

Abstract
Familial prion diseases are due to dominantly inherited, germline mutations in the PRNP gene that encodes the prion protein (PrP). The cellular mechanism underlying the pathogenic effect of these mutations remains uncertain. To investigate whether pathogenic mutations impair a normal, physiological activity of PrP, we have crossed Tg(PG14) mice, which express PrP with an octapeptide insertion associated with an inherited prion dementia, with Tg(PrPΔ32–134) mice. Tg(PrPΔ32–134) mice, which express an N-terminally truncated form of PrP, spontaneously develop a neurodegenerative phenotype that is stoichiometrically reversed by coexpression of wild-type PrP. We find that, at equivalent expression levels, PG14 PrP is significantly less efficient than wild-type PrP in suppressing the development of clinical symptoms and neuropathology in Tg(PrPΔ32–134) mice. Thus, our results suggest that some features of the neurological illness associated with inherited PrP mutations may be attributable to a loss of PrP neuroprotective function. This mechanism stands in contrast to the toxic gain-of-function mechanisms that are usually invoked to explain the pathogenesis of dominantly inherited neurodegenerative disorders.
Keywords: mutation, neuroprotection, octapeptide, prion, transgenic
Introduction
The prion disorders include kuru, Creutzfeldt-Jakob disease, Gerstmann-Sträussler-Scheinker disease and fatal familial insomnia in man, as well as scrapie, bovine spongiform encephalopathy and chronic wasting disease in animals (Prusiner, 2004). The central molecular event in prion diseases is the conformational conversion of PrPC, a normal cell-surface glycoprotein, into PrPSc, an abnormal isoform proposed to be infectious in the absence of nucleic acid (Prusiner, 1998; Weissmann, 2004). The precise structural differences between the two PrP isoforms remain to be defined, although it is clear that PrPSc contains significantly more β-sheet and is more protease-resistant and aggregated than PrPC. The conversion of PrPC to PrPSc is thought to involve a templating mechanism in which the two forms physically interact.
A subset of prion diseases is inherited in an autosomal dominant fashion. This group, which includes 10% of the cases of Creutzfeldt–Jakob disease as well as all cases of Gerstmann-Sträussler-Scheinker disease and fatal familial insomnia, are linked to germline mutations in the gene (designated PRNP) on chromosome 20 that encodes PrP (Kong et al, 2004). Point mutations occur in the C-terminal half of the PrP molecule. Insertional mutations consist of 1–9 additional copies of an octapeptide repeat that is normally present in five copies in the N-terminal half of the protein.
The mechanism by which PrP mutations cause familial prion diseases has been enigmatic. One hypothesis invokes a toxic gain-of-function mechanism in which mutant PrP misfolds into a PrPSc-like conformation that interferes with essential cellular processes or activates cell death pathways (Harris and True, 2006). Toxic gain-of-function is commonly invoked to explain the phenotypes of other dominantly inherited neurodegenerative disorders that are characterized by protein misfolding and aggregation, including Alzheimer's, Huntington's, and Parkinson's diseases (Taylor et al, 2002).
However, this mechanism may not be applicable to all familial prion diseases, based on studies of how different disease-associated mutations affect the molecular properties of PrP. Some mutations markedly destabilize recombinant PrP, favoring increased formation of partially folded intermediates and assembly into PrPSc-like aggregates (Riek et al, 1998; Liemann and Glockshuber, 1999; Vanik and Surewicz, 2002; Apetri et al, 2004, 2005). However, other mutations have very little effect on the stability or folding kinetics of PrP (Swietnicki et al, 1998; Liemann and Glockshuber, 1999; Apetri et al, 2004). The NMR-derived structure of one such mutant (E200K) is virtually indistinguishable from that of wild-type PrP (Zhang et al, 2000), and the same result is predicted for several other mutants (Riek et al, 1998). In addition, there is a wide variation in the biochemical properties of different mutant PrP molecules expressed in cultured cells and brain tissue, with some mutants displaying little or no aggregation and protease-resistance (Lehmann and Harris, 1996; Hegde et al, 1998; Priola and Chesebro, 1998). It is difficult to explain the pathogenicity of these latter mutants on the basis of the accumulation of toxic protein aggregates.
An alternative hypothesis is that some pathogenic mutations impair a physiological function normally performed by PrPC (Harris and True, 2006). This idea has received relatively little attention, largely because the normal cellular function of PrPC has remained mysterious. However, several lines of evidence have emerged recently suggesting that PrPC may function as a cytoprotective molecule, rescuing cells from apoptotic and other stresses (reviewed by Roucou and LeBlanc, 2005). A particularly compelling example of this protective activity is observed in transgenic mice that express the PrP paralog, Doppel (Dpl) (Rossi et al, 2001; Yamaguchi et al, 2004), or certain N-terminally deleted forms of PrP, including Δ32–134 and Δ32–121 (Shmerling et al, 1998), Δ94–134 (Baumann et al, 2007), and Δ105–125 (Li et al, 2007). Strikingly, coexpression of wild-type PrP ameliorates or completely abrogates the neurodegenerative phenotypes of these mice. Suppression of neurodegeneration by wild-type PrP is dose-dependent and is inversely related to the severity of the phenotype induced by the toxic protein.
Tg(PrPΔ32–134) and related mice provide sensitive, in vivo systems to assay the neuroprotective activity of PrPC. By coexpressing PrP test molecules in these animals, the ability of the test molecules to rescue the neurodegenerative phenotype can be assessed. In the present paper, we have crossed Tg(PrPΔ32–134) mice with Tg(PG14) mice. Tg(PG14) mice express a mutant form of PrP (designated PG14) carrying a nine-octapeptide insertion that is associated with an inherited prion dementia in humans (Owen et al, 1992; Duchen et al, 1993; Krasemann et al, 1995). These animals recapitulate several of the essential features of inherited human prion diseases, including progressive ataxia, neuronal loss, astrocytosis, and accumulation of an abnormally folded form of mutant PrP (Chiesa et al, 1998, 2000). We find that PG14 PrP is significantly less efficient than wild-type PrP in suppressing the neurodegenerative phenotype of Tg(PrPΔ32–134) mice. Thus, our results suggest that some features of the neurological illness associated with the PG14 mutation may be attributable to a loss of PrP neuroprotective function.
Results
Expression of PG14 PrP ameliorates clinical symptoms in Tg(F35) mice less efficiently than wild-type PrP
Tg(F35+/0)/Prn-p0/0 mice expressing PrPΔ32–134 in the absence of endogenous PrP develop ataxia, tremor, kyphosis, and weight loss beginning at 28±2 days of age (Figure 1A), with the illness progressing to the terminal stage by 83±3 days of age (Figure 1B). As reported earlier (Shmerling et al, 1998), coexpression of wild-type PrP from a single, endogenous PrP allele in Tg(F35+/0)/Prn-p+/0 mice completely abrogates clinical symptoms (Figure 1A and B).
Figure 1.
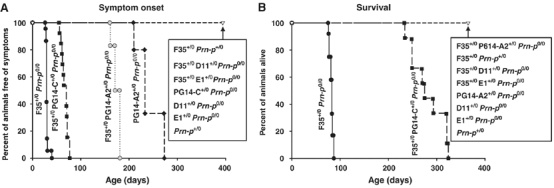
PG14 PrP does not fully suppress clinical symptoms in Tg(F35) mice. Onset of symptoms (A) and survival (B) were monitored in mice of the following genotypes, with the number of animals indicated in parentheses: F35+/0 Prn-p0/0 (14); F35+/0 PG14-A2+/0 Prn-p0/0 (11); F35+/0 Prn-p+/0 (9); F35+/0 PrP-E1+/0 Prn-p0/0 (8); F35+/0 PG14-C+/0 Prn-p0/0 (14); F35+/0 D11+/0 Prn-p0/0 (12); PG14-A2+/0 Prn-p0/0 (18); PG14-C+/0 Prn-p0/0 (14); D11+/0 Prn-p0/0 (11); E1+/0 Prn-p0/0 (10); Prn-p+/0 (13).
To test the ability of PG14 PrP to rescue the neurodegenerative phenotype induced by PrPΔ32–134, we crossed Tg(F35) mice with Tg(PG14-A2) mice. The latter animals express PG14 PrP at levels that are similar by Western blotting to that of endogenous PrP in Prn-p+/0 mice (Figure 2; lanes 3, 5, 10 and 12). In contrast to wild-type PrP, PG14 PrP only partially ameliorated the clinical phenotype of Tg(F35) mice. Tg(F35+/0/PG14-A2+/0)/Prn-p0/0 animals eventually became ill at 169±10 days (Figure 1A), although their symptoms were milder than those observed in Tg(F35+/0)/Prn-p0/0 mice, and none of the animals died during the period of observation (400 days) (Figure 1B). As expected (Chiesa et al, 1998), the presence of the PG14 transgene by itself caused the mice to become ill. However, the ages at symptom onset (248±27 days) (Figure 1A) and death (448±35 days; Chiesa et al, 2000) were much greater in Tg(PG14-A2+/0)/Prn-p0/0 mice than in Tg(F35+/0)/Prn-p0/0 animals, allowing the partial rescuing effect of the PG14 transgene on the F35 phenotype to be clearly appreciated.
Figure 2.
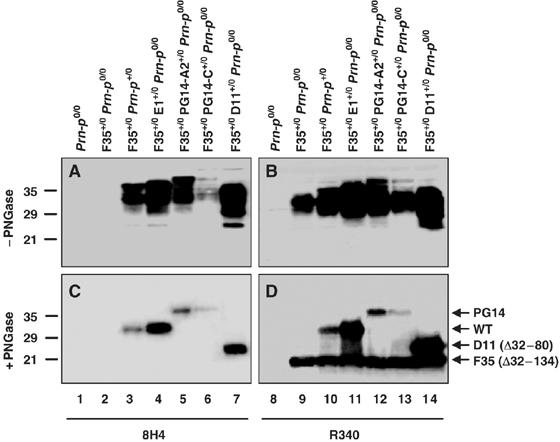
Coexpression of wild-type or other mutant PrP molecules does not alter the amount of F35 PrP. Brain homogenates were treated without (A, B) or with (C, D) PNGase F to remove N-linked oligosaccharides, and were then subjected to Western blotting with anti-PrP antibody 8H4 (lanes 1–7) or R340 (lanes 8–14). Equivalent amounts of protein were loaded in each lane. 8H4 detects wild-type, PG14, and D11 PrP, but not F35 PrP. R340 recognizes all PrP species. Arrows to the right of (D) indicate the positions of the PrP forms. Molecular size markers are given in kDa.
The transgenes in both Tg(PG14) and Tg(F35) mice are controlled by a half-genomic PrP promoter lacking intron 2, which contains an enhancer element specific for cerebellar Purkinje cells (Borchelt et al, 1996; Fischer et al, 1996). Thus, both transgenes are expressed together in all neurons, including cerebellar granule cells, where endogenous PrP is normally produced, with the exception of cerebellar Purkinje cells. To confirm that lack of Purkinje cell expression does not account for the reduced rescue activity of PG14 PrP compared to endogenous wild-type PrP, we crossed Tg(F35+/0) mice with Tg(E1+/0) mice, which express wild-type PrP from the same half-genomic promoter used in Tg(PG14) mice (Chiesa et al, 1998). We observed complete clinical rescue in the resulting Tg(F35+/0/E1+/0)/Prn-p0/0 offspring, similar to what was found when wild-type PrP was supplied by the endogenous Prn-p allele (Figure 1A and B).
To determine if the rescuing activity of PG14 PrP was dose-dependent, we crossed Tg(F35) mice with Tg(PG14-C) mice. Tg(PG14-C+/0) animals express PG14 PrP at ∼20% of the PrP levels in Prn-p+/0 mice (Figure 2; lanes 6 and 13), and never become clinically ill or develop neuropathology (Chiesa et al, 1998). Tg(F35+/0/PG14-C+/0)/Prn-p0/0 mice first developed symptoms at 67±7 days of age, ∼100 days earlier than Tg(F35+/0/PG14-A2+/0)/Prn-p0/0 animals, and became terminally ill at 281±33 days (Figure 1A and B). Thus, lowering the level of PG14 PrP reduced the extent of clinical rescue. This result demonstrates that a reduction of several-fold in the amount of functional PrP is readily detected by a substantial decrease in the clinical incubation time.
We confirmed by Western blotting that coexpression of wild-type PrP (either endogenous or transgenically encoded) did not alter the level of PrPΔ32–134 in Tg(F35) mice carrying either the endogenous Prn-p gene, or the WT-E1, PG14-A2 or PG14-C transgenes (Figure 2, lanes 9–13). This result rules out downregulation of the F35 transgene as an explanation for the rescuing effect of wild-type and PG14 PrP.
Expression of PG14 PrP does not prevent granule cell degeneration in Tg(F35) mice
We analyzed the effect of PG14 PrP expression on neuropathology in Tg(F35) mice. A prominent feature of these animals is massive degeneration of cerebellar granule cells (Shmerling et al, 1998). Granule cell loss was noticeable in clinically ill Tg(F35+/0)/Prn-p0/0 mice at 45 days of age (Figure 3A), and was even more prominent at 72 days, when the mice were terminal (Figure 3B and C). Granule cell loss was more severe in the anterior lobules of the cerebellum (II-VII) than in the posterior lobules (VIII-X). Mice carrying both the F35 and PG14 transgenes (Tg(F35+/0 /PG14-A2+/0)/Prn-p0/0) also showed substantial granule cell loss in the anterior cerebellar lobules at 45 and 72 days, although it was less severe than in Tg(F35+/0)/Prn-p0/0 animals and did not progress appreciably from 72 days to 300 days, consistent with the protracted clinical course in these animals (Figure 3D–G). Importantly, mice carrying only the PG14 transgene (PG14-A2+/0/Prn-p0/0) showed no changes in granule cell density up to 300 days (Figure 3L–O), making it possible to attribute the granule cell depletion seen in Tg(F35/PG14) mice to the F35 transgene. As negative controls, Tg(F35) mice coexpressing wild-type PrP, either from the endogenous Prn-p allele (F35+/0/Prn-p+/0) (Figure 3H–K) or from the E1 transgene (data not shown) displayed no pathological abnormalities up to 300 days of age.
Figure 3.
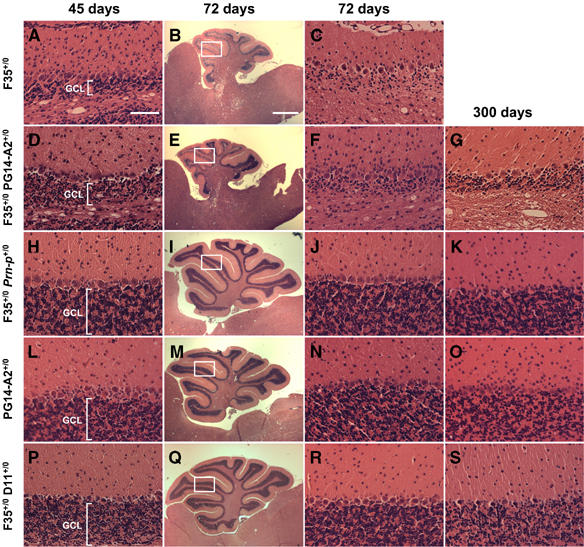
Expression of PG14 PrP delays but does not prevent degeneration of cerebellar granule cells in Tg(F35) mice. Sections from the cerebellar cortex of 45 day-old mice (A, D, H, L, P), 72 day-old mice (B, C, E, F, I, J, M, N, Q, R), and 300-day-old mice (G, K, O, S) of the following genotypes were stained with hematoxylin and eosin: F35+/0 Prn-p0/0 (A–C); F35+/0 PG14-A2+/0 Prn-p0/0 (D–G); F35+/0 Prn-p+/0 (H–K); PG14-A2+/0 Prn-p0/0 (L–O); F35+/0 D11+/0 Prn-p0/0 (P–S). The granule cell layer (GCL) is labeled in (A), (D), (H), (L), and (P). The white rectangles in panels (B), (E), (I), (M), and (Q) indicate the regions of lobule IV/V that are shown at higher magnification in the other panels. In F35+/0 Prn-p0/0 mice, loss of granule neurons is evident at 45 days (A) and is nearly complete by 72 days (B, C). Coexpression of PG14 PrP mitigates but does not prevent granule cell loss (D–G). In contrast, wild-type PrP (H-K) as well as PrPΔ32–80 (P-S) completely abrogate granule cell degeneration. Scale bar in (A) (applicable to A, C, D, F, G, H, J, K, L, N, O, P, R, S)=100 μm. Scale bar in (B) (applicable to E, I, M, and Q)=1 mm.
Expression of PG14 PrP does not ameliorate white matter pathology in Tg(F35) mice
Recently, white matter lesions have been described in Tg(F35) mice, suppression of which by oligodendrocyte-specific expression of wild-type PrP dramatically increased lifespan (Radovanovic et al, 2005). This pathology was characterized by vacuolar degeneration of white matter regions of the brain and spinal cord, accompanied by axonal loss and deterioration of myelin sheaths. In terminally ill Tg(F35+/0)/Prn-p0/0 mice at 72 days of age, extensive vacuolation was observed in the white matter of the cerebellum in both hematoxylin/eosin-stained paraffin sections, and in toluidine blue-stained, semithin plastic sections (Figure 4A and B). Strikingly, mice expressing both the F35 and PG14 transgenes showed the same degree of vacuolar degeneration in the cerebellar white matter at 72 days (Figure 4C and D), long before these animals developed symptoms (at 169±10 days) and despite the fact that they survived for >400 days. In contrast, coexpression of wild-type PrP completely abrogated white matter degeneration in Tg(F35) mice (Figure 4E and F). In addition, Tg(PG14) mice without the F35 transgene showed no white matter pathology (Figure 4G and H). Thus, even though PG14 PrP displays partial ability to rescue loss of cerebellar granule neurons, the mutant protein lacks substantial activity against white matter degeneration.
Figure 4.
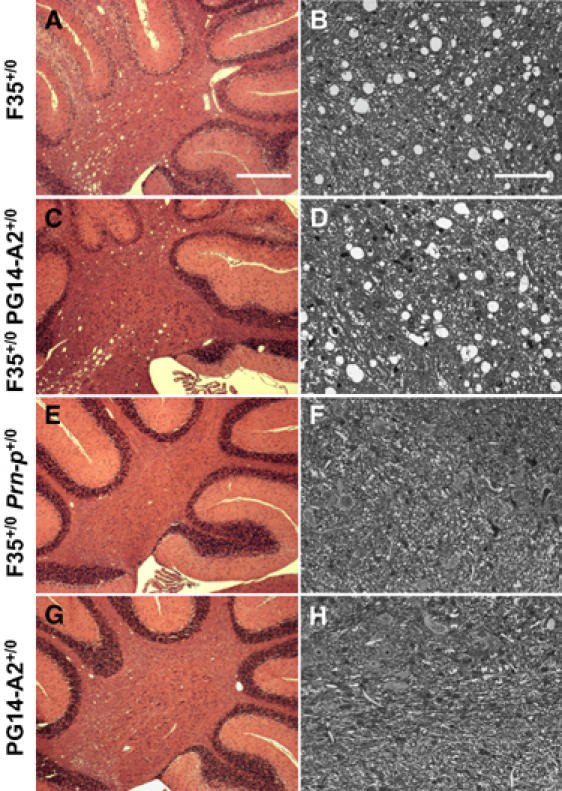
PG14 PrP does not suppress white matter pathology in the cerebellum of Tg(F35) mice. Paraffin sections stained with hematoxylin and eosin (A, C, E, G) and semithin plastic sections stained with toluidine blue (B, D, F, H) were prepared from the cerebella of 72-day-old mice of the following genotypes: F35+/0 Prn-p0/0 (A, B); F35+/0 PG14-A2+/0 Prn-p0/0 (C, D); F35+/0 Prn-p+/0 (E, F); PG14-A2+/0 Prn-p0/0 (G, H). The semithin sections were taken from the cerebellar white matter. White matter vacuolation is present to a similar extent in F35+/0 Prn-p0/0 mice (A, B) and F35+/0 PG14-A2+/0 Prn-p0/0 mice (C, D). Scale bar in (A) (applicable to C, E, G)=500 μm. Scale bar in (B) (applicable to D, F, H)=100 μm.
PG14 PrP does not prevent astrogliosis in Tg(F35) mice
Prominent astrocytosis with hypertrophy of Bergmann glial cells is apparent in the cerebellar cortex of Tg(F35) mice by 45 days of age, based on staining with antibody to glial fibrillary acidic protein (GFAP) (Figure 5A). Introduction of the PG14 transgene does not ameliorate this effect (Figure 5B). As expected, neither F35+/0/Prn-p+/0 nor PG14-A2+/0/Prn-p0/0 mice showed an astrocytic reaction at this stage (Figure 5C and D).
Figure 5.
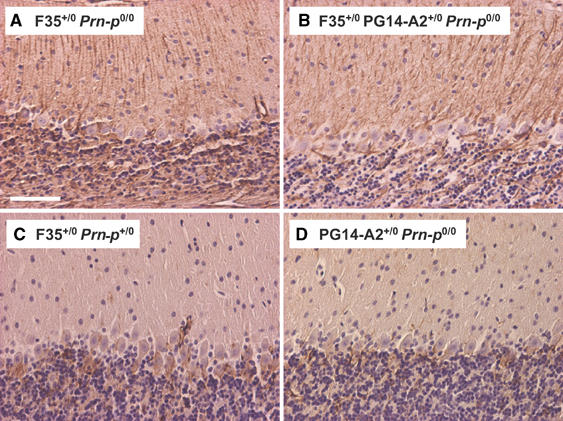
PG14 PrP does not prevent astrocytic reaction in Tg(F35) mice. Sections of cerebellum from 45-day-old mice of the following genotypes were stained with antibody to GFAP: F35+/0 Prn-p0/0 (A); F35+/0 PG14-A2+/0 Prn-p0/0 (B); F35+/0 Prn-p+/0 (C); PG14-A2+/0 Prn-p0/0 (D). F35+/0 Prn-p0/0 mice (A) show prominent GFAP staining with hypertrophy of radial Bergmann glial fibers in the molecular layer. Coexpression of wild-type PrP (C) but not PG14 PrP (B) prevents this pathology. No significant GFAP staining is seen in PG14-A2+/0 Prn-p0/0 mice at this age (D). Scale bar in (A) (applicable to B–D)=100 μm.
Octapeptide deletion does not alter PrP rescue activity
The experiments described above show that expansion of the octapeptide repeat region of PrP appears to adversely affect the ability of PrP to protect against disease caused by expression of PrPΔ32–134. To investigate the requirement for the octapeptide repeats in the rescuing activity of PrP, we crossed Tg(F35) mice with Tg(D11) mice, which express a form of PrP (Δ32–80) lacking four of the five repeats (Fischer et al, 1996). Tg(F35+/0/D11+/0)/Prn-p0/0 mice remained healthy (Figure 1A) and displayed a normal lifespan (Figure 1B). Moreover, no granule cell loss or white matter degeneration were observed in cerebellar sections from these mice as late as 300 days of age (Figure 3P–S and data not shown). Coexpression of PrPΔ32–80 did not alter the expression level of PrPΔ32–134 (Figure 2, lane 14), indicating that downregulation of the latter protein is not involved in the phenotypic rescue. These data show that the rescuing activity of PrP is independent of the first four octapeptide repeats.
Discussion
Several different mutations in the gene encoding PrP are associated with familial prion diseases, but the mechanisms by which these mutations cause pathology remain unclear. The experiments described here were undertaken to test whether an octapeptide insertion mutation (PG14) impairs an experimentally assayable physiological activity of PrP, namely its ability to suppress the neurodegenerative phenotype of Tg(F35) mice that express an N-terminally deleted form of PrP (Δ32–134). We found that although PG14 PrP was able to delay the onset of clinical illness in this mouse model and slow the progression of neuronal loss, its rescue activity at equivalent expression levels was inferior to that of wild-type PrP. Moreover, PG14 PrP did not ameliorate the early appearance of white matter pathology or astrocytosis in Tg(F35) mice. Our results, in conjunction with other data, suggest that a loss-of-function mechanism may contribute to the phenotype of some inherited prion diseases.
Why is PG14 PrP deficient in rescue activity?
Presumably, insertion of additional octapeptide repeats reduces the ability of PrP to interact with components of the cellular pathways responsible for suppression of PrPΔ32–134 neurotoxicity. Several different models have been proposed to explain the neurotoxicity of PrPΔ32–134 and other N-terminally deleted forms of PrP, but they each involve competition between wild-type and deleted PrP for binding to a hypothetical signal-transducing molecule (Shmerling et al, 1998; Baumann et al, 2007; Li et al, 2007). One hypothesis, therefore, is that the PG14 mutation obliterates or interferes with an essential site, presumably encompassing the octapeptide repeats, which is required for binding of PrP to the signal transducer. Consistent with this proposal, octapeptide insertions have been reported to alter the binding of antibodies and glycosaminoglycans to the N-terminus of PrP (Yin et al, 2006). Arguing against such a mechanism, however, is our observation that PrPΔ32–80, which is missing four of the five octapeptide repeats, retains full rescue activity in Tg(F35) mice (albeit at a supraphysiological expression level). A similar result has been reported for PrPΔ32–93, which is missing all of the repeats (Shmerling et al, 1998).
An alternative hypothesis is that the PG14 mutation acts by promoting aggregation of PrP, thereby reducing its ability to interact with the hypothetical signal transducer or other binding partners essential for its normal activity. Consistent with this idea, octapeptide insertions have been shown to promote self-association of PrP, enhance formation of amyloid fibrils, and increase the rate of conversion to PrPSc in an in vitro assay (Zahn, 2003; Leliveld et al, 2006; Moore et al, 2006; Yu et al, 2007). Moreover, there is a correlation between the number of octapeptide repeats and the degree of aggregation (Priola and Chesebro, 1998; Yu et al, 2007). Thus, PG14 PrP, which harbors the largest octapeptide insertion thus far described in human patients, is particularly aggregation-prone compared to other mutants when expressed in cultured cells and yeast (Lehmann and Harris, 1996; Li and Harris, 2005). In the brains of Tg(PG14) mice, the mutant protein spontaneously forms β-sheet-rich aggregates consisting of 20–30 molecules, and accumulates as punctuate deposits in a synaptic pattern (Chiesa et al, 1998, 2003). This aggregated form of PG14 PrP is not infectious in animal transmission experiments (Chiesa et al, 2003) or in vitro conversion assays (E Biasini, R Chiesa, DA Harris, manuscript in preparation), and we have postulated that it represents an example of a toxic, noninfectious species common to many prion diseases (Chiesa and Harris, 2001).
It is noteworthy that PG14 PrP retains some ability to suppress the Tg(F35) phenotype, delaying symptom onset, prolonging lifespan, and slowing granule cell degeneration. This residual activity may be attributable to the proportion of the mutant protein (20–30%), which is not aggregated and displays the same biochemical properties as wild-type PrP (Chiesa et al, 2003) (E Biasini, R Chiesa, DA Harris, manuscript in preparation). In contrast, PG14 PrP had no apparent effect on the evolution of white matter pathology in the cerebellum, which appeared in young Tg(F35)/Tg(PG14) animals as early as it did in Tg(F35) mice. This observation may indicate that suppression of axon and myelin damage requires a higher level of functional PrP than suppression of granule cell death.
Loss of PrP neuroprotection as a pathogenic mechanism
The results presented here suggest that part of the pathogenicity of PG14, and perhaps other disease-associated mutations, may be attributable to the mutant proteins being deficient in a neuroprotective activity normally possessed by wild-type PrP. Consistent with this suggestion, there is a growing body of evidence indicating that PrP can protect cells from several kinds of internal or environmental stresses (reviewed by Roucou and LeBlanc, 2005). For example, PrP overexpression rescues cultured neurons and some mammalian cell lines from proapoptotic stimuli, including Bax expression, serum withdrawal, and cytokine treatment (Kuwahara et al, 1999; Bounhar et al, 2001; Roucou et al, 2003, 2005; Diarra-Mehrpour et al, 2004). In addition, endogenous PrP protects cultured neurons against oxidative stress, and brain tissue against ischemia or hypoxia in vivo (Brown et al, 2002; McLennan et al, 2004; Spudich et al, 2005). In a particularly relevant example, we have shown that PrP suppresses Bax-induced cell death in the yeast Saccharomyces cerevisiae. The presence of the PG14 mutation abolishes this activity, concomitant with conversion of the protein to an aggregated and protease-resistant state (Li and Harris, 2005). This result directly demonstrates that aggregated PG14 PrP lacks cytoprotective activity. Similarly, two other disease-associated mutations (T183A and D178N) have been reported to partially or completely abolish the ability of PrP to rescue cultured human neurons from Bax-induced apoptosis (Bounhar et al, 2001). Taken together, these results suggest that some pathogenic mutations impair the antiapoptotic activity of PrP.
Two published studies have examined the functional activity in transgenic mice of PrP carrying another mutation, E200K, which is linked to familial Creutzfeldt–Jakob disease. In one study, mice expressing E200K PrP were crossed with Nagasaki Prn-p0/0 mice expressing Dpl. It was found that the mutant PrP efficiently suppressed the Dpl-induced neurodegenerative phenotype (Atarashi et al, 2003). In the second study, E200K PrP was reported to rescue the electrophysiological abnormalities seen in brain slices from Zurich I Prn-p0/0 mice (Asante et al, 2004). These results would seem to indicate that E200K, in contrast to PG14, does not impair the physiological activity of PrP. However, it is noteworthy that E200K transgenic mice do not become ill like Tg(PG14) mice, and the mutant PrP found in their brains is not aggregated or protease-resistant like PG14 PrP (Telling et al, 1996; Rosenmann et al, 2001). Thus, the functional activity of E200K PrP in transgenic mice may result from the large proportion of the protein that remains soluble and protease sensitive in the brains of these animals. In contrast, E200K PrP from the brains of affected patients is aggregated and protease resistant (Rosenmann et al, 2001), possibly due to the longer time course of disease development in humans compared to mice. Thus, the pathogenicity of E200K PrP in humans could result from loss of function due to protein aggregation, as we postulate for PG14 PrP.
Two considerations would seem to argue against a loss-of-function mechanism in prion diseases. First, genetic ablation of PrP expression, either prenatally (Büeler et al, 1992; Manson et al, 1994) or postnatally (Mallucci et al, 2002), has relatively little phenotypic effect, and does not produce any features of a prion disease. Thus, loss of PrPC function cannot, by itself, account for prion-induced neurodegeneration. However, it is possible that a loss of function mechanism exacerbates pathology caused by a toxic gain-of-function or other mechanisms. For example, a cytoprotective activity of PrPC that is dispensable under normal conditions may become essential in the disease state due to cellular or organismal stress. Toxicity of PrP aggregates coupled with loss of PrP neuroprotective function may account for the two independent pathological components that we have described in Tg(PG14) mice: Bax-independent synaptic loss, and Bax-dependent granule cell apoptosis (Chiesa et al, 2005).
Loss of PrPC function as a pathogenic mechanism also appears to be incompatible with the dominant mode of inheritance of familial prion diseases. However, PrPSc or mutant PrP may sequester wild-type PrPC into aggregates that lack functional activity, thereby producing a dominant-negative effect (Chen et al, 1997). It is noteworthy that a loss-of-function effect has been proposed as a causative factor in Huntington's disease, another autosomal dominant disorder (Cattaneo et al, 2001; Ross, 2004). Also, yeast prions such as [PSI+] and [URE3] are dominantly inherited, even though the phenotypes they cause are due a loss of function of the respective proteins due to aggregation (Shorter and Lindquist, 2005).
If loss of PrPC neuroprotective activity contributes to the pathology seen in Tg(PG14) mice, one would predict that coexpression of wild-type PrP in these animals would ameliorate the disease phenotype. We previously reported that there was no significant difference in age at symptom onset between Tg(PG14) mice on the Prn-p0/0 and Prn-p+/+ backgrounds (Chiesa et al, 2000), apparently arguing against a mitigating effect of wild-type PrP. However, we have not carefully compared the pathological abnormalities seen in Tg(PG14) mice on the two genetic backgrounds. In addition, we have not analyzed the effects of overexpressing wild-type PrP in Tg(PG14) mice (via a second transgene), which might be necessary to produce an observable neuroprotective effect. Such experiments are now in progress.
Determining which mechanism is responsible for the pathogenicity of PrP mutations has important therapeutic implications. If pathology is attributable to a loss of PrP function, then augmenting expression of wild-type PrP may suppress the development of neurodegeneration (although it might accelerate generation of PrPSc). Conversely, suppression of PrP expression, a strategy that has been proposed for preventing or treating prion diseases (Pfeifer et al, 2006; Mallucci et al, 2007), may have detrimental consequences due to loss of the neuroprotective activity of PrPC.
Materials and methods
Mice
Mice expressing PrPΔ32–134 (line F35) (Shmerling et al, 1998) were obtained from Adriano Aguzzi (University of Zurich, Switzerland). Prn-p0/0 mice (Büeler et al, 1992) and mice expressing PrPΔ32–80 (line D11) (Shmerling et al, 1998) were obtained form Charles Weissmann (The Scripps Research Institute, FL). Construction of Tg(PG14) mice (lines A2 and C) (Chiesa et al, 1998) and Tg (WT) mice (line E1) (Chiesa et al, 1998) has been described previously. All mice were maintained on a C57BL/6J × CBA/J hybrid strain background. Tg(PG14-A2), Tg(PG14-C), Tg(D11), and Tg(E1) mice were propagated on a Prn-p0/0 background and Tg(F35) mice on a Prn-p+/0 background. Mice were intercrossed to produce the genotypes referred to in the text. The presence of each transgene was determined by PCR analysis of tail DNA using primers described previously (Chiesa et al, 1998; Shmerling et al, 1998).
Western blotting
Western blots of brain homogenates were performed as described previously (Chiesa et al, 1998). For enzymatic deglycosylation, denatured proteins were incubated at 37°C for 2 h with PNGase F (New England Biolabs, Ipswich, MA) according to the manufacturer's instructions. Blots were developed with either rabbit polyclonal anti-PrP antiserum R340 (Brandner et al, 1996) or mouse monoclonal anti-PrP antibody 8H4 (Zanusso et al, 1998).
Histopathology
Mice were perfusion fixed with 4% paraformaldehyde in PBS (pH 7.2), and then brains were paraffin embedded and cut into 2-μm sagittal sections. Sections were stained with hematoxylin and eosin, or with an antibody to GFAP (Biogenex, San Ramon, CA) followed by visualization using the peroxidase-anti-peroxidase technique. For preparation of semithin plastic sections, brain tissue was fixed in 4% paraformaldehyde/3% glutaraldehyde and embedded in Epon. One micron sections were cut, and stained with toluidine blue for viewing by light microscopy.
Acknowledgments
We thank A Aguzzi for supplying Tg(F35) mice, and C Weissmann for providing Tg(D11) and Prn-p0/0 mice. We also thank C Weissmann for R340 antibody and M-S Sy for 8H4 antibody. We are grateful to C Adles and S Deng for mouse colony maintenance and genotyping. This work was supported by grants from the NIH to DAH (NS040975) and BG (P30 AG10133). SJB was supported by the Medical Scientist Training Program at Washington University (NIH Grant T32GM07200). The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any Agency determination or policy.
References
- Apetri AC, Surewicz K, Surewicz WK (2004) The effect of disease-associated mutations on the folding pathway of human prion protein. J Biol Chem 279: 18008–18014 [DOI] [PubMed] [Google Scholar]
- Apetri AC, Vanik DL, Surewicz WK (2005) Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein 90–231. Biochemistry 44: 15880–15888 [DOI] [PubMed] [Google Scholar]
- Asante EA, Li YG, Gowland I, Jefferys JG, Collinge J (2004) Pathogenic human prion protein rescues PrP null phenotype in transgenic mice. Neurosci Lett 360: 33–36 [DOI] [PubMed] [Google Scholar]
- Atarashi R, Nishida N, Shigematsu K, Goto S, Kondo T, Sakaguchi S, Katamine S (2003) Deletion of N-terminal residues 23-88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced neurodegeneration. J Biol Chem 278: 28944–28949 [DOI] [PubMed] [Google Scholar]
- Baumann F, Tolnay M, Brabeck C, Pahnke J, Kloz U, Niemann HH, Heikenwalder M, Rülicke T, Bürkle A, Aguzzi A (2007) Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 26: 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL (1996) A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal Biomol Eng 13: 159–163 [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A (2001) Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem 276: 39145–39149 [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996) Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 379: 339–343 [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L (2002) Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67: 211–224 [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C (1992) Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature 356: 577–582 [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S (2001) Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci 24: 182–188 [DOI] [PubMed] [Google Scholar]
- Chen SG, Parchi P, Brown P, Capellari S, Zou WQ, Cochran EJ, Vnencakjones CL, Julien J, Vital C, Mikol J, Lugaresi E, Autilio-Gambetti L, Gambetti P (1997) Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nature Med 3: 1009–1015 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Drisaldi B, Quaglio E, Migheli A, Piccardo P, Ghetti B, Harris DA (2000) Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc Natl Acad Sci USA 97: 5574–5579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Harris DA (2001) Prion diseases: what is the neurotoxic molecule? Neurobiol Dis 8: 743–763 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA (2005) Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA 102: 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA (1998) Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21: 1339–1351 [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Quaglio E, Drisaldi B, Si-Hoe SL, Takao M, Ghetti B, Harris DA (2003) Molecular distinction between pathogenic and infectious properties of the prion protein. J Virol 77: 7611–7622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S (2004) Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res 64: 719–727 [DOI] [PubMed] [Google Scholar]
- Duchen LW, Poulter M, Harding AE (1993) Dementia associated with a 216 base pair insertion in the prion protein gene: clinical and neuropathological features. Brain 116: 555–567 [DOI] [PubMed] [Google Scholar]
- Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C (1996) Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J 15: 1255–1264 [PMC free article] [PubMed] [Google Scholar]
- Harris DA, True HL (2006) New insights into prion structure and toxicity. Neuron 50: 353–357 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, Defea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR (1998) A transmembrane form of the prion protein in neurodegenerative disease. Science 279: 827–834 [DOI] [PubMed] [Google Scholar]
- Kong Q, Surewicz WK, Petersen RB, Zou W, Chen SG, Gambetti P, Parchi P, Capellari S, Goldfarb L, Montagna P, Lugaresi E, Piccardo P, Ghetti B. (2004) Inherited prion diseases. In Prion Biology and Diseases, Prusiner SB (ed) Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press pp 673–775 [Google Scholar]
- Krasemann S, Zerr I, Weber T, Poser S, Kretzschmar H, Hunsmann G, Bodemer W (1995) Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Mol Brain Res 34: 173–176 [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T (1999) Prions prevent neuronal cell-line death. Nature 400: 225–226 [DOI] [PubMed] [Google Scholar]
- Lehmann S, Harris DA (1996) Mutant and infectious prion proteins display common biochemical properties in cultured cells. J Biol Chem 271: 1633–1637 [DOI] [PubMed] [Google Scholar]
- Leliveld SR, Dame RT, Wuite GJ, Stitz L, Korth C (2006) The expanded octarepeat domain selectively binds prions and disrupts homomeric prion protein interactions. J Biol Chem 281: 3268–3275 [DOI] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA (2007) Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105-125. EMBO J 26: 548–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Harris DA (2005) Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem 280: 17430–17434 [DOI] [PubMed] [Google Scholar]
- Liemann S, Glockshuber R (1999) Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 38: 3258–3267 [DOI] [PubMed] [Google Scholar]
- Mallucci GR, Ratte S, Asante EA, Linehan J, Gowland I, Jefferys JG, Collinge J (2002) Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO J 21: 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci GR, White MD, Farmer M, Dickinson A, Khatun H, Powell AD, Brandner S, Jefferys JG, Collinge J (2007) Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 53: 325–335 [DOI] [PubMed] [Google Scholar]
- Manson JC, Clarke AR, Hooper ML, Aitchison L, McConnell I, Hope J (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol Neurobiol 8: 121–127 [DOI] [PubMed] [Google Scholar]
- McLennan NF, Brennan PM, McNeill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE (2004) Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol 165: 227–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RA, Herzog C, Errett J, Kocisko DA, Arnold KM, Hayes SF, Priola SA (2006) Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci 15: 609–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen F, Poulter M, Collinge J, Leach M, Lofthouse R, Crow TJ, Harding AE (1992) A dementing illness associated with a novel insertion in the prion protein gene. Mol Brain Res 13: 155–157 [DOI] [PubMed] [Google Scholar]
- Pfeifer A, Eigenbrod S, Al-Khadra S, Hofmann A, Mitteregger G, Moser M, Bertsch U, Kretzschmar H (2006) Lentivector-mediated RNAi efficiently suppresses prion protein and prolongs survival of scrapie-infected mice. J Clin Invest 116: 3204–3210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Chesebro B (1998) Abnormal properties of prion protein with insertional mutations in different cell types. J Biol Chem 273: 11980–11985 [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95: 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB (ed) (2004) Prion Biology and Diseases. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A (2005) Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci 25: 4879–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K (1998) Prion protein NMR structure and familial human spongiform encephalopathies. Proc Natl Acad Sci USA 95: 11667–11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmann H, Talmor G, Halimi M, Yanai A, Gabizon R, Meiner Z (2001) Prion protein with an E200K mutation displays properties similar to those of the cellular isoform PrPC. J Neurochem 76: 1654–1662 [DOI] [PubMed] [Google Scholar]
- Ross CA (2004) Huntington's disease: new paths to pathogenesis. Cell 118: 4–7 [DOI] [PubMed] [Google Scholar]
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, Aguzzi A, Weissmann C (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J 20: 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A (2005) Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ 12: 783–795 [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC (2003) Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem 278: 40877–40881 [DOI] [PubMed] [Google Scholar]
- Roucou X, LeBlanc AC (2005) Cellular prion protein neuroprotective function: implications in prion diseases. J Mol Med 83: 3–11 [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C (1998) Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 93: 203–214 [DOI] [PubMed] [Google Scholar]
- Shorter J, Lindquist S (2005) Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 6: 435–450 [DOI] [PubMed] [Google Scholar]
- Spudich A, Frigg R, Kilic E, Kilic U, Oesch B, Raeber A, Bassetti CL, Hermann DM (2005) Aggravation of ischemic brain injury by prion protein deficiency: role of ERK-1/-2 and STAT-1. Neurobiol Dis 20: 442–449 [DOI] [PubMed] [Google Scholar]
- Swietnicki W, Petersen RB, Gambetti P, Surewicz WK (1998) Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem 273: 31048–31052 [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296: 1991–1995 [DOI] [PubMed] [Google Scholar]
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB (1996) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10: 1736–1750 [DOI] [PubMed] [Google Scholar]
- Vanik DL, Surewicz WK (2002) Disease-associated F198S mutation increases the propensity of the recombinant prion protein for conformational conversion to scrapie-like form. J Biol Chem 277: 49065–49070 [DOI] [PubMed] [Google Scholar]
- Weissmann C (2004) The state of the prion. Nat Rev Microbiol 2: 861–871 [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Sakaguchi S, Shigematsu K, Okimura N, Katamine S (2004) Doppel-induced Purkinje cell death is stoichiometrically abrogated by prion protein. Biochem Biophys Res Commun 319: 1247–1252 [DOI] [PubMed] [Google Scholar]
- Yin S, Yu S, Li C, Wong P, Chang B, Xiao F, Kang SC, Yan H, Xiao G, Grassi J, Tien P, Sy MS (2006) Prion proteins with insertion mutations have altered N-terminal conformation and increased ligand binding activity and are more susceptible to oxidative attack. J Biol Chem 281: 10698–10705 [DOI] [PubMed] [Google Scholar]
- Yu S, Yin S, Li C, Wong P, Chang B, Xiao F, Kang SC, Yan H, Xiao G, Tien P, Sy MS (2007) Aggregation of prion protein with insertion mutations is proportional to the number of inserts. Biochem J 403: 343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn R (2003) The octapeptide repeats in mammalian prion protein constitute a pH-dependent folding and aggregation site. J Mol Biol 334: 477–488 [DOI] [PubMed] [Google Scholar]
- Zanusso G, Liu D, Ferrari S, Hegyi I, Yin X, Aguzzi A, Hornemann S, Liemann S, Glockshuber R, Manson JC, Brown P, Petersen RB, Gambetti P, Sy MS (1998) Prion protein expression in different species: analysis with a panel of new mAbs. Proc Natl Acad Sci USA 95: 8812–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sonnichsen FD (2000) Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J Biol Chem 275: 33650–33654 [DOI] [PubMed] [Google Scholar]