through a signalling cascade involving the DDR signalling proteins ATM, p53 and p21 (Herbig et al, 2004). Taken together, these findings suggest that in late-passage cultures of primary human cells, it is the subset of very short telomeres—devoid of most of their telomeric repeat sequences—which triggers DNA-damage foci formation and terminal cell-cycle arrest.
Here, by using the technique of ChIP-chip (chromatin immunoprecipitation) in combination with array-CGH (comparative genome hybridisation), we have analysed human senescent fibroblasts to study the molecular changes in chromatin that result from a DDR at uncapped telomeres. Our high-resolution analyses with whole-genome tile-path arrays provide precise data about the spreading distances in mammalian cells for various DDR factors, indicating that modification of the nucleosomal fibre flanking DSBs is not homogenous. We discuss the relevance of our findings in relation to telomere uncapping in senescence and in regard to the factors governing γH2AX and DDR factor distribution in senescent and proliferating cells.
Results
DNA-damage foci in senescent cells are ATM dependent, dynamic and reversible
To characterise the DDR as part of the senescent programme in the human fibroblasts used in this study, we first quantified DNA-damage foci in proliferating and senescent BJ fibroblasts. Whereas 25% of proliferating BJ cells displayed colocalising foci for γH2AX and 53BP1, most of these cells only had one prominent focus and very few had two or more foci (Figure 1A). By contrast, more than 90% of senescent BJ cells exhibited prominent DNA-damage foci, and most of these cells had between two and six foci per cell (Figure 1A). Although we cannot exclude clustering of telomeres and the calculated numbers might therefore be too conservative, these observations support the hypothesis that only relatively few uncapped telomeres are sufficient to initiate senescence (Zou et al, 2004). When we treated senescent cells for 24 h with potent small-molecule inhibitors of the ATM protein kinase (Hickson et al, 2004)—the major kinase responsible for DSB signalling—around 75% of senescent cells lost their DNA-damage foci and the remaining foci were small and difficult to detect (Figure 1B; senescent cells treated with the ATM inhibitor did not incorporate BrdU, indicating that inhibition of ATM was not sufficient to trigger cell-cycle re-entry—data not shown). Thus, as concluded previously by Herbig et al (2004), uncapped telomeres signal mainly through ATM. After removal of the ATM inhibitor for a further 48 h, senescent cells regained their foci, demonstrating that the DDR in senescent cells is continuously active, dynamic and reversible (Figure 1B). As these phenomena were also observed for cells that had been senescent in culture for more than 6 months (data not shown), we conclude that under these conditions, uncapped telomeres represent unrepairable DSBs that engage and maintain constantly active DNA-damage signalling.
Figure 1.
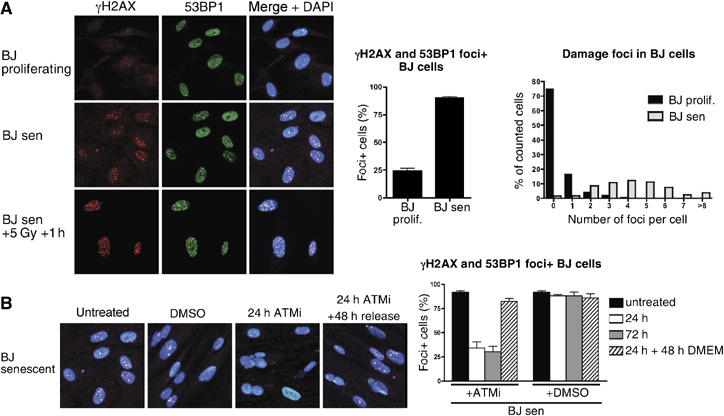
DNA-damage foci in senescent cells are ATM dependent, dynamic and reversible. (A) BJ fibroblasts were grown to senescence, fixed, incubated with antibodies against γH2AX (red) and 53BP1 (green) and processed for immunofluorescence microscopy. DNA was stained with DAPI (blue). Proliferating cells were ∼30 population doublings away from senescence. As a control, senescent BJ cells were irradiated with 5 Gy and incubated for 1 h before fixation. Cells with colocalising foci of γH2AX and 53BP1 were scored as foci-positive cells; the average of six independent experiments is shown (65–170 cells scored per sample). The average number of foci per cell was scored from 640 and 381 cells for proliferating and senescent cells, respectively. (B) Senescent BJ fibroblasts were treated with an ATM inhibitor (ATMi) at 10 μM for 24 or 72 h, or mock treated with DMSO and released into medium without the ATMi for 48 h. Representative images show a merge of γH2AX (red), 53BP1 (green) and DAPI. Cells with colocalising foci of γH2AX and 53BP1 were scored as foci-positive cells. Results show the average of at least three independent experiments (50–110 cells scored per sample).
Only a subset of telomeres signals in senescent fibroblasts
To identify the subset of telomeres being recognised as DNA damage during replicative senescence, we compared senescent cells from the two different fibroblast cell lines—BJ and MRC5—and performed ChIP in combination with high resolution array-CGH analysis (ChIP-chip; d'Adda di Fagagna et al, 2003). Thus, γH2AX was immunoprecipitated from cross-linked chromatin, DNA was purified, differentially labelled with fluorescent Cy dyes and hybridised to a whole-genome large-insert clone tile-path array covering all human chromosomes (Fiegler et al, 2006), resulting in the first whole genome-wide analysis of γH2AX distribution in senescent cells, at a resolution of approximately 100 kb. As shown in Figure 2A and B, prominent γH2AX signals were observed at a variety of telomeres, including those of chromosomes 1, 11 and 17 (data from the full set of chromosomes are provided in Supplementary Figures S1 and S3). Notably, whereas most of the telomeres displayed strong γH2AX enrichment, some, such as chromosome 1q and 17p, showed no or very weak signals, indicating that only a subset of telomeres elicits a detectable DDR in these senescent fibroblasts, as suggested previously (Zou et al, 2004). Furthermore, we found that BJ and MRC5 cells displayed reproducible differences in the subset of telomeres with detectable γH2AX signals, indicating that telomere shortening can differ in different cell lines. These differences are apparent in the graphical representation of the signalling intensity at individual telomeres (Figure 2E and F) and were reproduced in replicate experiments. It was also evident from these analyses that γH2AX enrichments within chromosomes were generally low, suggesting that the majority of DNA-damage signals in these senescent cells originated from telomeres. However, some hotspots for γH2AX signals were reproducibly observed within chromosomes, some of which could be attributed to fragile sites (http://www.gdb.org/gdb-bin/genera/generaSF/hgd/FragileSite). In addition, we obtained a very similar signalling pattern when we carried out ChIP-chip experiments with antibodies directed against the DDR protein 53BP1 (Figure 2C and D; data for the full set of chromosomes are shown in Supplementary Figures S2 and S4). These findings are therefore in line with data from immunofluorescence analyses of senescent cells (Figure 1) and cells treated with DNA-damaging agents (Bekker-Jensen et al, 2006), indicating that spreading of γH2AX and 53BP1 take place over similar distances along the chromosome (also, see below).
Figure 2.
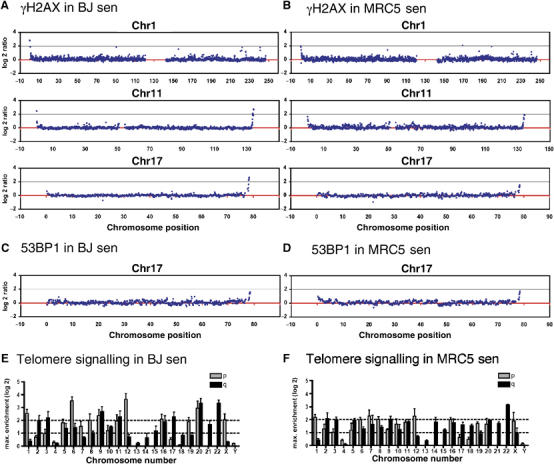
Only a subset of telomeres signals in senescent fibroblasts. (A–D) BJ and MRC5 fibroblasts were grown to senescence and cross-linked with formaldehyde. Soluble chromatin was prepared and used for chromatin immunoprecipitation (ChIP) with antibodies against γH2AX or 53BP1. Purified DNA from the immunoprecipitation and input fraction was differentially labelled with fluorescent Cy dyes and hybridised to a human tile-path DNA microarray (ChIP-chip). Enrichment of factors is plotted as the log2 of the ratio of the immunoprecipitated versus input fraction. Selected chromosomes are shown for senescent BJ cells (A, C) and senescent MRC5 cells (B, D). Analyses for γH2AX are shown in (A) and (B), with those for 53BP1 in (C) and (D). Graphs of γH2AX IPs represent averages of biological duplicate experiments with each colour-reversal hybridisation. The graphs for 53BP1 ChIP-chip represent the average of one experiment done in colour-reversal hybridisation (full data sets of all chromosomes are shown in the Supplementary Figures S1–S4). (E, F) The maximum signalling intensities at each telomere (p- and q-arm shown as grey and black boxes, respectively) are plotted for senescent BJ (E) and senescent MRC5 (F) cells. Data represent the average of three independent experiments.
The subset of γH2AX-positive telomeres correlates with average telomere length in pre-senescent cells
Next, we addressed whether the telomeric regions displaying enrichment of γH2AX during senescence might correspond to those telomeres that were particularly short in the cell line. Thus, we prepared metaphase spreads from BJ and MRC5 cultures about one population doubling away from complete arrest, and then stained telomeres quantitatively (Q-FISH) and identified individual telomeres by spectral karyotyping (SKY) analysis (see representative images in Figure 3A and B). Quantification of such images revealed that telomere lengths differed between various chromosomes, with a subset displaying significantly shorter than average telomeres (bars marked with an asterix in the histograms for Figure 3A and B). Furthermore, there were differences in the patterns of telomere length distributions between BJ and MRC5 cells, confirming the conclusions drawn from Figure 2 that telomere shortening can differ from one cell line to another. To investigate a possible correlation between short telomeres and signalling strength at senescence, we plotted telomere length against the maximum signalling ratio of each telomere (Figure 3A and B). For both BJ and MRC5 fibroblasts, strong signalling was only observed for short telomeres (upper left quadrant), whereas weak γH2AX signals were observed for long telomeres (lower right quadrant), linking short telomeres with those that signal a DDR at senescence. The observation of short telomeres with low damage signalling intensity (bottom left quadrant) could be explained by the fact that signalling was present but was not detected in the ChIP-chip analyses because the spreading region was too short to be detected by the arrays used. Alternatively, it could be that short telomeres do not always initiate a DDR or that some short telomeres are relatively impervious to further shortening.
Figure 3.
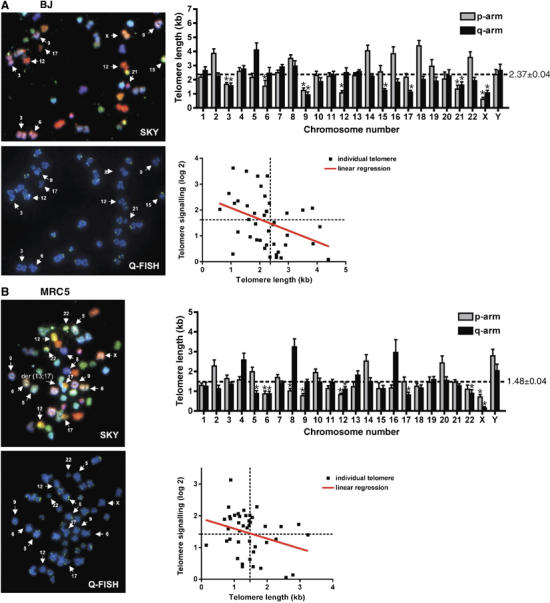
The subset of γH2AX-positive telomeres correlates with average telomere-length in pre-senescent cells. Fibroblasts were grown close to senescence, metaphase spreads were prepared and hybridised with telomeric fluorescent probe (Q-FISH) and chromosomes were identified by SKY. Representative images and a selection of identified chromosomes are shown for BJ cells (A) and MRC5 cells (B). A fusion between chromosome 13 and 17 in an MRC5 cell is marked in (B) as der(13;17). Average telomere length (kb) for p- and q-arms was calculated for each chromosome from 7 to 13 metaphases, and mean and standard errors are represented in the histograms. Bars with an asterix represent significantly short telomeres compared to average telomere length of all chromosomes. Telomere length in pre-senescent cells was plotted against signalling of each telomere in fully senescent cells (data from Figure 2E and F for BJ and MRC5 cells, respectively). The red line shows the linear regression of all data points; the dotted lines represent the average length in kb (X-axis) and the average enrichment of γH2AX on all telomeres as log2 ratio (Y-axis).
Telomere uncapping and DNA-damage signalling occur at low levels in proliferating cells
As controls for the ChIP-chip assays described above, we analysed proliferating BJ cells that were about 30 population doublings away from senescence and therefore were not expected to yield telomeric DDR signals. Surprisingly, however, weak but reproducible telomeric γH2AX enrichments were found in these cells; and moreover, the telomeres displaying γH2AX enrichment corresponded to those signalling in senescent cell populations (see Figure 4A for chromosomes 5 and 17 and full set of data in Supplementary Figure S6). Although the enrichments were much weaker than in senescent cells, they were clearly distinguishable from the low background levels within chromosomes. We reasoned that these signals might either reflect a DDR resulting from transient uncapping of telomeres during replication—as was proposed from ChIP studies of synchronised cells (Verdun et al, 2005)—or might represent a subpopulation of early senescent cells in the population of proliferating cells. To distinguish between these two alternatives, we analysed hTERT-immortalised BJ cells with long telomeric repeats and BJ cells arrested in G0 by serum starvation (efficacy of G0 arrest was verified by lack of BrdU incorporation and FACS analysis; Supplementary Figure S5). Notably, the telomeric γH2AX signals were not observed in the telomerase-expressing, immortalised cells (BJ-hTERT; Figure 4A and Supplementary Figure S8), indicating that cells with long telomeres do not accumulate damage at telomeres and making it likely that the weak signals we detected in proliferating cells were a consequence of telomere shortening. Furthermore, the telomeric γH2AX signals were observed when BJ cells were induced to enter G0 (Figure 4A and Supplementary Figure S7), supporting the above conclusion and indicating that the signals seen in proliferating cells do not arise through transient telomere uncapping during replication. Taken together, the above data suggest that primary cells stochastically encounter problems during telomere replication and that there might be a low but significant proportion of senescent cells in the proliferating BJ cultures, as far as 30 doublings away from full senescence. Indeed, we observed cells displaying β-galactosidase activity at pH 6 (a biomarker for cellular senescence; Dimri et al, 1995) even in early population doubling cultures, with the proportion of such cells gradually rising with increasing population doubling (Figure 4B). These findings therefore argue that the telomeric signals seen in Figure 4A are a result of stochastic telomere shortening and entrance into senescence within the cell population.
Figure 4.
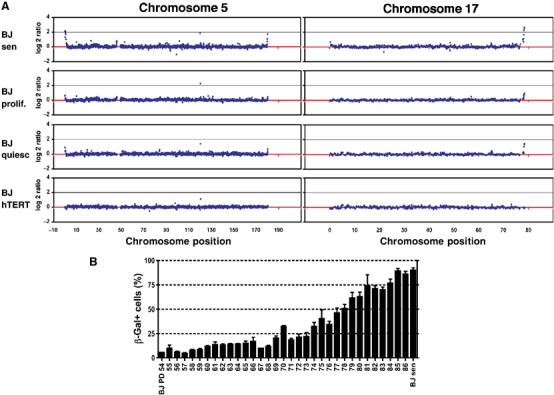
Telomere uncapping and DNA-damage signalling occur at low levels in proliferating cells. (A) BJ cells were grown as described in Materials and Methods. Cells were cross-linked with formaldehyde, processed for ChIP with an antibody against γH2AX and hybridised as described in Figure 2. Selected plots for chromosomes 5 and 17 are shown for BJ-senescent (BJsen), BJ-proliferating (BJ prolif.; around PD60), BJ-quiescent (BJ quiesc) and BJ-hTERT cells. (B) BJ cells were grown on coverslips and fixed at different population doublings between PD54 and PD87 (full senescence) and stained for senescence-associated β-Gal activity at pH 6. The percentage of β-Gal-positive cells is plotted against population doubling. Error bars represent standard deviation of three or more dishes, and more than 100 cells were counted per dish.
γH2AX, 53BP1 and MDC1 spread to similar extents into the subtelomeric regions of senescent cells
To characterise the extent of spreading of different DDR factors in BJ and MRC5 cells, we generated a telomeric microarray covering 2 Mb of each of the four subtelomeric regions of chromosome arms 7q, 8q, 12p and 20q; in each case, spotting PCR products of 1 kb in length spaced at ∼10 kb intervals. We then used these arrays in ChIP-chip analyses with samples generated with antibodies directed against γH2AX, 53BP1 and MDC1. In senescent BJ cells, enrichment of all three factors was strong on telomeres 12p and 20q, but weak or absent on 7q and 8q (Figure 5A). The spreading pattern was, however, different in MRC5 cells, where all three factors were strongly enriched at telomeres 7q and 12p (Figure 5B). These data therefore confirm the tile-path array results shown in Figure 2 and Supplementary Figure S1 and highlight the fact that different subsets of telomeres signal in the two different fibroblast cell lines. Interestingly, the spreading patterns for γH2AX, 53BP1 and MDC1 were very similar (see for example, telomere 7q in MRC5 cells) and reached into the subtelomeric regions to equivalent extents. This strongly suggests that the three factors spread to essentially the same extent, implying close proximity or interaction of these factors on nucleosomal fibres flanking uncapped telomeres. Notably, the spreading distance measured in such assays was found to reach up to ∼450 kb, which is considerably further than has been estimated in S. cerevisiae, where phosphorylated histone H2A has been detected only up to ∼25 kb distal to an HO-induced DSB (Shroff et al, 2004).
Figure 5.
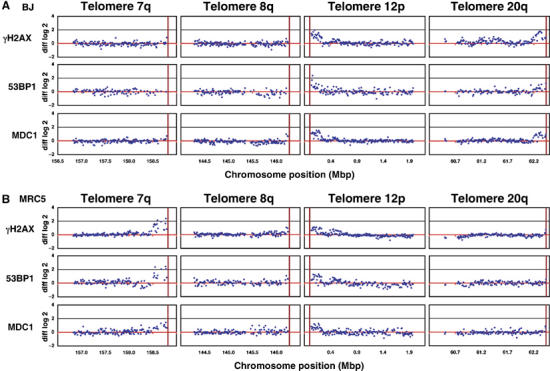
γH2AX, 53BP1 and MDC1 spread to similar extents into the subtelomeric regions of senescent cells. BJ and MRC5 fibroblasts were grown to senescence. ChIP experiments were performed with antibodies against γH2AX (rabbit or mouse), 53BP1 (rabbit) and MDC1 (two different sheep antibodies). The purified DNA derived from immunoprecipitation and input fraction was differentially labelled with fluorescent Cy dyes and hybridised to a custom microarray covering 2 Mb of telomeres 7q, 8q, 12p and 20q at a 10 kb resolution. Enrichment of factors was calculated as the log2 of the ratio of the immunoprecipitated versus input fraction. ChIP-chip of proliferating cells was carried out in parallel and graphs show subtracted log2 ratios of senescent minus proliferating cells (diff log2). All graphs show averaged data from dye-swap hybridisations for BJ (A) and MRC5 (B). The red vertical bar depicts the end of the respective chromosome.
High-resolution analysis of γH2AX spreading
To map the spreading pattern of γH2AX in more detail, we prepared γH2AX chromatin immunoprecipitations from senescent BJ cells and hybridised these to a custom-designed oligonucleotide array, where 3 Mb for each of a range of human telomere sequences were covered by oligonucleotides at an average spacing of 230 bp (NimbleGen Systems Inc., as oligonucleotides were picked from repeat-masked sequences, not all telomeres were covered because subtelomeric regions are repeat rich). Results of selected telomeres are shown in Figure 6A, with the full set of data shown in Supplementary Figures S9A and B. Perhaps surprisingly, whereas telomeres 12p, 20q and 22q showed a continuous distribution of γH2AX spreading, other telomeres—such as 6p, 9q and 11q—exhibited multiple peaks. Furthermore, telomere 6p, which signalled strongly even in proliferating cells (Supplementary Figure S6, see also Discussion), displayed multiple peaks of γH2AX enrichment spanning more than 1.5 Mb. This could indicate that the distribution of γH2AX is not homogeneous along the chromosome, that some regions of the nucleosomal fibre are refractory to H2AX phosphorylation, or that large subtelomeric fragments were lost in subsets of cells, with signalling being initiated at these break sites.
Figure 6.
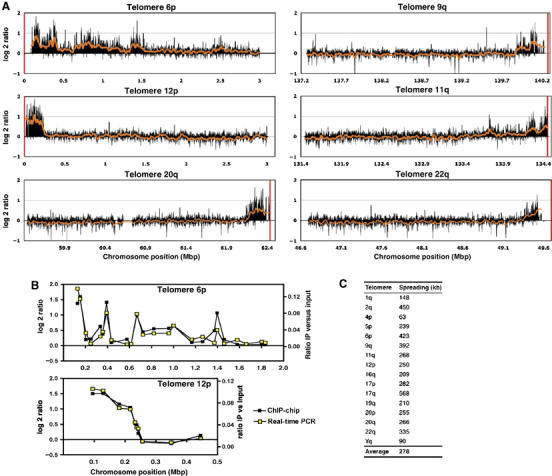
DDR factors spread up to 570 kb in mammalian cells. (A) Senescent BJ fibroblasts were processed for ChIP-chip experiments with antibodies against γH2AX. Labelling and hybridisation were performed by NimbleGen Systems Inc. (www.nimblegen.com). The oligo-array covered 3 Mb of most human telomeres and 5 Mb of telomere 6p with oligos spaced at a mean interval of 230 bp. Representative graphs of one experiment are shown in (A). Black bars show the average of five neighbouring data points, the red curve represents averaging of 100 neighbouring data points and the red vertical bar depicts the end of the respective chromosome. The full set of data is shown in Supplementary Figures S9A and B. (B) Enrichment of γH2AX (immunoprecipitation versus input) was assayed on telomeres 6p and 12p by real-time PCR with 24 and 10 primer pairs, respectively. Real-time PCR was carried out in duplicate and the averaged results (yellow open squares) are plotted together with the corresponding enrichment values (black squares) obtained from the ChIP-chip analysis with the oligo-arrays (A). (C) The extent of γH2AX spreading was determined for selected telomeres. The spreading boundary was defined as the transition point, where the log2 ratio fell to background levels. For telomere 6p, the multiple peaks inside the subtelomeric region were not considered for measuring the spreading distance.
To validate the above results obtained with the ChIP-chip analyses, we amplified the ChIP samples by real-time PCR with primer pairs covering the subtelomeric region of telomeres 6p and 12p (Figure 6B). The enrichment of γH2AX (IP versus input) obtained with real-time PCR (yellow open squares) matched very well to the data produced from the ChIP-chip studies (black squares), showing the reproducibility of our results with two different approaches.
Finally, to further define the spreading distances revealed by the above analyses, we generated data sets by averaging 100 neighbouring points (red line in Figure 6A), and determined the spreading boundary for selected telomeres as the transition point, where the log2 ratio was higher than the background scattering (tabulated in Figure 6C). The spreading distances thus obtained varied from 63 to 568 kb, with an average spreading distance of 280 kb (for telomere 6, there were also additional peaks observed between 0.7 and 1.5 Mb). These data therefore confirmed the conclusions drawn from the data in Figure 5, and reveal that the spreading distance of γH2AX in mammals is considerably longer than for H2A phosphorylation in S. cerevisiae, but is shorter than previous estimations made with human cell lines (Rogakou et al, 1998).
Discussion
Use of ChIP-chip to study DSB-induced chromatin alterations in mammalian cells
In this study, we applied a ChIP-chip approach to investigate the molecular consequences of DNA damage on chromatin structure. For our analyses, we employed senescent primary cells, which display a high degree of genome stability, but possess uncapped telomeres that initiate a DDR, making this a good model system to study molecular events triggered by DSBs. This study of senescent cells has revealed insights not only into how the DDR impacts on chromatin, but also into telomere-initiated senescence with respect to individual telomeres.
Spreading of mammalian DDR factors along chromatin fibres
We used a custom telomeric array, comprised of PCR products, that covers four telomeres at 10 kb resolution as well as a custom-designed oligonucleotide array (NimbleGen Systems Inc.) covering 3 Mb of most telomeres at a mean resolution of 230 bp. Thus, limited by the average fragment size of the sonicated chromatin, we were able to measure γH2AX at most human telomeres at a resolution of about 1 kb. Perhaps surprisingly, the spreading distances observed were not uniform, but ranged from 60 to 570 kb for the different subtelomeric regions analysed. These spreading distances are therefore considerably longer than those documented in yeast, where phosphorylated H2A spreads to approximately 25 kb to each side of a HO-induced DSB (Shroff et al, 2004). A possible explanation for this difference is that γH2AX spreading in mammalian cells is promoted and amplified by the γH2AX-binding factor MDC1, which has no clear counterpart in yeast (Stucki et al, 2005; Lou et al, 2006; Stucki and Jackson, 2006). Alternatively or in addition, it is possible that the number of phosphorylated γH2AX molecules produced in response to a DSB is similar in yeast and mammals, but that not every H2AX molecule becomes phosphorylated (Pilch et al, 2003). Because signalling events emanating from uncapped telomeres bear all the major hallmarks of a bona fide DDR, and as telomere-induced foci are likely to represent unrepairable DSBs, we anticipate that the spreading distances we have obtained will be in the same range when DNA damage takes place at other sites in the mammalian genome. Notably, however, the values we obtained are considerably shorter than those estimated for mammalian γH2AX from two-dimensional electrophoresis data, which suggested that regions up to 3 × 106 kb are produced in various human cell lines (Rogakou et al, 1998). Further work will be required to determine whether this discrepancy reflects real differences between γH2AX spreading in subtelomeric regions and interstitial chromosomal loci or is due to inaccuracies in previous estimates—for example because of technical limitations or possibly due to variations in H2AX distribution throughout the genome (see below). In regard to these issues, we note that one exception in our studies was telomere 6p, where enrichment of γH2AX was observed up to 1.5 Mb. This suggests that, at least in some cases, γH2AX can be generated well over 1 Mb away from the initiating DNA lesion, although at this time we are unable to exclude the possibility that this might instead reflect the occurrence of multiple, alternative sites for chromosomal DSB generation in the 6p subtelomeric region.
If H2AX phosphorylation can occur over hundreds of kilobases of chromatin, what then prevents γH2AX spreading throughout the damaged chromosome and why is it that spreading takes place to different extents in different subtelomeric regions? Although our analyses have so far been unable to identify any clear physical or sequence-composition signatures that might correspond to the boundary regions of γH2AX spreading, it is possible that such features do exist. Alternatively, the extent of spreading could be affected by the initial chromatin structure of the region in question. Notably, a recent study analysed H2AX distribution patterns with high-resolution confocal microscopy and concluded that H2AX is not distributed randomly throughout bulk chromatin, but is rather deposited in distinct regions of the genome and absent from others (Bewersdorf et al, 2006). It will therefore be interesting to compare the H2AX distribution at telomeres 5p, 6p, 9q and 11q and see whether any variations correlate with the different extents of γH2AX spreading we observed. In this regard, it is tempting to speculate that the multiple subtelomeric peaks of γH2AX that we have observed might reflect the existence of relatively H2AX-dense and H2AX-poor regions of the genome. It is also possible that spreading could be influenced by the histone variant H2AZ, which is distributed nonrandomly throughout the yeast genome (Raisner et al, 2005; Zhang et al, 2005). Alternatively, it is possible that γH2AX spreading is influenced by higher-order chromatin structure, an issue of particular relevance to our work, because mammalian subtelomeric regions display features of constitutive heterochromatin (Gonzalo et al, 2006). ChIP-chip studies with antibodies against heterochromatin components and/or with H2AX-specific antibodies, may shed light on these issues.
Molecular architecture of the DNA-damage modified nucleosomal fibre
When we used arrays covering four telomeres to compare the spreading of γH2AX with that of the DDR factors 53BP1 and MDC1, we found that the spreading distances were very similar. This strongly suggests that these factors are in close proximity on nucleosomal fibres flanking DSBs. Whereas the γH2AX C-terminus was shown to interact with the tandem-BRCT domain of MDC1 with high affinity, it is at present not clear what precisely controls the association of 53BP1 with damaged chromatin. Whereas retention of 53BP1 in DNA-damage foci requires γH2AX and MDC1 (Celeste et al, 2002; Ward et al, 2003; Bekker-Jensen et al, 2005), it is clear that methylated lysine residues in histone proteins play a major role in 53BP1 recruitment to DNA-damage sites (Huyen et al, 2004; Botuyan et al, 2006). Whether the link between γH2AX/MDC1 and 53BP1 in chromatin flanking DSBs is direct or indirect is, therefore, yet to be established.
Cell line specific telomere shortening correlates with signalling of uncapped telomeres
By comparing the telomeres enriched for γH2AX and 53BP1 in BJ and MRC5 fibroblasts, we observed not only a difference in the subset of uncapped telomeres that signals in the two cell lines, but also observed a correlation of these subsets with individual telomere lengths. Therefore, these findings provide support for a model in which only the shortest telomeres become uncapped in fibroblasts undergoing replicative senescence, and also link telomere attrition to the DDR at the level of individual chromosomes. Our data stand in agreement with a study that analysed selected telomeres in BJ fibroblasts (Zou et al, 2004). Notably, this study reported telomere 6p to be short in pre-senescent BJ cells, providing support for our finding that this telomere strongly triggers DDR signalling in proliferating BJ cell populations. The observation that the telomere lengths and the subset of signalling telomeres were different in BJ and MRC5 cells was not surprising as variations of telomere length between individuals and between maternal and paternal telomeres have been reported previously (Londono-Vallejo et al, 2001; Graakjaer et al, 2003). It is also possible that the observed differences might be caused by endogenous cellular stress, which affects the speed of telomere attrition (Lorenz et al, 2001; Von Zglinicki, 2003).
Telomeric DNA-damage signalling in proliferating cells
When we analysed proliferating cells that were about 30 population doublings away from senescence, we routinely observed significant signatures for telomeric DNA-damage signalling, although the magnitude of signalling was considerably lower than that occurring in senescent cell populations. These findings are reminiscent of those reported in a recent study by Verdun et al (2005), where the authors postulated that functional telomeres are transiently recognised as DSBs after replication. However, as we found that quiescent cells (in the G0 phase of the cell cycle) showed the same telomere-damage pattern, the phenomenon we observed is most likely not due to transient uncapping during S-phase. Instead, it is more likely that the damage signal we observed reflected a subpopulation of cells within the proliferating cell culture undergoing early senescence. It is generally accepted that telomere-initiated senescence is a stochastic process, rather than a ‘clock-based' mechanism that shortens telomeres at a defined rate (Von Zglinicki, 2003). After 87 population doublings, the majority of BJ cells are senescent; however, similar to observations made with other cell lines (Hastings et al, 2004), we found low but significant numbers of senescent BJ cells after only 60 population doublings. Senescence in young proliferating BJ cells might be caused by problems during replication of short telomeres, as we found that hTERT immortalised cells with long telomeres exhibited no detectable telomeric damage signals at all. Interestingly, proliferating BJ cells exhibited a major enrichment peak at telomere 6p (Supplementary Figure S6), but this was not observed in hTERT immortalised BJ cells or in proliferating MRC5 cells (data not shown), suggesting that telomere 6p is prone to be uncapped specifically in BJ cells in the absence of telomerase.
Concluding remarks
We have presented the first genome-wide high-resolution data about the extent of spreading of DDR factors in mammalian cells. Although we are unable to rule out the possibility of variations in the extent and distribution of other chromosomal breaks in subtelomeric regions, our findings strongly support the idea that γH2AX spreading is not uniform, but is modulated in a manner suggesting that H2AX distribution or its ability to be phosphorylated is not equal between different regions of the genome. Future studies will be required to identify the underlying molecular mechanisms for these phenomena, to establish precisely how DDR chromatin spreading is controlled, and how and why there are dramatic differences in the extent of DDR factor spreading between yeast and mammalian cells.
Materials and methods
Mammalian cell culture
BJ and MRC5 human primary fibroblasts and immortalised BJ-hTERT cells were grown in DMEM with 10% fetal bovine serum and PSQF at 37°C/5% CO2. Senescence was evaluated by failure to reach confluence after 4 weeks in culture from the last 1:2 passage, failure of >95% cells to incorporate BrdU after a 24-h period and expression of senescence-associated β-Gal in >95% of cells. For quiescence, cells were serum starved for 3 days in DMEM/0.5%FBS/PSQF. Senescent cells were treated for 24 h with an inhibitor specific for ATM (Hickson et al, 2004) at 10 μM and released into fresh medium.
Indirect immunofluorescence microscopy
Cells were grown on coverslips, fixed for 10 min on ice with ice-cold methanol/acetone. Nonspecific interactions were blocked by incubation for 60 min in PBS with 5% FBS. Fixed cells were incubated with primary antibodies overnight at 4°C, washed with PBS, incubated with Alexa Fluor secondary antibodies (Molecular Probes) for 45 min at room temperature and washed with PBS. Antibodies used for immunofluorescence were obtained from Upstate biotechnology (γH2AX mouse monoclonal) and Novus (53BP1 rabbit). Images were obtained using a Nikon Eclipse E800 with a BioRad Radiance 2100 laser set-up and BioRad LaserSharp software.
ChIP
In vivo cross-linking, chromatin purification and immunoprecipitations were as described previously (Orlando et al, 1997). The average fragment size of soluble chromatin fragments after sonication was ∼750 bp. Commercial antibodies used for ChIP were obtained from Upstate biotechnology (γH2AX mouse monoclonal and rabbit polyclonal antibodies) and Novus (rabbit 53BP1). Sheep antisera to MDC1 have been described in Goldberg et al (2003). Two different antibodies for each of γH2AX and MDC1 were used to confirm specificity in the ChIP experiments.
Array-CGH
Whole-genome tile path arrays were prepared as described previously (Fiegler et al, 2006) and printed at the UCSF microarray facility (http://cc.ucsf.edu/microarray). For telomeric arrays, 1 kb amino-modified PCR products covering 2 Mb of telomeres 7q, 8q, 12p and 20q at 10 kb resolution were printed onto CodeLink activated slides (GE Healthcare Life Sciences) by the Wellcome Trust Sanger Institute Microarray Facility (http://www.sanger.ac.uk/Projects/Microarrays/; for details, see Supplementary Information). The custom designed oligo-array (NimbleGen Systems Inc., www.nimblegen.com) covered 3 Mb of each human chromosome end with the exception of 5 Mb of telomere 6p. A repeat masked sequence of NCBI build 36, hg18, was used to select 390 000 isothermal oligos of 50 to 75 bp spaced at a mean interval of 230 bp. Array-CGH was performed as described previously with slight modifications (Fiegler et al, 2006; for details, see Supplementary Information). For oligonucleotide arrays, amplified ChIP samples were labelled and hybridised by NimbleGen Systems Inc. (www.nimblegen.com).
Data analysis
Arrays were scanned with an Agilent scanner (Agilent Technologies, UK) and images quantified by GenePix version 6.0 software (GRI, UK). Fluorescent ratios were normalised to the median ratio of intensities of all autosomal clones on the array. For tile-path arrays, the data of the colour-reversal experiments and replicate experiments were averaged and data were plotted as the log2 ratio. For telomeric arrays, the data of colour reversal and replicate experiments were averaged and data points that were outside a confidence interval of 50% were excluded. The difference of the log2 ratio of senescent minus proliferating cells (diff log2) was calculated and plotted. For NimbleGen oligo-arrays, the data were averaged over a window of N data points (either 5 or 100). Given a window size of N, for each data point, an average was calculated including the point and N/2 data points on each side (where possible). For all figures, NCBI build 36, hg18 was used.
Staining for senescent associated β-Gal activity
BJ cells were grown on coverslips and fixed in formaldehyde buffer at different population doublings between PD50 and PD87 (full senescence) and stained for senescence-associated β-Gal activity at pH 6 (Dimri et al, 1995) with a senescence cell histochemical staining kit (Sigma Cat No CS0030-1KT). Coverslips were mounted on slides with DAPI mounting medium and the percentage of β-Gal-positive cells was scored under a fluorescence/DIC microscope.
Real-time PCR
Real-time PCR was carried out using the SYBR green PCR system (Applied Biosystems); purified DNA samples of the ChIP fraction and the input DNA samples were amplified together with serial dilutions of genomic DNA on an ABI Prism 7000 sequence detector system (Applied Biosystems). HPLC-purified primer pairs were selected from a repeat masked sequence (NCBI build 36, hg18) and tested for their specificity. Primer pairs are listed in Supplementary data.
Telomere length measurements by Q-FISH and spectral karyotyping
Metaphases from pre-senescent BJ and MRC5 cells were prepared for Q-FISH, hybridised with a telomeric probe and analysed as described previously (Herrera et al, 1999; Samper et al, 2000; McIlrath et al, 2001). For details, see Supplementary data.
Supplementary Material
Supplementary Figures
Supplementary Figures
Supplementary Figures Legend
Supplementary Materials
Acknowledgments
We thank members of the Jackson laboratory for their suggestions and advice; S Gravel, P Huertas, K Miller and K Dry for critical reading of the manuscript; J Coates for help with tissue culture; S Polo for advice with real-time PCR; M Gilchrist for bioinformatics and help with analysis of microarray data; R Andrews for amplicon and primer design for the telomeric arrays; G Saretzki and T von Zglinicki for cell lines; P Reaper and V Smits for pilot studies and the design of telomeric arrays; and D Albertson, D Pinkel, R Davis, G Hamilton and the UCSF Comprehensive Cancer Center Microarray Core Facility for whole-genome tiling path array spotting. This work was made possible through core funding by Cancer Research UK and the Wellcome Trust, and was supported by Cancer Research UK and grant no. PA00A-111456 from the Swiss National Foundation to AM. MA Blasco's laboratory is funded by the Spanish Ministry of Education and Culture, the Regional Government of Madrid, European Union (TELOSENS FIGH-CT-2002-00217, INTACT LSHC-CT-2003-506803, ZINCAGE FOOD-CT-2003-506850, RISC-RAD FI6R-CT-2003-508842, MOL CANCER MED LSHC-CT-2004-502943) and the Josef Steiner Award 2003.
References
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J (2006) Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J (2005) Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol 170: 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewersdorf J, Bennett BT, Knight KL (2006) H2AX chromatin structures and their response to DNA damage revealed by 4Pi microscopy. Proc Natl Acad Sci USA 103: 18137–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6: 611–622 [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127: 1361–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A (2002) Genomic instability in mice lacking histone H2AX. Science 296: 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP (2003) A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198 [DOI] [PubMed] [Google Scholar]
- D'Adda Di Fagagna F, Teo SH, Jackson SP (2004) Functional links between telomeres and proteins of the DNA-damage response. Genes Dev 18: 1781–1799 [DOI] [PubMed] [Google Scholar]
- de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Celeste A, Nussenzweig A (2003) Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle 2: 426–427 [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of the genome. DNA Repair (Amst) 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Fiegler H, Redon R, Andrews D, Scott C, Andrews R, Carder C, Clark R, Dovey O, Ellis P, Feuk L, French L, Hunt P, Kalaitzopoulos D, Larkin J, Montgomery L, Perry GH, Plumb BW, Porter K, Rigby RE, Rigler D, Valsesia A, Langford C, Humphray SJ, Scherer SW, Lee C, Hurles ME, Carter NP (2006) Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res 16: 1566–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg M, Stucki M, Falck J, D'Amours D, Rahman D, Pappin D, Bartek J, Jackson SP (2003) MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421: 952–956 [DOI] [PubMed] [Google Scholar]
- Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8: 416–424 [DOI] [PubMed] [Google Scholar]
- Graakjaer J, Bischoff C, Korsholm L, Holstebroe S, Vach W, Bohr VA, Christensen K, Kolvraa S (2003) The pattern of chromosome-specific variations in telomere length in humans is determined by inherited, telomere-near factors and is maintained throughout life. Mech Ageing Dev 124: 629–640 [DOI] [PubMed] [Google Scholar]
- Hastings R, Qureshi M, Verma R, Lacy PS, Williams B (2004) Telomere attrition and accumulation of senescent cells in cultured human endothelial cells. Cell Prolif 37: 317–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585–621 [DOI] [PubMed] [Google Scholar]
- Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but Not p16(INK4a). Mol Cell 14: 501–513 [DOI] [PubMed] [Google Scholar]
- Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA (1999) Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J 18: 2950–2960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64: 9152–9159 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T (2005) POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J 24: 2667–2678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432: 406–411 [DOI] [PubMed] [Google Scholar]
- Khanna KK, Jackson SP (2001) DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet 27: 247–254 [DOI] [PubMed] [Google Scholar]
- Londono-Vallejo JA, DerSarkissian H, Cazes L, Thomas G (2001) Differences in telomere length between homologous chromosomes in humans. Nucleic Acids Res 29: 3164–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Saretzki G, Sitte N, Metzkow S, von Zglinicki T (2001) BJ fibroblasts display high antioxidant capacity and slow telomere shortening independent of hTERT transfection. Free Radic Biol Med 31: 824–831 [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J (2006) MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21: 187–200 [DOI] [PubMed] [Google Scholar]
- Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J (2004) Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 23: 2674–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens UM, Chavez EA, Poon SS, Schmoor C, Lansdorp PM (2000) Accumulation of short telomeres in human fibroblasts prior to replicative senescence. Exp Cell Res 256: 291–299 [DOI] [PubMed] [Google Scholar]
- McIlrath J, Bouffler SD, Samper E, Cuthbert A, Wojcik A, Szumiel I, Bryant PE, Riches AC, Thompson A, Blasco MA, Newbold RF, Slijepcevic P (2001) Telomere length abnormalities in mammalian radiosensitive cells. Cancer Res 61: 912–915 [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R (1997) Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11: 205–214 [DOI] [PubMed] [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM (2003) Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 81: 123–129 [DOI] [PubMed] [Google Scholar]
- Raisner RM, Hartley PD, Meneghini MD, Bao MZ, Liu CL, Schreiber SL, Rando OJ, Madhani HD (2005) Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123: 233–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297: 547–551 [DOI] [PubMed] [Google Scholar]
- Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA (2000) Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep 1: 244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD (2000) p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Wright WE (2004) Telomeres are double-strand DNA breaks hidden from DNA damage responses. Mol Cell 14: 420–421 [DOI] [PubMed] [Google Scholar]
- Shroff R, Arbel-Eden A, Pilch D, Ira G, Bonner WM, Petrini JH, Haber JE, Lichten M (2004) Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 14: 1703–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421: 961–966 [DOI] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64: 2390–2396 [DOI] [PubMed] [Google Scholar]
- Stucki M, Jackson SP (2006) gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5: 534–543 [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123: 1213–1226 [DOI] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T (2003) DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556 [DOI] [PubMed] [Google Scholar]
- Verdun RE, Crabbe L, Haggblom C, Karlseder J (2005) Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 20: 551–561 [DOI] [PubMed] [Google Scholar]
- Von Zglinicki T (2003) Replicative senescence and the art of counting. Exp Gerontol 38: 1259–1264 [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, Jorda KG, Chen J (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 278: 19579–19582 [DOI] [PubMed] [Google Scholar]
- Zhang H, Roberts DN, Cairns BR (2005) Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123: 219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
- Zou Y, Sfeir A, Gryaznov SM, Shay JW, Wright WE (2004) Does a sentinel or a subset of short telomeres determine replicative senescence? Mol Biol Cell 15: 3709–3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figures
Supplementary Figures Legend
Supplementary Materials