

Abstract
Increased glycogen synthase kinase-3 (GSK-3) activity is believed to contribute to the etiology of chronic disorders like Alzheimer's disease and diabetes, thus supporting therapeutic potential of GSK-3 inhibitors. However, sustained GSK-3 inhibition might induce tumorigenesis through β-catenin-APC dysregulation. Besides, sustained in vivo inhibition by genetic means (constitutive knock-out mice) revealed unexpected embryonic lethality due to massive hepatocyte apoptosis. Here, we have generated transgenic mice with conditional (tetracycline system) expression of dominant-negative-GSK-3 as an alternative genetic approach to predict the outcome of chronic GSK-3 inhibition, either per se, or in combination with mouse models of disease. By choosing a postnatal neuron-specific promoter, here we specifically address the neurological consequences. Tet/DN-GSK-3 mice showed increased neuronal apoptosis and impaired motor coordination. Interestingly, DN-GSK-3 expression shut-down restored normal GSK-3 activity and re-established normal incidence of apoptosis and motor coordination. These results reveal the importance of intact GSK-3 activity for adult neuron viability and physiology and warn of potential neurological toxicity of GSK-3 pharmacological inhibition beyond physiological levels. Interestingly, the reversibility data also suggest that unwanted side effects are likely to revert if excessive GSK-3 inhibition is halted.
Keywords: apoptosis, conditional, dominant negative, GSK-3, transgenic
Introduction
Glycogen synthase kinase-3 (GSK-3) is a serine/threonine kinase that is present in most tissues and that is particularly abundant in the central nervous system (CNS) (Woodgett, 1990). There are two isoforms of the enzyme termed GSK-3α and GSK-3β that are encoded by two different genes (Woodgett, 1990). GSK-3 is known to participate in multiple signaling pathways coupled to receptors for a variety of signaling molecules such as insulin or wnt among many others (Jope and Johnson, 2004). Regulation of GSK-3 is characterized by the fact that the enzyme is active in resting conditions with activation of the different signaling pathways, leading to GSK-3 inhibition by its phosphorylation on the Ser21 or Ser9 residue of GSK-3α and GSK-3β, respectively (Grimes and Jope, 2001). GSK-3 phosphorylation substrates include cytoskeletal proteins, transcription factors, and metabolic regulators, thus leading to a prominent role of GSK-3 in cellular architecture, gene expression, cell division and fate decision, and apoptosis among others (Grimes and Jope, 2001; Jope and Johnson, 2004).
Aberrantly increased GSK-3 activity is believed to play a key role in the pathogenesis of chronic metabolic disorders like type-II diabetes (Eldar-Finkelman, 2002), as well as of CNS conditions such as mood disorders and Alzheimer's disease (Avila et al, 2004; Jope and Johnson, 2004). With regard to GSK-3 and neurodegeneration, increased GSK-3 activity has been reported to result in neuronal apoptosis and GSK-3 inhibitors have been shown to exert antiapoptotic and neuroprotective effects in many different cell and mouse models (Pap and Cooper, 1998; Hetman et al, 2000; Beurel and Jope, 2006). Accordingly, potent and specific GSK-3 inhibitors are currently under development in view of their therapeutic potential (Cohen and Goedert, 2004; Frame and Zheleva, 2006; Gould et al, 2006). However, there are two main concerns that counteract the predicted usefulness of GSK-3 inhibitor therapies. On the one hand, the prominent role of GSK-3 in the adenomatous polyposis coli (APC)–β-catenin destruction complex implies that inhibition of GSK-3 could possibly lead to tumor promotion through the activation of β-catenin (Polakis, 2000). On the other hand, despite the above mentioned predicted antiapoptotic effect of GSK-3 inhibition, in vivo sustained inhibition by genetic means (constitutive knock-out mice) revealed an unexpected embryonic lethality due to massive hepatocyte apoptosis (Hoeflich et al, 2000). In this regard, pharmacological inhibition of GSK-3 in cell lines has also been shown to facilitate apoptosis triggered by certain stimuli (Beyaert et al, 1989; Song et al, 2004; Beurel and Jope, 2006).
In order to explore the consequences of sustained GSK-3 inhibition in adult tissues, here we have generated transgenic mice carrying a transgene encoding a dominant negative (DN) form of GSK-3β (DN-GSK-3) under control of a tetracycline-controlled conditional promoter. Due to the binary nature of the conditional transgenic system, these mice become a powerful tool because transgene transactivation can be directed to different tissues upon crossing with any of the available driver mice that express tTA (Tet-Off) under control of different promoters (Lewandoski, 2001).
To specifically address the neurological consequences of a sustained decrease in GSK-3 activity, we used driver mice with postnatal neuron-specific expression (CamKII-tTA mice; Mayford et al, 1996). Here, we report that sustained expression of DN-GSK-3 in the resulting double transgenic mice (Tet/DN-GSK-3 mice) leads to decreased GSK-3 activity and concomitant decreased phosphorylation of the microtubule-associated protein tau, a well-characterized GSK-3 substrate. Interestingly, Tet/DN-GSK-3 mice grew normally and no evidence of tumor formation was obtained either after gross anatomical examination or after analysis of brain morphology. However, apoptosis was detected in Tet/DN-GSK-3 mice in brain regions involved in motor control. Accordingly, Tet/DN-GSK-3 mice also showed a behavioral deficit in motor coordination. Finally, DN-GSK-3 transgene shutdown by doxycycline administration resulted in normal GSK-3 activity and in full reversal of the motor and of the neuronal apoptosis phenotypes.
Results
Mouse design
Constitutive knock-out of GSK-3β in mice is known to result in embryonic lethality (Hoeflich et al, 2000). As an alternative genetic approach to explore the consequences of a sustained decrease in GSK-3 activity in adult tissues, we decided to generate mice with conditional transgenic expression of a dominant-negative (DN) form of GSK-3. More precisely, the K85R mutant form of GSK-3β (DN-GSK-3) was chosen in view of its previously shown efficacy in decreasing GSK-3 activity (Dominguez et al, 1995).
The tetracycline-regulated system can be used for conditional gene expression in mice (Gingrich and Roder, 1998; Lewandoski, 2001). By using this system, we have previously generated inducible mouse models of neurodegenerative diseases (Yamamoto et al, 2000; Lucas et al, 2001). Similarly, here we decided to generate mice with the DN-GSK-3 transgene linked to a tetracycline responsive (tetO) promoter (Figure 1A). These mice can then be bred with mice with tissue-specific expression of the tetracycline transactivator (tTA, also known as Tet-Off). Then, double transgenic mice will express DN-GSK-3 in a tetracycline-repressible manner (Figure 1B). There are many available mouse lines expressing tTA (Lewandoski, 2001). Here, we aimed to explore the neurological consequences of sustained GSK-3 inhibition by using mice that express tTA under control of a postnatal neuron-specific promoter (Cam-KII-tTA mice; Mayford et al, 1996).
Figure 1.
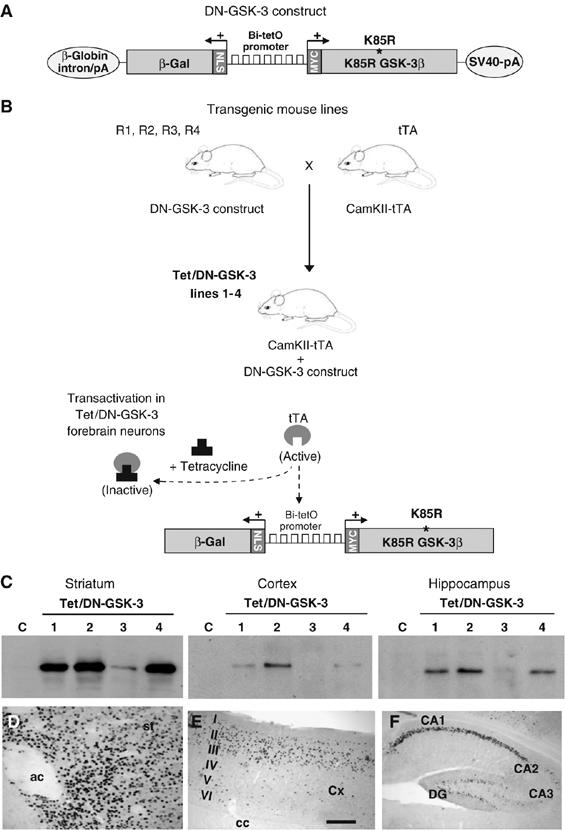
Generation of Tet/DN-GSK-3 mice and mapping of transgene expression. (A) Diagram showing the structure of the DN-GSK-3 construct used for transgenesis. The bidirectional Bi-tetO promoter is followed in one direction by the K85R-GSK-3β (DN-GSK-3) cDNA sequence with a myc epitope, and by the β-galactosidase (β-gal) sequence with a nuclear localization signal (NLS) in the other direction. (B) Generation of the conditional transgenic Tet/DN-GSK-3 mice (lines 1–4) by breeding the four DN-GSK-3 founder mice (R1, R2, R3 and R4 mice) with mice expressing tTA under control of the CamKIIα promoter (tTA mice). (C) Western blot detection of β-gal in striatum, cortex and hippocampus of 2-month-old Tet/DN-GSK-3 (R1, R2, R3 or R4) mice and control mice. (D–F) β-gal immunohistochemistry on sagittal brain sections from a 2-month-old Tet/DN-GSK-3 mouse (line 2). (D) striatum, (E) cortex and (F) hippocampus. ac, anterior commisure; Cx, cortex, I–VI, cortical layers; cc, corpus callosum; DG, dentate gyrus. Scale bar in panel E corresponds to 300 μm in panels (E, F), and to 200 μm in panel (D).
To generate the DN-GSK-3 construct for transgenesis, the sequence of the K85R mutant form of GSK-3β was cloned into a cassette containing the bidirectional tetO (Bi-tetO) promoter linked to a β-galactosidase (β-gal) reporter in divergent orientation (Figure 1A). Our previous experience in generating conditional transgenic mice with the tet-regulated system indicates that the site of insertion and/or the copy number of the tetO construct influences the final pattern and level of transactivation by tTA (Yamamoto et al, 2000; Lucas et al, 2001). The β-gal reporter sequence in the DN-GSK-3 construct permits, on one hand, a quick analysis of the pattern of transgene expression in the double transgenic (Tet/DN-GSK-3) mice from the different founder lines by X-gal staining or by immunohistochemistry against β-gal. On the other hand, it also allows to test the efficiency of transgene silencing after tetracycline administration (Yamamoto et al, 2000; Diaz-Hernandez et al, 2005; Engel et al, 2006).
Generation of mice with conditional expression of DN-GSK-3 in forebrain neurons
Four independent transgenic mouse founder lines were obtained that carried the DN-GSK-3 construct and they were termed R1, R2, R3 and R4 (Figure 1B). To study the effect of sustained GSK-3 inhibition in adult neurons, these mouse lines were then bred with the Cam-KII-tTA mouse line. In good agreement with the postnatal nature of the driver neuronal promoter, the percentage of double transgenic mice was close to the expected 25% for all founder lines (R1: 29/151, R2: 56/177, R3: 37/167, R4: 54/200 in the first analyzed litters).
The level and pattern of expression of the reporter transgene β-gal was then analyzed in 2-month-old Tet/DN-GSK-3 mice by Western blot and by immunohistochemistry. Tet/DN-GSK-3 mice resulting from line R3 showed the lowest level of transactivation by Western blot (Figure 1C) and, as evidenced by immunohistochemistry, this was restricted to the ventral striatum (data not shown). Lines R1, R2 and R4, on the other hand, led to β-gal expression in a spatial pattern very similar to that of endogenous CamKIIα, with expression evident in cortex, hippocampus, striatum, amygdala, and olfactory bulb neurons (Figure 1C–F and data not shown). Since CamKIIα is known to be expressed to a lesser extent in spinal cord (Liang et al, 2004), we also analyzed this structure, but no expression was detected either by Western blot, immunohistochemistry, or X-Gal staining (Supplementary Figure 1). Similarly and as expected, no transgene expression was detected in other brain regions with no expression of CamKIIα, such as cerebellum, brainstem, or thalamus nor in peripheral tissues (Supplementary Figure 1 and data not shown). Expression was highest in the striatum, where the vast majority of neurons showed transgene expression (Figure 1C and D). On the other hand, in the cortex and in the hippocampus, expression was restricted to certain neuronal subpopulations (Figure 1E and F). More precisely, cortical expression was located mainly in layer II–III neurons and hippocampal expression was essentially restricted to neurons in the CA1 field. In good agreement with the β-gal immunohistochemistry data, striatum was the only analyzed brain region, where increased total GSK-3β (endogenous plus transgenic DN-GSK-3β; 0.9 fold increase, P<0.05) was detected by Western blot (Figure 2A), and where some neurons expressed DN-GSK-3β above threshold for detection by immunofluorescence with anti-myc antibody (data not shown). Since R1, R2, and R4 Tet/DN-GSK-3 mice gave a similar pattern and level of transactivation, the three lines were analyzed in parallel. For all the biochemical, histological, and behavioral determinations, the three lines exhibited very similar phenotypes and we will refer to them indistinguishably as Tet/DN-GSK-3 mice.
Figure 2.
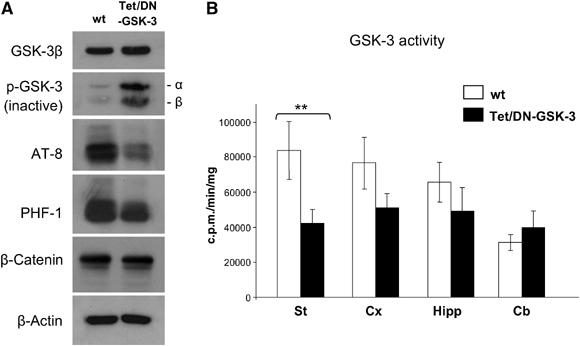
Decreased GSK-3 activity in Tet/DN-GSK-3 mice. (A) Western blot detection of GSK-3β, phosphorylated GSK-3 (pSer21/9 GSK-3α/β), AT-8 and PHF-1 phospho-tau epitopes, and β-catenin levels in homogenates from striatum of Tet/DN-GSK-3 mice and wild-type (wt) littermates. (B) In vitro GSK-3 activity assay performed on striatum, cortex, hippocampus and cerebellum homogenates from Tet/DN-GSK-3 mice and wt littermates (**P<0.001).
Tet/DN-GSK-3 mice grew normally and they showed no differences in body weight with respect to their wild-type littermates. Tet/DN-GSK-3 mice also showed a normal lifespan and no evidence of tumor formation was obtained either after gross anatomical examination or after analysis of brain morphology.
Reduced GSK-3 activity in the striatum of Tet/DN-GSK-3 mice
To verify that the forebrain expression of DN-GSK-3 resulted in decreased GSK-3 activity, we performed GSK-3 enzymatic activity assays on brain lysates as well as Western blot determination of the phosphorylated forms of GSK-3 and of its well-established substrate tau. In good agreement with the fact that striatum was the brain region with the highest level of transgene expression, GSK-3 activity was significantly reduced on striatal homogenates of Tet/DN-GSK-3 mice compared with those of wild-type mice (Figure 2B). A tendency toward decreased activity was observed also in cortical and hippocampal homogenates, but this did not reach statistical significance (Figure 2B). Similarly, no decrease was observed in non-expressing brain regions such as cerebellum.
We then analyzed by Western blot the striatal level of the inactive forms of GSK-3 that results from phosphorylation on Ser9 of the β isoform and on Ser21 of the α isoform (p-GSK-3) (Figure 2A). Interestingly, expression of DN-GSK-3β resulted in a dramatic increase (3.2-fold, P<0.001) not only in phospho-Ser9-GSK-3β but also in phospho-Ser21-GSK-3α. Since total GSK-3α levels are not altered in Tet/DN-GSK-3 mice (data not shown), this strongly suggests that the observed net reduction in GSK-3 activity is probably due in part to the previously reported mechanism that amplifies GSK-3 inhibition (Jope, 2003; Zhang et al, 2003). This mechanism is based on increased phosphorylation at the Ser21/9 residues in response to sustained inhibition of GSK-3 either by pharmacological or genetic means, even if the latter is performed in a single isoform (e.g. GSK-3β knock-out fibroblasts; Hoeflich et al, 2000). A moderate increase in the levels of inactive Ser21/9 phospho-GSK-3 was also observed by Western blot in the other two analyzed transgene expressing brain regions, hippocampus, and cortex. However, as for the GSK-3 activity assay data, the cortical and hippocampal increases in Ser21/9 phospho-GSK-3 did not reach statistical significance after normalizing for total GSK-3 levels (data not shown). In good agreement with the marked striatal decrease in GSK-3 activity, the level of phosphorylation of tau was reduced in striatal homogenates of Tet/DN-GSK-3 mice. This was evidenced by Western blot with the AT-8 (72% reduction, P<0.01) and the PHF-1 (46% reduction, P<0.01), antibodies (Figure 2A) that recognize two independent phospho-epitopes that are known to be phosphorylated by GSK-3 (Lovestone et al, 1994). We also analyzed the level of β-catenin that is also a GSK-3 substrate. Since β-catenin phosphorylation by GSK-3 favors its degradation by the proteasome (Aberle et al, 1997), we reasoned that its levels might be increased in Tet/DN-GSK-3 mice. However, as shown in Figure 2A, β-catenin levels were not changed in these mice.
We then explored by immunofluorescence the tissue distribution of the detected decrease in tau phosphorylation (Supplementary Figure 2). As expected, given the axonal localization of tau, the decrease in phospho-tau staining occurs diffusely throughout the striatal neurophil. This strongly suggests that the decrease takes place predominantly in the axons of medium size spiny neurons, that represent more than 90% of the neurons in the striatum (see staining with the DARPP-32 marker in Supplementary Figure 2B and E) and that have long axons that project outside the striatum. However, the somas of few large interneurons were also detected by PHF-1 immunofluorescence and the intensity of this staining was diminished in Tet/DN-GSK-3 mice (Supplementary Figure 2A, D, G, and I). By double labeling immunofluorescence we were able to identify these neurons as a subset of the choline acetyltransferase (ChAT)-positive large interneurons (Supplementary Figure 2G–J). In summary, decreased tau phosphorylation seems to take place mainly in the axons of medium size spiny neurons and also in a subset of ChAT-positive large interneurons.
Apoptosis detection in the striatum and cortex of Tet/DN-GSK-3 mice
Since apoptosis of liver cells was the most prominent phenotype in GSK-3β knock-out mice (Hoeflich et al, 2000), we wondered whether expression of DN-GSK-3 in neurons of Tet/DN-GSK-3 mice might also result in apoptosis. In this regard, although no obvious atrophy takes place in the brain of Tet/DN-GSK-3 mice, in immunofluorescence experiments we observed that some myc-positive neurons showed fragmented nuclei (not shown). To explore in a quantitative manner the incidence of neuronal apoptosis in the forebrain of Tet/DN-GSK-3 mice, we performed cleaved caspase-3 and TUNEL stainings. As anticipated, Tet/DN-GSK-3 mice showed a marked increase in the number of cleaved caspase-3-positive cells in the striatum (Figure 3B–D). Although decreased enzymatic activity was detected only in the striatum, we also analyzed apoptosis in the other brain regions with robust transgene expression (cortex and hippocampus), because apoptosis might take place in specific neuronal subpopulations that show relatively higher expression of the transgene an/or that are particularly vulnerable to apoptosis triggered by decreased GSK-3 activity. In fact, the number of cleaved caspase-3 positive cells was also significantly increased in the cortex of Tet/DN-GSK-3 mice (Figure 3A and D). As expected, this takes place in the external layers (Figure 3A), where transgene expressing neurons are located (see Figure 1E). No significant difference in the number of cleaved caspase-3-positive cells was detected in the hippocampus or in non-expressing regions such as cerebellum (Figure 3D). Besides, we performed double immunofluorescence experiments that confirmed that caspase-3-positive cells were also detected with anti β-gal (Figure 3E and F) and with anti-Neu-N (not shown) antibodies, thus proving that the detected dying cells are transgene expressing neurons. Similar results were obtained with TUNEL staining (Figure 3G and H).
Figure 3.
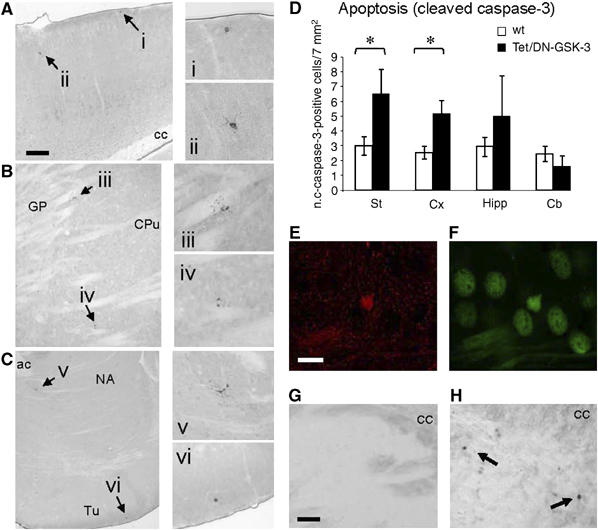
Apoptosis detection in striatum and cortex of Tet/DN-GSK-3 mice. (A–D) Immunohistochemical detection and quantification of cleaved caspase-3-positive cells. (A–C) Low-magnification pictures of showing cleaved caspase-3-positive cells (indicated by arrows) in cortex (A), dorsal striatum (B) and ventral striatum (C) of a Tet/DN-GSK-3 mouse. (i–vi) Magnifications of the cleaved caspase-3-positive cells shown in panels (A–C). Scale bar in panel (A) corresponds to 200 μm in panels (A–C). (D) Histogram showing the incidence of cleaved caspase-3 immunopositive cells in striatum, cortex, hippocampus and cerebellum of Tet/DN-GSK-3 mice and their wild-type (wt) littermates. Data are presented as the mean±s.e.m. number of immunopositive cells per 7 mm2 in a 30 μm section (n=9, *P<0.05). (E–F) Double labeling immunofluorescence with anti-cleaved caspase-3 (E) and anti-β-gal (F) antibodies in the striatum of a Tet/DN-GSK-3 mouse. Scale bar in panel (E) corresponds to 10 μm in panels (E and F). (G, H) TUNEL staining in the striatum of a wt (G) and of a Tet/DN-GSK-3 (H) mouse. Scale bar in panel (G) corresponds to 80 μm in panels (G, H); cc, corpus callosum; GP, globus pallidus; CPu, caudate putamen; ac, anterior commisure; NA, nucleus accumbens; Tu, olfactory tubercle.
Tet/DN-GSK-3 mice show reduced motor coordination
The striatum and the cerebral cortex, the brain regions where increased apoptosis was detected in Tet/DN-GSK-3 mice, are part of the basal ganglia circuit involved in motor control. For this reason we decided to analyze Tet/DN-GSK-3 mice in various tests of motor coordination. We first analyzed the Tet/DN-GSK-3 mice in the rotarod apparatus (Figure 4A–C). After pretraining of mice at constant speed, the rotarod was set to accelerate from 4 to 40 r.p.m. over 5 min and mice were tested four times at 1-h intervals. As shown in Figure 4A, Tet/DN-GSK-3 mice showed a marked deficit in the three first accelerating trials. On average, across the four tests, only 38.6±4.6% of Tet/DN-GSK-3 mice versus 58.8±4.1% of the wild-type mice remained on rod when it reached maximal speed (Figure 4B), and the total time on rod was 262.1±12.2 s for wild type and 187.66±13.4 s for Tet/DN-GSK-3 mice (P<0.007; Figure 4C). The motor coordination deficit in Tet/DN-GSK-3 mice was confirmed in the vertical pole test that measures the time needed to descend along a rough-surfaced pole and detects striatal-dependent motor deficits (Matsuura et al, 1997). As shown in Figure 4D, Tet/DN-GSK-3 mice required almost double amount of time to descend as compared with the wild-type mice (43.15±9.44 versus 25.36±3.62 s; P<0.03). We then performed analysis of stride length in the footprint test. As shown, in Figure 4E, Tet/DN-GSK-3 mice also showed a reduction in stride length (56.31±0.87 versus 53.17±1.07 mm; P<0.03) and higher maximal difference on stride length (23.56±1.95 versus 28.82±1.41 mm; P<0.02), thus further demonstrating a deficit in motor coordination. Finally, to confirm the specificity of the observed motor coordination deficit, we analyze the general motor activity of Tet/DN-GSK-3 mice in the open field test (Figure 4A). Interestingly, no difference between control and Tet/DN-GSK-3 mice was observed either during the first minute (transferal arousal) or in the rest of the testing session.
Figure 4.
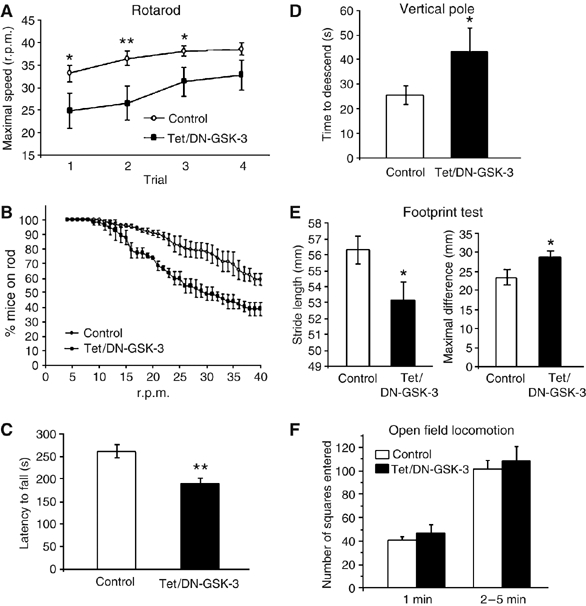
Tet/DN-GSK-3 mice show impaired motor coordination. Motor behavior tests were performed on 3-month-old control and Tet/DN-GSK-3 mice. (A–C) Rotarod. (A) Performance in the four accelerating trials. (B) Average percentage of mice staying on rod in the four trials as the rod accelerates. (C) Histogram showing the mean±s.e.m. latencies to fall from rod in the four trials per genotype (*P<0.05). (D) Time to descend in the vertical pole test (*P<0.03). (E) Average stride length (*P<0.03) and maximal difference in stride length (*P<0.02) in the footprint test. (F) Open field locomotion test. The number of squares entered was counted during the 5 min testing period. Performance in the first minute is also depicted to distinguish the effect of the transferal arousal from the total activity.
Reduced dopamine-dependent behavior in Tet/DN-GSK-3 mice
The motor deficit in Tet/DN-GSK-3 mice might be explained at least in part by the increased incidence of apoptosis in basal ganglia regions, cortex and striatum. On the other hand, dopamine (DA) neurotransmission from midbrain to the striatum is a key determinant of the activity of striatal neurons and, as a consequence, of motor behavior. Interestingly, striatal GSK-3 activity has recently been shown to be an important mediator of DA action on striatal function and behavior (Beaulieu et al, 2004). It is therefore possible that the observed motor deficit also results from decreased DA-dependent striatal function and behavior in Tet/DN-GSK-3 mice. To explore whether Tet/DN-GSK-3 mice indeed show reduced striatal activation and reduced motor activity in response to DA, wild-type and Tet/DN-GSK-3 mice were challenged with amphetamine to induce a classic DA-dependent behavioral response. As shown in Figure 5A, in vivo microdialysis revealed that systemic administration of amphetamine (2.5 mg/kg, i.p.) induced a similar increase in extracellular DA levels in both groups of mice. Two-way ANOVA revealed a significant effect of amphetamine (F1,15=3.76, P<0.00002), but no significant effect of genotype or genotype × amphetamine interaction. The maximal effect on striatal DA levels was 224±41% of baseline in wild-type mice and 229±39% in transgenic mice. However, despite equivalent striatal DA release, this amphetamine challenge increased locomotor activity in wild-type but not Tet/DN-GSK-3 mice (Figure 5B). In good agreement, the amphetamine-induced expression of c-Fos (a marker of neuronal activity) in striatum was markedly lower in Tet/DN-GSK-3 (Figure 5C–F). Altogether, these results indicate that (a) the dopaminergic (presynaptic) component of the nigrostriatal pathway is unaltered in Tet/DN-GSK-3 mice and (b) the reduction of DA-dependent behaviors in transgenic mice can be attributed to an attenuation of postsynaptic dopaminergic signalling resulting from the stratial expression of DN-GSK-3.
Figure 5.
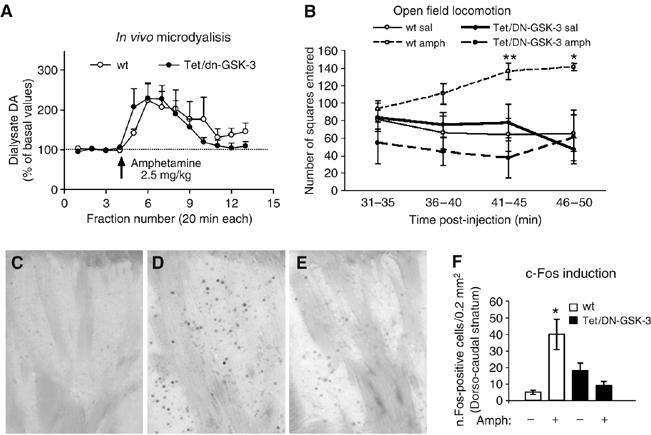
Reduced striatal c-Fos induction and reduced motor activity in response to a dopaminergic challenge. Amphetamine (2.5 mg/kg, i.p.) was administered to induce DA release in the striatum. (A) In vivo microdialysis: wild-type (wt) and Tet/DN-GSK-3 mice (n=4 and 8, respectively) were stereotaxically implanted with microdialysis probes in the striatum. Dialysate fractions were collected every 20 min and the concentration of DA in dialysate samples was determined by HPLC. (B) Open field locomotion 31–50 min after saline or amphetamine i.p. injection to wt and Tet/DN-GSK-3 mice (n=4 and 8, respectively). (C–E) Representative images of immunohistochemistry of c-Fos in the striatum after 90 min of a saline injection to a wt mouse (C) or amphetamine (2.5 mg/kg i.p) injections to a wt mouse (D) and a Tet/DN-GSK-3 mouse. (F) Histogram showing the quantification of the mean±s.e.m. number of c-Fos immunoreactive nuclei in the striatum of saline or amphetamine treated wt and Tet/DN-GSK-3 mice (*P<0.05).
Reversal of motor deficit and of neuronal death in doxycycline-treated Tet/DN-GSK-3 mice
Selective GSK-3 inhibitors are under development for the treatment of chronic neurodegenerative and metabolic conditions (Cohen and Goedert, 2004). However, the above described toxicity for adult basal ganglia neurons elicited by DN-GSK-3 expression suggests possible motor side effects if selective GSK-3 inhibitors are given at excessive doses. This prompted us to explore whether the neuronal apoptosis and motor-deficit phenotype was susceptible to revert upon cessation of transgene expression by giving tetracyclines to symptomatic Tet/DN-GSK-3 mice. For this, we first verified that doxycycline administration results in efficient transgene shut-down in Tet/DN-GSK-3 mice as a first step before assessing whether restoration of normal GSK-3 levels would also result in normal GSK-3 activity. As expected, according to our previous experience in shutting down transgene expression in similar conditional mouse models (Yamamoto et al, 2000; Diaz-Hernandez et al, 2005; Engel et al, 2006), 6 weeks of a high dose of doxycycline (2 mg/ml in the drinking water) or 4 months of a low dose (0.5 mg/ml) led to undetectable levels of the reporter transgene expression in treated (Gene-Off) Tet/DN-GSK-3 mice (Figure 6A and B). Similarly, GSK-3β and p-GSK-3 levels were no longer increased respect to non-transgenic littermates (data not shown).
Figure 6.
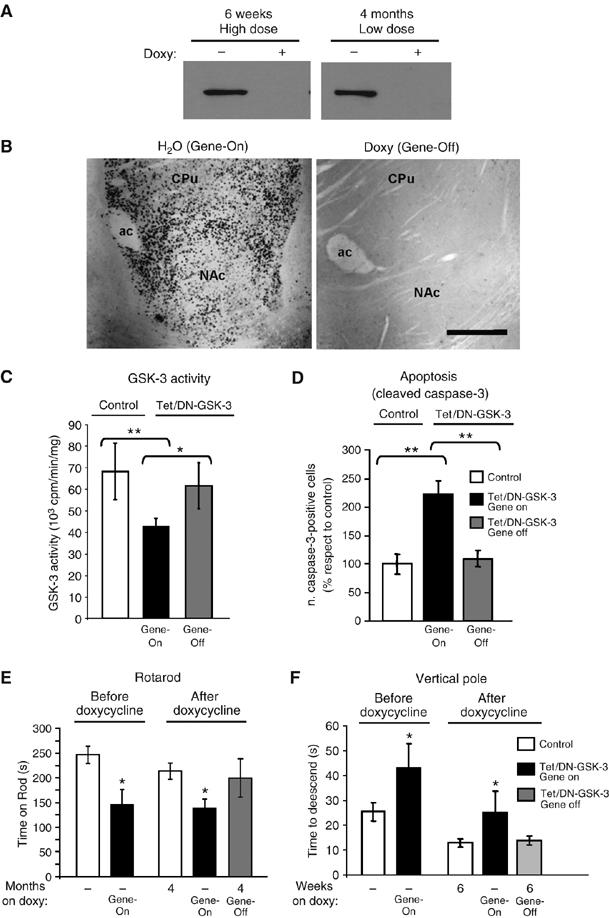
Effects of DN-GSK-3 expression revert after doxycycline treatment. (A) Western blot detection of β-gal in the striatum of Tet/DN-GSK-3 mice either untreated or doxycycline-treated for 6 weeks with the high-dose and for 4 months with the low-dose paradigms. (B) Immunohistochemistry of β-gal in the striatum of Tet/DN-GSK-3 mice either untreated (Gene-ON) or treated (Gene-OFF) with the high-doxycycline paradigm. Scale bar corresponds to 150 μm. (C) In vitro GSK-3 activity assay on striatal homogenates. (*P<0.05; **P<0.001). (D) Immunohistochemical detection of cleaved caspase-3-positive cells in the striatum. The number of immunopositive cells in striatum per 30 μm section from either gene-On or Gene-Off Tet/DN-GSK-3 mice was quantified and normalized with respect to control mice (**P<0.001). (E) Performance on accelerating rotarod of wild-type and Tet/DN-GSK-3 mice at the age of 3 months, without pharmacological intervention and at the age of 7 months after the 4 month low-doxycycline paradigm. (*P<0.01). (F) Performance on vertical pole of wild-type and Tet/DN-GSK-3 mice at the age of 3 months, without pharmacological intervention and at the age of 4.5 months after the 6 week high-doxycycline paradigm. (*P<0.01).
We then analyzed whether GSK-3 activity was susceptible to return to normal levels after doxycycline treatment. For this, untreated (Gene-On) and treated (Gene-Off) Tet/DN-GSK-3 mice and their respective control littermates were analyzed by in vitro GSK-3 enzymatic assays on striatal homogenates. As shown in Figure 6C, Gene-Off Tet/DN-GSK-3 mice showed significantly increased GSK-3 activity compared to Gene-On Tet/DN-GSK-3 mice and they were indistinguishable from their control littermates. This demonstrates that restoration of normal GSK-3 levels also results in normal GSK-3 activity.
This prompted us to analyze whether restoration of normal GSK-3 activity would also prevent the increased incidence of neuronal apoptosis detected in Tet/DN-GSK-3 mice. As shown in Figure 6D, the increase in the number of cleaved caspase-3-positive neurons observed in gene-On Tet/DN-GSK-3 mice was no longer detected in gene-Off Tet/DN-GSK-3 mice.
Finally, the potential reversibility of the Tet/DN-GSK-3 mouse motor phenotype was also explored. As shown in Figures 4A–C and 6E, 3-month-old Tet/DN-GSK-3 mice show a motor deficit in the rotarod apparatus. We then split the Tet/DN-GSK-3 mice shown in Figure 6E and their respective control littermates into two groups. One group was maintained without any pharmacological intervention and the other was given doxycycline in the low-dose administration paradigm (see Materials and methods) previously shown to efficiently stop transgene expression without affecting rotarod performance in control mice (Diaz-Hernandez et al, 2005). Untreated (Gene-On) and treated (Gene-Off) Tet/DN-GSK-3 mice and their respective control littermates were then retested every month in the accelerating rotarod test. Despite a tendency for improved rotarod performance over time after 1, 2 or 3 months of doxycycline treatment, this did not reach statistical significance (data not shown). However, as shown in Figure 6E, after 4 months, Gene-off Tet/DN-GSK-3 mice showed a significantly better performance in rotarod compared to Gene-on Tet/DN-GSK-3, and they were indistinguishable from wild-type mice. Similarly, 6 weeks of 2 mg/ml doxycycline also resulted in reversal of the deficit observed in the vertical pole test and the average stride length was no longer significantly different between untreated (Gene-On) and treated (Gene-Off) Tet/DN-GSK-3 mice (Figure 6F and data not shown). Together, these results suggest that potential motor side effects of excessive pharmacological GSK-3 inhibition might be susceptible to revert.
Discussion
By generating conditional transgenic mice with expression of DN-GSK-3 in adult forebrain neurons, here we show that a sustained reduction in GSK-3 activity is deleterious for striatal and cortical neurons, as evidenced by histological detection of apoptosis and by a concomitant deficit in motor coordination. Interestingly, by shutting down DN-GSK-3 expression in symptomatic mice, we found that both the motor-impairment phenotype and the neuronal toxicity were susceptible to revert upon restoration of normal GSK-3 activity.
Extensive data have proposed GSK-3 inhibitors as neuroprotective and antiapoptotic agents (Frame and Cohen, 2001; Grimes and Jope, 2001; Beurel and Jope, 2006). Supporting reports explore many different apoptotic stimuli, including trophic support withdrawal, PI3-kinase inhibition, DNA damage, mitochondrial toxins, hypoxia/ischemia and glutamate excitotoxicity, among others (Pap and Cooper, 1998; Hetman et al, 2000; Beurel and Jope, 2006). However, decreased GSK-3 activity by disruption of the murine GSK-3β gene was also known to result in embryonic lethality caused by massive apoptosis in liver during mid-gestation (Hoeflich et al, 2000). Our results of neuronal apoptosis in striatum and cortex of Tet/DN-GSK-3 mice further confirm that chronically decreased GSK-3 activity compromises cell viability in vivo and extends this finding to adult tissues and post-mitotic cells such as neurons. In this regard, there is an apparent discrepancy between the regional extent of GSK-3 inhibition and of detection of apoptosis in Tet/DN-GSK-3 mice because apoptosis is found in cortex, where no significant decrease of GSK-3 activity is detected by enzymatic assays or by Ser21/9GSK-3 Western blot in cortical homogenates. This can be explained by the fact that, in the cortex only a fraction of neurons express the transgene. More precisely, expression is restricted to certain neurons within layers I–III. It is in these layers that increased apoptosis is detected by techniques with cellular resolution (e.g. cleaved caspase-3 staining). However, cortical homogenates for biochemical measurement of GSK-3 activity include layers IV–VI, as well as other non-expressing cells in layers I–III. Therefore, enzymatic activity measurements in cortical samples, despite showing a tendency towards decreased activity, do not detect the inhibition in specific transgene expressing neurons due to a dilution effect of those neurons within the whole homogenate.
There are also some reports of GSK-3 inhibitor treatment resulting in facilitation of apoptosis. More precisely, in apoptosis triggered in cultured cells by TNFα (Beyaert et al, 1989; Hoeflich et al, 2000) or, including in neurons, by agonistic anti-Fas antibodies (Song et al, 2004; Beurel and Jope, 2006). Interestingly, an explanation for this paradoxical role of GSK-3 activity and inhibition on apoptosis regulation has recently been proposed (Beurel and Jope, 2006). It seems that inhibition of GSK-3 activity facilitates apoptosis executed through the extrinsic (involving stimulation of death domain containing receptors) pathway, but prevents apoptosis execution if it is elicited through the intrinsic (mitochondria mediated) pathway. In view of this, it is therefore likely that embryonic hepatocytes and adult cortical and striatal neurons have in common a tonic influence of extracellular signals able to stimulate death domain containing receptors leading to a situation in which intact GSK-3 activity would be required to prevent apoptosis execution.
The results reported here are indicative of the effects of sustained GSK-3 inhibition and, to some extent, they may have implications when considering the potential side effects and the potential therapeutic efficacy of chronic administration of the potent and selective GSK-3 inhibitors that are currently under development for treatment of chronic conditions such as Alzheimer's disease, mood disorders and diabetes (Cohen and Goedert, 2004; Frame and Zheleva, 2006; Gould et al, 2006). However, it should also be noticed that drug administration can vary greatly from genetic inhibition in terms of the efficacy, retention and clearance and the specificity of treatments. Regarding possible side effects, as above mentioned, one of the a priori concerns is the tumorigenic potential of chronic GSK-3 inhibition (Polakis, 2000). In this regard, we did not find any evidence of tumor formation in Tet/DN-GSK-3 mice. However, this is not surprising, since transgene expression in these mice is restricted to neurons that are post-mitotic cells. Breeding DN-GSK-3 mice with driver mice expressing tTA under control of broader expression promoters will give a more comprehensive view of the tumorigenesis risk. Apoptosis, the other predicted potential side effect, is confirmed in neurons of Tet/DN-GSK-3 mice and therefore suggests potential neurological consequences of chronic GSK-3 inhibitor administration that are further supported by the motor phenotype. However, since the rational for using GSK-3 inhibitors arises from the concept of aberrantly increased GSK-3 activity contributing to the etiology of various disorders, it is likely that these inhibitors will prove effective and safe if they are given to a dose that decreases GSK-3 activity without lowering it beyond its normal level.
Regarding the value of Tet/DN-GSK-3 mice in predicting the therapeutic potential of inhibitors, this can be explored by combining these mice with animal models of the various related disorders. In the case of Alzheimer's disease, GSK-3 has been linked to β-amyloid (Aβ) production from its precursor APP (Sun et al, 2002; Phiel et al, 2003; Ryder et al, 2003), to mediate Aβ-induced toxicity (Takashima et al, 1993; Alvarez et al, 1999), to contribute to mutant Presenilin-1 (PS-1) toxicity (Takashima et al, 1998), and to tau hyperphosphorylation (Hanger et al, 1992; Lovestone et al, 1994). Since Tet/DN-GSK-3 mice express the transgene in neurons of the brain regions affected in Alzheimer's disease, it would be interesting to combine them with any of the many available mouse models of Alzheimer's disease including those with modified expression of APP, PS-1, tau (Lim et al, 2001; Wong et al, 2002), or combinations of these (Engel et al, 2005). If neuropathology and/or learning deficit improve in the mice resulting from combining Alzheimer's disease models with Tet/DN-GSK-3 mice, this would strongly support the therapeutic potential of GSK-3 inhibitors for this disease. However, to be predictive of the potential of GSK-3 inhibitors to treat metabolic disorders like diabetes, Tet/DN-GSK-3 mice should be generated with muscle and/or liver promoters to then be combined with chemical or transgenic models of diabetes.
The observed reduction in DA-dependent striatal induction of c-Fos and locomotor behavior in Tet/DN-GSK-3 mice fits well with the previously reported role of GSK-3 as an important mediator of DA actions in the striatum (Beaulieu et al, 2004). This, together with the here reported importance of intact GSK-3 activity for cortical and striatal neuron viability, may explain not only the motor deficit observed in Tet/DN-GSK-3 mice but also the tremor that appears as the most prominent motor side effect of the therapy of bipolar disorder with lithium, a well established GSK-3 inhibitor (Klein and Melton, 1996; Macritchie and Young, 2004).
In summary, the here reported generation and characterization of conditional transgenic mice with postnatal neuron expression of DN-GSK-3 revealed a key role of GSK-3 in adult neuron physiology and viability, and warns of potential neurological side effects of chronic administration of GSK-3 inhibitors. Interestingly, the reported reversibility data strongly support that such unwanted side effects are likely to revert if excessive GSK-3 inhibition is halted. Besides, the future combination of these DN-GSK-3 mice with mouse models of the various diseases linked to increased GSK-3 activity will help to elucidate the therapeutic potential of GSK-3 inhibitor therapies for each of these diseases.
Materials and methods
The Materials and methods are only described very briefly. For further information, see the Supplementary data section.
Generation of Tet/DN-GSK-3 mice
To generate Tet/DN-GSK-3β mice, wild-type GSK-3β sequence in the pBI-G-GSK-3 plasmid (Lucas et al, 2001) was replaced by the K85R-GSK-3β sequence from the XGSK3β-R85 plasmid (Munoz-Montano et al, 1999). The transgene fragment was microinjected into single cell FVB/N embryos. Four founder, mice were identified by PCR and each founder line was amplified by backcrossing with wild-type FVB/N mice. The CamkII-tTa mouse line (Mayford et al, 1996) is maintained in a mixed CBAxC57BL/6 background.
Doxycycline treatment
Tet/DN-GSK-3β mice and their control littermates were given doxycycline (Sigma) (2 mg/ml) in drinking water ad libitum for 6 weeks. We have previously shown that this paradigm results in complete shut-down of the transgene in a similar conditional mouse model that overexpresses wild-type GSK-3 (Lucas et al, 2001; Engel et al, 2006). To explore reversal of rotarod phenotype, doxycycline was given at 2 mg/ml for 1 week, 1 mg/ml for a second week and 0.5 mg/ml the rest of the treatment session up to 4 months. This paradigm also results in complete transgene shut-down in Tet/DN-GSK-3β mice, without worsening rotarod performance in wild-type mice (Diaz-Hernandez et al, 2005).
Immunohistochemistry and immunofluorescence
Brains were fixed in 4% paraformaldehyde in PBS and cryoprotected. Sagittal sections were pretreated for 30 min in 1% H2O2/PBS, followed by 1 h with 1% BSA, 5% FBS and 0.2% Triton X-100, incubated overnight with primary antibody and developed with the Elite Vectastain kit (Vector Laboratories) using diaminobenzidine (Sigma) and 0.003% H2O2. For immunofluorescence, after treatment with primary antibody, the sections were washed in PBS and incubated with the following secondary antibodies: goat anti-rabbit Alexa 488 (Invitrogen) and goat anti-mouse Alexa 594 (Invitrogen). Nuclei were stained by using DAPI (1:5000, Calbiochem).
TUNEL assay
Mice were anesthetized and transcardially perfused with 4% PFA in PBS for 10 min. Brains were post-fixed in 4% PFA for 2 h at 4°C and cryoprotected. Sections were treated following the protocol of the In Situ Cell Death Detection Kit, POD (Roche).
Western blot analysis
The protocols are described in detail in the Supplementary data section.
GSK-3 activity assay
Tissue was homogenized in 20 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM NaF, 1 mM VO4Na, 1% Triton 100-X, 1 mM EDTA, 1 mM EGTA and a cocktail of peptidase inhibitors (Roche). Supernatants were collected after centrifugation at 14 000 g for 15 min. The GS1 peptide (YRRAAVPPSPSLSRHSSPHQS*EDEE) with phosphorylated ser21 was used as substrate (Stambolic and Woodgett, 1994). Supernatants were incubated at 37°C with GS1 peptide and [γ-32P]ATP in 25 mM Tris–HCl pH 7.5, 1 mM DTT 10 mM MgCl2 and either 20 mM NaCl or 20 mM LiCl. The assays were stopped by spotting aliquots on P81 phosphocellulose as described (Engel et al, 2006).
Behavioral testing
Open field locomotion: Major locomotor activity was tested as in the SHIRPA protocol (Rogers et al, 1997). Briefly, the arena (55 × 33 × 18 cm) is divided in equally-dimensioned squares (about 11 cm). Locomotion activity (as number of square entered with all the four paws) is counted during 5 min.
Rotarod test was performed with accelerating rotarod apparatus (Ugo Basile, Comerio, Italy). Mice were pretrained during 2 days. Then, the rotarod was set to accelerate from 4 to 40 r.p.m. over 5 min and mice were tested four times. During accelerating trials, the latency to fall from the rotarod was measured.
Vertical pole test was performed as described (Matsuura et al, 1997). The mouse was placed head-upward on the top of a vertical rough-surfaced pole (diameter 1 cm; height 50 cm) and the time until it descended to the floor was recorded, with a maximum duration of 120 s.
Footprint pattern test was performed as described (Barlow et al, 1996). The back paws of each mouse were dipped into ink and it was placed at the entrance of a dark tunnel (9.2 × 6.3 × 35.5 cm). The footprints were recorded on paper. Stride lengths were determined by measuring the distance between each step on the same side of the body.
In vivo microdialysis
Microdialysis was performed as described (Díaz-Mataix et al, 2005). Anesthetized mice were stereotaxically implanted with microdialysis probes in the striatum at the following coordinates (in millimeters): AP+0.5, L −1.7, DV −4.5, according to the mouse atlas of Franklin and Paxinos (1997). Microdialysis was performed ∼20 h after surgery. Probes were perfused with artificial cerebrospinal fluid. Dialysates were collected every 20 min. After a 100 min stabilization, four fractions were collected for basal values before systemic administration of amphetamine. Then, dialysis samples were collected. The DA concentration in dialysates was determined by HPLC.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary data
Acknowledgments
We thank Drs MR Fernandez, J Díaz-Nido and F Wandosell for helpful discussion, and Dr Peter Davies for kindly providing the PHF-1 antibody. We also thank Oscar Pintado for transgene-DNA microinjection and Javier Palacín, Desiree Ruiz, Raquel Cuadros and Elena Langa for technical assistance. This work was supported by Spanish Ministry of Science and Education (Grants: SAF 2004-05525 to FA, SAF2006-02424 to JA, and SAF 2003-04144 and SAF 2006-05995 to JJL), Comunidad de Madrid, Fundación ‘La Caixa', Fundación Ramón Areces and Fondo de Investigaciones Sanitarias/isciii-CiberNed. Analía Bortolozzi is recipient of a Ramón y Cajal contract from MEC through IDIBAPS.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997) beta-catenin is a target for the ubiquitin–proteasome pathway. EMBO J 16: 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J (1999) Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett 453: 260–264 [DOI] [PubMed] [Google Scholar]
- Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84: 361–384 [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG (2004) Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA 101: 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Jope RS (2006) The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 79: 173–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyaert R, Vanhaesebroeck B, Suffys P, Van Roy F, Fiers W (1989) Lithium chloride potentiates tumor necrosis factor-mediated cytotoxicity in vitro and in vivo. Proc Natl Acad Sci USA 86: 9494–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Goedert M (2004) GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 3: 479–487 [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Torres-Peraza J, Salvatori-Abarca A, Moran MA, Gomez-Ramos P, Alberch J, Lucas JJ (2005) Full motor recovery despite striatal neuron loss and formation of irreversible amyloid-like inclusions in a conditional mouse model of Huntington's disease. J Neurosci 25: 9773–9781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Mataix L, Scorza MC, Bortolozzi A, Toth M, Celada P, Artigas F (2005) Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity. Role in atypical antipsychotic action. J Neruosci 25: 10831–10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez I, Itoh K, Sokol SY (1995) Role of glycogen synthase kinase 3 beta as a negative regulator of dorsoventral axis formation in Xenopus embryos. Proc Natl Acad Sci USA 92: 8498–8502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H (2002) Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med 8: 126–132 [DOI] [PubMed] [Google Scholar]
- Engel T, Hernandez F, Avila J, Lucas JJ (2006) Full reversal of Alzheimer's disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J Neurosci 26: 5083–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel T, Lucas JJ, Gomez-Ramos P, Moran MA, Avila J, Hernandez F (2005) Cooexpression of FTDP-17 tau and GSK-3beta in transgenic mice induce tau polymerization and neurodegeneration. Neurobiol Aging 27: 1258–1268 [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Zheleva D (2006) Targeting glycogen synthase kinase-3 in insulin signalling. Expert Opin Ther Targets 10: 429–444 [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (1997) The Mouse Brain in Stereotaxic Coordinates. Academic Press: San Diego [Google Scholar]
- Gingrich JR, Roder J (1998) Inducible gene expression in the nervous system of transgenic mice. Annu Rev Neurosci 21: 377–405 [DOI] [PubMed] [Google Scholar]
- Gould TD, Picchini AM, Einat H, Manji HK (2006) Targeting glycogen synthase kinase-3 in the CNS: implications for the development of new treatments for mood disorders. Curr Drug Targets 7: 1399–1409 [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS (2001) The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol 65: 391–426 [DOI] [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH (1992) Glycogen synthase kinase-3 induces Alzheimer's disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett 147: 58–62 [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000) Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR (2000) Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 406: 86–90 [DOI] [PubMed] [Google Scholar]
- Jope RS (2003) Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 24: 441–443 [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 29: 95–102 [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA (1996) A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 93: 8455–8459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M (2001) Conditional control of gene expression in the mouse. Nat Rev Genet 2: 743–755 [DOI] [PubMed] [Google Scholar]
- Liang D, Li X, Clark JD (2004) Increased expression of Ca2+/calmodulin-dependent protein kinase II alpha during chronic morphine exposure. Neuroscience 123: 769–775 [DOI] [PubMed] [Google Scholar]
- Lim F, Hernandez F, Lucas JJ, Gomez-Ramos P, Moran MA, Avila J (2001) FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol Cell Neurosci 18: 702–714 [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH, Latimer D, Davis DR, Anderton BH, Gallo JM, Hanger D, Mulot S, Marquardt B, Stabel S, Woodgett JR, Miller CCJ (1994) Alzheimer's disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol 4: 1077–1086 [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. Embo J 20: 27–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macritchie KAN, Young AH (2004) Adverse syndromes associated with lithium. In: Adverse Syndromes & Psychiatric Drugs. A Clinical Guide, Haddad P, Dursun S, Deakin B (eds), pp 89–109. Oxford University Press: USA [Google Scholar]
- Matsuura K, Kabuto H, Makino H, Ogawa N (1997) Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods 73: 45–48 [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER (1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678–1683 [DOI] [PubMed] [Google Scholar]
- Munoz-Montano JR, Lim F, Moreno FJ, Avila J, Diaz-Nido J (1999) Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer's disease. J Alzheimers Dis 1: 361–378 [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-Kinase/Akt cell survival pathway. J Biol Chem 273: 19929–19932 [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435–439 [DOI] [PubMed] [Google Scholar]
- Polakis P (2000) Wnt signaling and cancer. Genes Dev 14: 1837–1851 [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE (1997) Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome 8: 711–713 [DOI] [PubMed] [Google Scholar]
- Ryder J, Su Y, Liu F, Li B, Zhou Y, Ni B (2003) Divergent roles of GSK3 and CDK5 in APP processing. Biochem Biophys Res Commun 312: 922–929 [DOI] [PubMed] [Google Scholar]
- Song L, Zhou T, Jope RS (2004) Lithium facilitates apoptotic signaling induced by activation of the Fas death domain-containing receptor. BMC Neurosci 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Woodgett JR (1994) Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem J 303: 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Sato S, Murayama O, Murayama M, Park JM, Yamaguchi H, Takashima A (2002) Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci Lett 321: 61–64 [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B (1998) Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci USA 95: 9637–9641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci USA 90: 7789–7793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Cai H, Borchelt DR, Price DL (2002) Genetically engineered mouse models of neurodegenerative diseases. Nat Neurosci 5: 633–639 [DOI] [PubMed] [Google Scholar]
- Woodgett JR (1990) Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J 9: 2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57–66 [DOI] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS (2003) Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem 278: 33067–33077 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary data