

Summary
Resistance to camptothecin (CPT), a topoisomerase I (Top1) inhibitor, is frequently encountered in non-small cell lung cancer (NSCLC) and CPT resistance is linked with TDP1, an enzyme capable of cleaving the covalent linkage between stabilized Top1 with DNA. The aim of this study is to evaluate the in vivo expression level of TDP1, as well as parallel repair pathway components XPF and MUS81, in primary NSCLC. We collected thirty un-matched and four NSCLC samples matched with normal lung tissue and eight samples of non-neoplastic lung tissue from patients with and without lung cancer, and determined the protein expression of these three genes using Western blot and TDP1 activity by a specific enzymatic assay. Both TDP1 and XPF were overexpressed in over 50% of NSCLC tissues, with wide ranges of expression levels. MUS81 did not exhibit alteration in expression. Overexpression of TDP1 and XPF is common in NSCLC, and is therefore of interest as a possible contributor to drug resistance in NSCLC.
Keywords: Non-small cell lung cancer, Camptothecin resistance, Topoisomerase I-mediated DNA damage, Oxidative damage, Single-strand-break repair, Double-strand-break repair, Overexpression of repair genes, TDP1, XPF, MUS81
1. Introduction
NSCLC is commonly treated by surgery or surgery with adjuvant chemotherapy, and the unresectable disease is treated locally with radiation or radiation combined with chemotherapy. In either group, response to chemotherapy or radiotherapy varies greatly with different patients. In general, drug resistance may be related to tumor specific alterations within targeted therapeutic pathways. For example, overexpression of β-tubulin III may induce resistance to microtubule interacting drugs such as paclitaxel [1], and tamoxifen resistance is correlated with activation of PKA that causes conformational arrest of the estrogen receptor on the surface of a breast tumor [2]. For those therapies that induce DNA damage, overexpression of DNA repair genes involved in the targeted pathway may result in drug resistance. Overexpression of ERCC1, a gene involved in nucleotide excision repair (NER), is related with lower response to cisplatin therapy [3]. Topoisomerase I (Top1) inhibor, camptothecin (CPT) analogues such as irinotecan and topotecan are commonly used in combination with platinum based compounds for NSCLC [4, 5]. Drug resistance is frequently encountered, and studies on the underlying mechanisms have been carried out focusing on the repair pathways of Top1-mediated damage, and mounting evidence has pointed to the ability of TDP1 to induce drug resistance in experimental systems [6–8].
TDP1, a member of the phospholipase D family, is the only known enzyme capable of cleaving the phosphodiester bond between a tyrosyl residual of Top1 and the 3’ phosphate of DNA, which is formed when Top1 is stabilized by CPT [9–11]. TDP1 achieves this hydrolysis via attacking on the phosphate moiety of the substrate using its conserved histidine, releasing tyrosine and forming a phosphohistidine intermediate, which is further hydrolyzed, yielding a 3’ phosphate and the native enzyme [9, 12–14]. Some researches have found that overexpression of TDP1 in human cells caused significant reduction of DNA damage induced by CPT [6, 8], while a recent study demonstrated that a point mutation of TDP1 at its active site in SCAN1 patients caused CPT hypersensitivity [7]. These data suggest that TDP1 might be a factor in CPT resistance, and that pharmacologic inhibition of TDP1 may be useful in combination with CPT based therapy. However, the status of in vivo TDP1 expression and activity in normal tissue and primary tumors is unknown. Examination of the status of the TDP1-mediated repair pathway in NSCLC tissues may provide an indication of which Top1 repair pathway elements may contribute to drug resistance.
Other elements in the Top1 repair pathway include, XPF, which is involved in NER by forming a complex with ERCC1 to excise the damaged DNA strand 5’ from the DNA lesion [15], and MUS81. MUS81 is homologous to XPF, cleaves 3’ trapped DNA in a similar way, and can resolve Holliday junctions [16, 17]. XPF and MUS81 can be assumed to function in parallel to TDP1 in repairing human Top1 damage based on studies of their Saccharomyces cerevisiae homologs RAD1 and MUS81 [3, 18]. For XPF, although its relationship with drug resistance may be implied from the report about its partner ERCC1, its expression, as well as the expression of MUS81, has never been concordantly observed together with the expression of TDP1 in NSCLC. Knowledge of the status of these three genes may help to understand which pathway may contribute to drug resistance in Top1 inhibitors therapy, and may be candidates for combined therapy.
In this study, we collected a total of thirty-four matched and un-matched NSCLC tissues, and observed the expression and activity of TDP1. We found that the expression of TDP1 had increased in more than 50% of cancer tissues, and the activity of TDP1 had increased accordingly. We also observed the expression of XPF and MUS81 in these samples, and found that XPF expression had also increased in more than 50% cancer tissues and the overexpression of TDP1 and XPF did not occur simultaneously in the same patients. MUS81 expression level was not found significantly altered.
2. Materials and methods
2.1. Patients
Thirty NSCLC tissues, eight non-neoplastic lung tissues including five from margins of tumors, and four pairs of matched NSCLC tissues and normal lung tissues were collected from patients at Department of Surgery, School of Medicine, The Johns Hopkins University, after appropriate approval was obtained from the Johns Hopkins institutional review board. These tissues were snap frozen immediately after resection. Appropriate clinical information was abstracted via chart review according to previously approved protocol.
2.2. Human tissue extract
About 50 to 100 mg of above mentioned frozen tissues were ground in lysis buffer [150 mM NaCl, 1 mM KH2PO4, pH6.4, 5 mM MgCl2, 1 mM EGTA, supplemented with Complete ™ protease inhibitor cocktail tablets (Roche, Indianapolis, IN)] in cold mortar. The homogenized mixtures were further added with 0.4 M NaCl (final concentration) and incubated at 4ºC for 20 minutes before centrifuge. The supernatants were collected, and their protein concentrations were measured.
2.3. Western blotting
Aliquots (32 μg each) of tissue extracts were loaded onto 10% acrylamide gels. After each electrophoresis and transfer, TDP1, XPF and MUS81 were blotted respectively followed by detection of β-actin to confirm protein loading. The antibodies used were 1:1000-fold dilution of rabbit anti-TDP1 (Abcam, Inc, Cambridge, MA), 1:500-fold dilution of mouse anti-XPF (clone 218) (Trevigen, Gaithersberg, MD), 1:1000-fold dilution of mouse anti-MUS81 (ImmuQuest Ltd, Cleveland, UK), and 1:3000-fold dilution of mouse monoclonal anti-β-actin (Sigma, Saint Louis, MI). The blots were visualized using peroxidase substrate system (ECL Western blotting detection reagents, Amersham Biosciences UK Limited, Little Chalfont, Buckinghamshire, UK)
2.4. TDP1 enzymatic assay
The activity of TDP1 as a phosphodiesterase to cleave tyrosyl residue was examined as described [18–20]. Briefly, an 18-mer oligonucleotide that terminates in a 3’-phosphotyrosine (oHN279y, 5′-TCCGTTGAAGCCTGCTTTy-3′, kindly provided by Dr. Howard Nash, NIMH) was 5′-labelled with γ-[32P]-ATP using T4 polynucleotide kinase (New England Biolabs), and incubated with aliquots (4 or 0.08 μg) of tissue extracts or 9 ng of purified human Tdp1 (kindly provided by Dr. Howard Nash, NIMH) in a total of 12 μl reaction mix. The reaction contained 50 mM Tris-HCl, pH 8.0, 80 mM KCl, 2 mM EDTA, 1 mM dithiothreitol, 40 μg/mL BSA, and 5% glycerol. After 30-minute incubation at 28 °C, reactions were stopped by the addition of 5 μL formamide sequencing buffer. 6 μL aliquots were electrophoresed on a 12% (acrylamide : bis-acrylamide 19 : 1) sequencing gel. To visualize bands, gels were exposed to HyBlot CL™ autoradiography film (Denville, Scientific Inc.) at −80ºC for 2 hours. The lower bands in Figures 1D and 3B showed the conversion of tyrosyl-DNA substrate to phosphoryl- and hydroxyl-DNA, which demonstrated the unique activity of TDP1 in releasing tyrosine.
Figure 1.
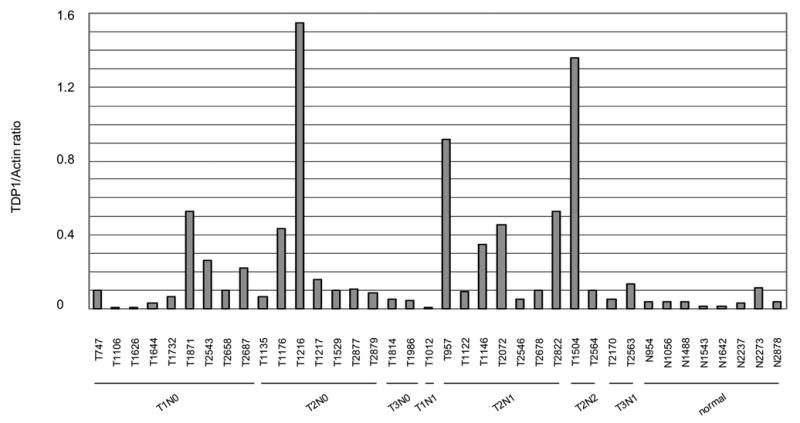
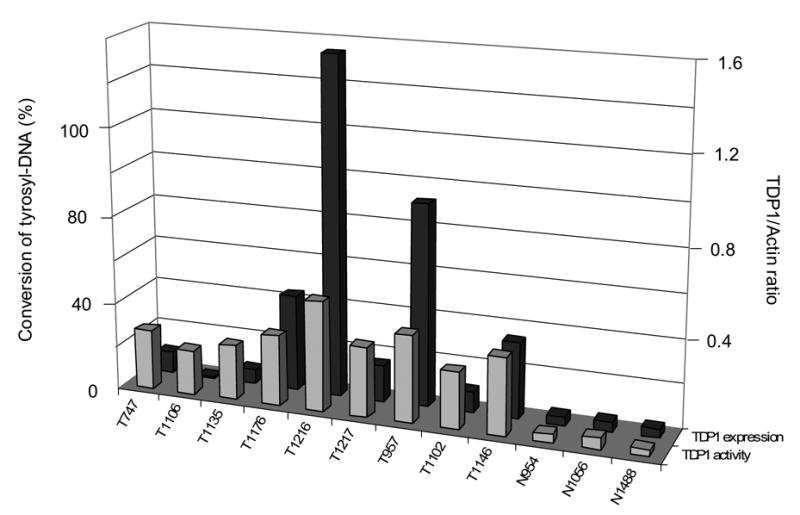
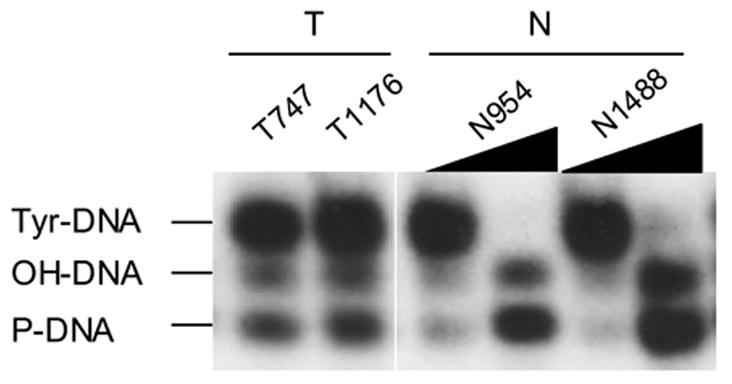
Expression and activity of TDP1 was increased in un-matched NSCLC. A: Quantitative analysis of the protein expression levels of TDP1 of thirty un-matched NSCLC versus eight non-neoplastic tissues, normalized with their expression of β-actin using ImageJ. The samples were run on different gels, and the blotting variations between gels were adjusted using the ratio of TDP1/Actin of N954 that was loaded on every gel. B: Western blotting of representative samples as shown in A. All samples were loaded with 32 μg of tissue extract. C: Quantification of TDP1 enzymatic activity of nine tumor samples versus three non-neoplastic tissues and the comparison with their protein expression levels. All samples were assayed with 80 ng of tissue extract. D: Electrophoresis of the reaction products of TDP1 enzymatic assay on a sequencing gel. The samples were representatives of C. Tumor samples were assayed with 80 ng of tissue extract, and non-neoplastic tissues were assayed with 80 ng and 4 μg of extract. The upper bands were the 18 mer Tyr-DNA substrate; the lower ones converted hydroxyl- and phosphoryl-DNA, respectively. A–D: N, non-neoplastic tissue; T, tumor. The lab ID number was used to name each sample.
Figure 3.
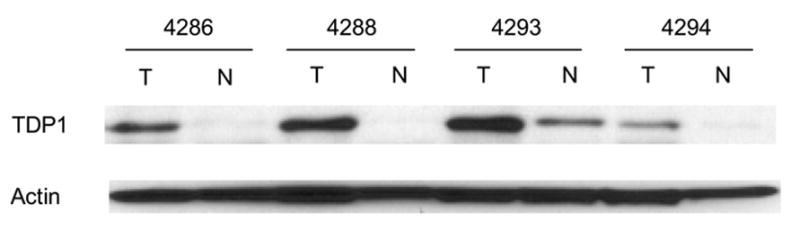
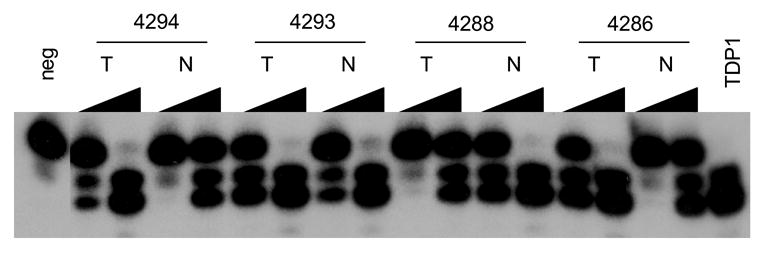
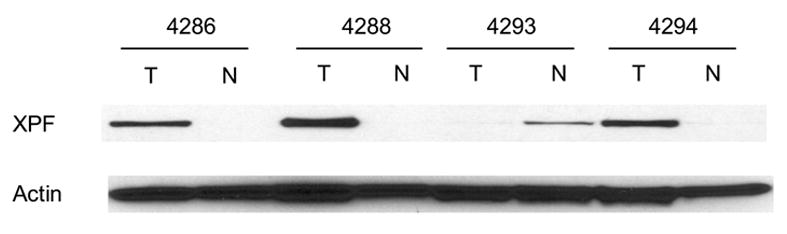
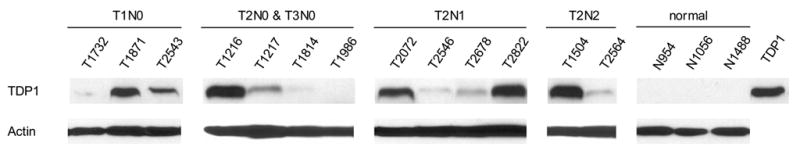
Increased expression and activity of TDP1and increased expression of XPF were cancer-tissue specific. A: Western blotting of TDP1 protein in four matched NSCLC. B: Electrophoresis of the reaction products of TDP1 enzymatic assay of the same samples. All samples were assayed with both 80 ng and 4 μg of tissue extract. C: Western blotting of XPF protein of the same samples. All samples in A and C were loaded with 32 μg of tissue extract. A–C: N, non-neoplastic tissue; T, tumor.
3. Results
To determine TDP1 expression in NSCLC, we collected thirty NSCLC tissues of diverse histology and stage microdissected to greater than 60% purity and eight non-neoplastic lung tissues including five tumor margins from different individuals as control (Table 1). We found that compared with the average expression level of control samples, TDP1 protein expression had increased in more than 50% of cancer samples by Western blot, with the levels ranging from 50% to over 30-fold after measurement using densitometry and normalization with β-actin (Fig. 1A, 1B and 2C). Of note, the increase in TDP1 was not correlated with clinical stage, histology and smoking history (Fig. 1A and Table 1). We selected nine tumor samples with adequate tissue and tested their TDP1 activity (Fig. 1C and 1D). In all the tumors examined, there were increased levels of conversion of tyrosyl-DNA to phosphoryl- and hydroxyl-DNA through TDP1 compared with control samples assayed with the same amount of cell extract. The increased activity of TDP1 generally correlated with level of expression. Remarkably, the samples that did not show increased level of TDP1 protein such as T1106 exhibited increased enzymatic activity compared with controls (Fig. 1C). This implied that TDP1 was not only induced but also activated in tumors.
Table 1.
Histology and smoking history.
| Clinicopathologic characteristics of the 34 NSCLC patients
| ||
|---|---|---|
| Characteristic | N (%) | |
| Sex | ||
| Male | 20 | |
| Female | 14 | |
| Age, y* | 61 (41–84) | |
| Stage | ||
| T1N0 | 10 (29) | |
| T2N0 | 8 (24) | |
| T3N0 | 2 (6) | |
| T1N1 | 1 (3) | |
| T1N2 | 2 (6) | |
| T2N1 | 7 (21) | |
| T3N1 | 2 (6) | |
| T2N2 | 2 (6) | |
| Histology | ||
| Adenocarcinoma | 21 | |
| Squamous cell carcinoma | 10 | |
| Large cell carcinoma | 3 | |
| Smoker | 27 (79) | |
| non-smoker or unknown | 7 (21) | |
Age is reported as mean (range).
Figure 2.
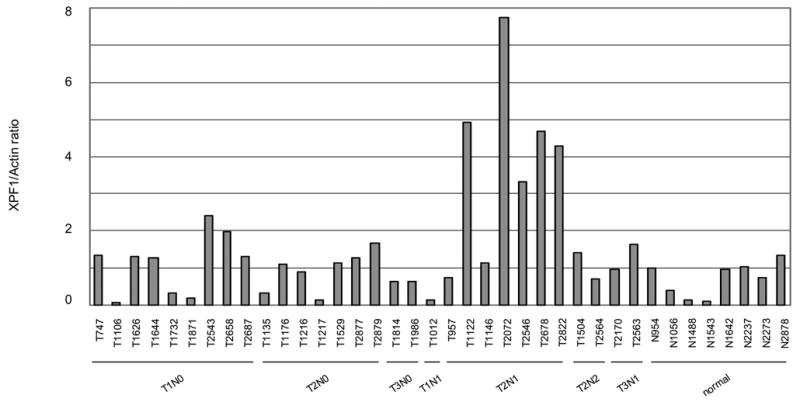
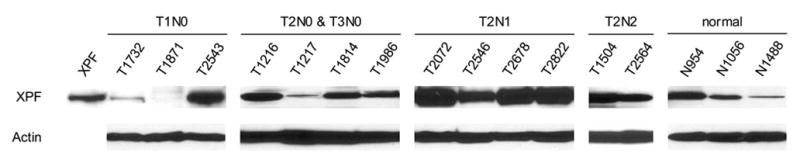
Expression of XPF was increased in un-matched NSCLC. A: Quantitative analysis of the protein expression levels of XPF of thirty un-matched NSCLC versus eight non-neoplastic tissues. Normalization was done as described in Fig. 1A. B: Western blotting of representative samples as shown in A. All samples were loaded with 32 μg of tissue extract. All samples were loaded with 32 μg of tissue extract. C: Comparison of the fold changes of overexpressed TDP1 and XPF relative to the average expression level of non-neoplastic tissues. A–C: N, non-neoplastic tissue; T, tumor.
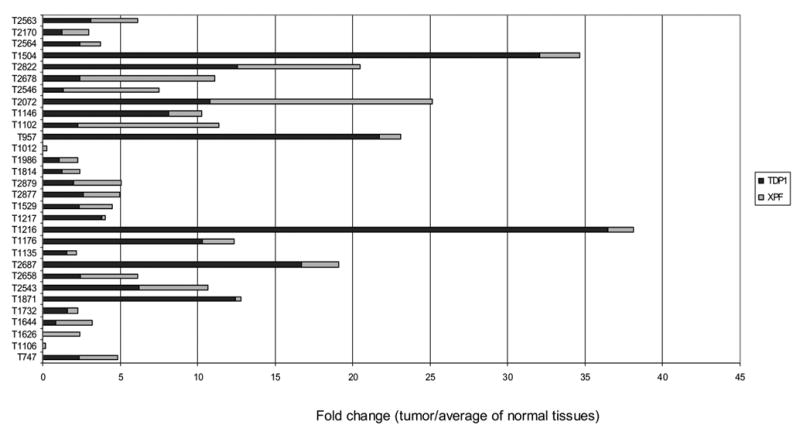
XPF protein expression level was increased in more than 50% of thirty NSCLC samples compared with the average expression level of control samples on Western blots, but compared with that of TDP1, showed less dramatic fold changes, from around 50% to 10-fold as measured using densitometry and normalized with β-actin (Fig. 2A, 2B and 2C). XPF overexpression also showed no correlation with clinical stage, histology and smoking status (Fig. 2A and Table 1). We compared the fold changes of TDP1 and XPF overexpressed relative to normal tissues side by side, and found that in most samples TDP1 showed a greater extent of overexpression than XPF, and interestingly, those samples that highly expressed TDP1 did not exhibit high levels of XPF (Fig. 2C). We measured the protein expression level of MUS81 in nine tumors using Western blot, but did not find significant changes (Fig. 4A).
Figure 4.
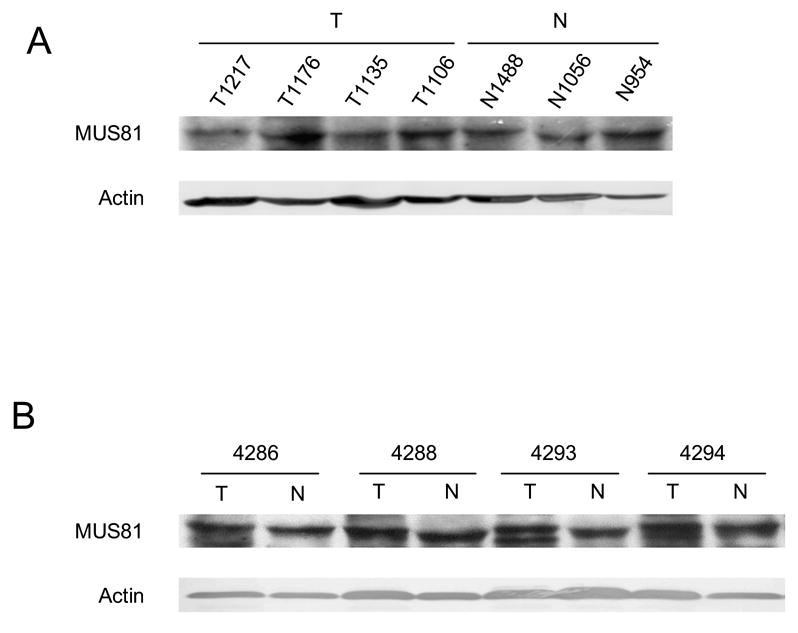
Expression of MUS81 in NSCLC. A: Western blotting of four representative tumor samples versus three non-neoplastic tissues. B: Western blotting of four matched NSCLC. All samples in A and B were loaded with 32 μg of tissue extract.
In order to determine whether the increased expression of TDP1 and XPF and the increased activity of TDP1 was cancer-patient specific or cancer-tissue specific, we further collected and evaluated four pairs of cancer versus normal tissues from the same NSCLC patients. The cancers were at stages T1N0, T2N0 and T1N2, respectively (Table 1). Consistent with our expectation, all tumor tissues showed increased level of TDP1 expression and activity when compared to normal (Fig. 3A, 3B). XPF overexpression was also found in three out of the four paired tumor samples (Fig. 3C). Therefore, the increased expression of TDP1 and XPF and the increased activity of TDP1 was indeed a specific feature of the cancer tissues rather than a patient-dependent phenomenon. XPF versus TDP1 expression status in samples #4286, 4293 and 4294 might again imply the inverse correlation of these two proteins, i.e., when one protein was at a higher level, the other one was expressed relatively less. Consistent with the previous observation, MUS81 did not exhibit expression alteration in the paired normal and cancer tissues (Fig. 4B).
4. Discussion
Understanding of the mechanism of drug resistance in therapy of NSCLC is necessary for development of novel therapeutic strategies, and investigators have correlated upregulation of several well-recognized repair genes with drug resistance [3, 21, 22]. CPT induced DNA damage through topoisomerase I-DNA complex stabilization is a unique type of DNA damage, and resistance to CPT therapy has been linked with TDP1 activity [6–8]. Our report is the first one that reports expression and activity of TDP1 in primary NSCLC. We have shown that TDP1 overexpression was present in a majority of tumors when compared to normal controls as well as matched lung tissue, and was unrelated to clinical variables, albeit in a relatively modestly sized cohort. Only one patient in our cohort had undergone radiation or chemotherapy prior to operation, therefore it is unlikely that TDP1 overexpression is a result of selective pressures from radiation or chemotherapeutic administration.
Our study also discovered that expression of XPF had increased in NSCLC tissues. Interestingly, although both TDP1 and XPF contribute to the repair of Top1-mediated damage, TDP1 showed a greater extent of fold change of overexpression, and the overexpression of these two genes seemed inversely correlated. This observation may support the conclusion of a study of Saccharomyces cerevisiae [18] that these two genes function independently of each other in repair of Top1 damage, since a coordinate correlation might be seen if they were in the same pathway. As a specific repair enzyme for the Top1-DNA complex, TDP1 is probably the cells’ first choice for such damages and therefore is more intensely upregulated in most NSCLC. However, since there are several redundant pathways for the repair of Top1 damage [18, 20], it is possible that in certain circumstances, other pathways such as XPF may dominate the repair and be upregulated more even in the presence of TDP1. It is not clear why, if human MUS81 functioned in parallel toTDP1 as it does in yeast, the gene is not correspondingly increased in primary lung cancer. However, this might be explained if MUS81 is not limiting for repair because it is present in excess of its partner subunit, Eme1. Another possibility is that MUS81 may be controlled or induced by a diverse set of regulatory elements. Further work may be required to evaluate whether XPF and MUS81 indeed function synergistically in repair of Top1 damage in humans.
It is well recognized that DNA repair genes are stability genes or caretakers whose inactivation causes mutations in other genes to occur at a higher rate [23]. These genes are usually expected to have reduced expression and activity in cancer cells when they are involved in carcinogenesis. In our study the overexpression of TDP1 might be a physiologic compensation for increased repair demands due to faulty DNA repair or simple elevated mitotic rate. TDP1 may be particularly advantageous in that it can repair multiple forms of DNA damage [18, 24].
Firstly, TDP1 has been shown to be an important player in both double-strand-break repair (DSBR) and single-strand-break repair (SSBR). Top1-DNA complexes can be converted into irreversible double-strand-breaks (DSBs) when collide with replication forks [25, 26]. These complexes trap the 3’ ends of DSBs, so need to be removed before the recombination (HR) pathway for DSBR take over the repair [20, 27]. In fact, a yeast study has placed TDP1 in a RAD52-dependent DSBR pathway [20]. Single-strand-breaks (SSBs) can form when Top1-DNA complexes collide with transcription machinery or are flanked by some types of DNA lesions [28, 29]. TDP1 has been considered to play an important part in the process of SSBR [28, 30, 31] through resolving trapped Top1 residual and interacting with components of SSBR such as XRCC and DNA ligase IIIα.
In addition, TDP1’s ability to remove 3’ phosphoglycolates (PGs) formed by free radical-mediated oxidative damage [18, 24] gives TDP1 an important role in oxidative damage repair. Oxidative damage can be generated exogenously by tobacco, ionizing radiation, or xenobiotic agents such as bleomycin, neocarzinostatin, and mitomycin C, and intrinsically by enzymes of immune system as part of antimicrobial or antiviral response. Oxidative DNA damage may result in lesions such as 8-hydroxy-2’-deoxyguanosine (8-OH-dG), apurinic/apyrimidinic (AP) sites, SSBs, and DSBs. An 8-OH-dG lesion can be repaired by OGG1, a well-studied repair gene whose impaired activity was found correlated with risk of lung cancer [32]. AP sites are usually repaired by endonuclease Ape1, whose polymorphisms are identified as being correlated with increased lung cancer risk [33]. The major structural alterations in SSBs and DBSs, are caused by the loss of a nucleoside moiety or degradation of a sugar residual. Among the free ends, 3’PG is most refractory, and it is found that this unusual lesion can only be processed by relatively few enzymes, including TDP1 [18, 24]. In fact, the nuclear extract of cells from SCAN1 patients showed no capacity to cleave a typical 3’PG in vitro, implying that TDP1 might be the only enzyme capable of repairing this type of DNA damage. However, in spite of the importance of the enzymatic activity of TDP1, the lack of radiosensitivity of SCAN1 patients suggested that other unknown TDP1-independent pathways completed the task of 3’-PG repair [34].
TDP1 has recently reported to be able to cleave Top2-mediated damage [35]. Top2 inhibitors such as etoposide and adriamycin are frequently used in cancer chemotherapy, and if drug resistance is attributable to increased TDP1 expression and activity, the development of a TDP1 inhibitor in combination with Top2 inhibitors would be more attractive.
Finally, TDP1 has recently been shown to be able to cleave additional 3’ adducts from DNA. These activities include limited DNA and RNA 3’-exonuclease activities to remove a 3’ abasic site, an artificial 3’-biotin adduct from the DNA, and surprisingly, a phosphohistidine substrate accumulated by a mutant form of TDP1 with DNA [12]. Overexpression of TDP1 in cancer may imply therefore not only the resistance to CPT, but possibly a wide range of other drugs as well.
It is not clear whether expression of TDP1 and XPF are limited only in lung cancer. Further study is required to test the organ specificity of overexpression status of these genes, and determine whether measurement of expression level would help to tailor the CPT therapy of other solid tumors. This study also provides a possible explanation at a molecular level for variation in response to CPT treatment, and implies that there is a possibility for overcoming CPT resistance by developing a TDP1 inhibitor for those patients with high levels of TDP1. In order to definitively determine the responsibility of overexpression of TDP1 for CPT resistance, we expect further experiments be done by making primary cell lines from those cancerous tissues expressing different levels of TDP1, and testing in vitro the resistance of these cell lines to CPT. If the CPT resistance is consistent with the expression level of TDP1, a genetically engineered TDP1 inhibitor can be used to knockdown the expression of TDP1 and observe whether those resistant cells can become sensitive. Regardless, these data provide an insight into the in vivo status of the major repair pathway of CPT induced DNA damage in NSCLC patients.
Acknowledgments
We are grateful to Dr. Howard Nash (National Institute of Mental Health) for his critical reading and valuable comments for the manuscript.
Footnotes
Conflict of interest statement
None declared
Grant sponsor: NIDCR 1R01DE015939-01 [J.C.], NCI 5P50CA096784-03 Head and Neck SPORE [C.L. D.S., S.B., W.H.W., J.C.] Dr. Califano is a Damon Runyon-Lilly Clinical Investigator supported by the Damon Runyon Cancer Research Foundation (CI-#9) and a Clinical Innovator Award from the Flight Attendant Medical Research Institute
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mozzetti S, Ferlini C, Concolino P, Filippetti F, Raspaglio G, Prislei S, et al. Class iii beta-tubulin overexpression is a prominent mechanism of paclitaxel resistance in ovarian cancer patients. Clin Cancer Res. 2005;11:298–305. [PubMed] [Google Scholar]
- 2.Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after pka activation in breast cancer. Cancer Cell. 2004;5:597–605. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Lord RV, Brabender J, Gandara D, Alberola V, Camps C, Domine M, et al. Low ercc1 expression correlates with prolonged survival after cisplatin plus gemcitabine chemotherapy in non-small cell lung cancer. Clin Cancer Res. 2002;8:2286–91. [PubMed] [Google Scholar]
- 4.Pizzolato JF, Saltz LB. The camptothecins. Lancet. 2003;361:2235–42. doi: 10.1016/S0140-6736(03)13780-4. [DOI] [PubMed] [Google Scholar]
- 5.Stewart DJ. Update on the role of topotecan in the treatment of non-small cell lung cancer. Oncologist. 2004;9 (Suppl 6):43–52. doi: 10.1634/theoncologist.9-90006-43. [DOI] [PubMed] [Google Scholar]
- 6.Nivens MC, Felder T, Galloway AH, Pena MM, Pouliot JJ, Spencer HT. Engineered resistance to camptothecin and antifolates by retroviral coexpression of tyrosyl DNA phosphodiesterase-i and thymidylate synthase. Cancer Chemother Pharmacol. 2004;53:107–15. doi: 10.1007/s00280-003-0717-6. [DOI] [PubMed] [Google Scholar]
- 7.Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ. Scan1 mutant tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity. Embo J. 2005;24:2224–33. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, et al. Tdp1 overexpression in human cells counteracts DNA damage mediated by topoisomerases i and ii. J Biol Chem. 2004;279:55618–25. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 9.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase tdp1 is a member of the phospholipase d superfamily. Proc Natl Acad Sci U S A. 2001;98:12009–14. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a tyr-DNA phosphodiesterase that repairs topoisomerase i complexes. Science. 1999;286:552–5. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 11.Raymond AC, Burgin AB., Jr Tyrosyl-DNA phosphodiesterase (tdp1) (3′-phosphotyrosyl DNA phosphodiesterase) Methods Enzymol. 2006;409:511–24. doi: 10.1016/S0076-6879(05)09030-0. [DOI] [PubMed] [Google Scholar]
- 12.Interthal H, Chen HJ, Champoux JJ. Human tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem. 2005;280:36518–28. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for tdp1 assembled from vanadate, DNA, and a topoisomerase i-derived peptide. Chem Biol. 2003;10:139–47. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 14.Davies DR, Interthal H, Champoux JJ, Hol WG. Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (tdp1) from vanadate and tungstate-inhibited structures. J Mol Biol. 2002;324:917–32. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- 15.Houtsmuller AB, Rademakers S, Nigg AL, Hoogstraten D, Hoeijmakers JH, Vermeulen W. Action of DNA repair endonuclease ercc1/xpf in living cells. Science. 1999;284:958–61. doi: 10.1126/science.284.5416.958. [DOI] [PubMed] [Google Scholar]
- 16.Kaliraman V, Mullen JR, Fricke WM, Bastin-Shanower SA, Brill SJ. Functional overlap between sgs1-top3 and the mms4-mus81 endonuclease. Genes Dev. 2001;15:2730–40. doi: 10.1101/gad.932201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen XB, Melchionna R, Denis CM, Gaillard PH, Blasina A, Van de Weyer I, et al. Human mus81-associated endonuclease cleaves holliday junctions in vitro. Mol Cell. 2001;8:1117–27. doi: 10.1016/s1097-2765(01)00375-6. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Pouliot JJ, Nash HA. Repair of topoisomerase i covalent complexes in the absence of the tyrosyl-DNA phosphodiesterase tdp1. Proc Natl Acad Sci U S A. 2002;99:14970–5. doi: 10.1073/pnas.182557199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang SW, Burgin AB, Jr, Huizenga BN, Robertson CA, Yao KC, Nash HA. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type i topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–9. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase i covalent complexes in saccharomyces cerevisiae. Genes Cells. 2001;6:677–87. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 21.Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R. Xrcc3 induces cisplatin resistance by stimulation of rad51-related recombinational repair, s-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther. 2005;314:495–505. doi: 10.1124/jpet.105.084053. [DOI] [PubMed] [Google Scholar]
- 22.Furuchi T, Nitta K, Takahashi T, Naganuma A. Overexpression of ssl2p confers resistance to adriamycin and actinomycin d in saccharomyces cerevisiae. Biochem Biophys Res Commun. 2004;314:844–8. doi: 10.1016/j.bbrc.2003.12.160. [DOI] [PubMed] [Google Scholar]
- 23.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 24.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, Povirk LF. Conversion of phosphoglycolate to phosphate termini on 3′ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase htdp1. J Biol Chem. 2002;277:27162–8. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- 25.Strumberg D, Pilon AA, Smith M, Hickey R, Malkas L, Pommier Y. Conversion of topoisomerase i cleavage complexes on the leading strand of ribosomal DNA into 5′-phosphorylated DNA double-strand breaks by replication runoff. Mol Cell Biol. 2000;20:3977–87. doi: 10.1128/mcb.20.11.3977-3987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsao YP, Russo A, Nyamuswa G, Silber R, Liu LF. Interaction between replication forks and topoisomerase i-DNA cleavable complexes: Studies in a cell-free sv40 DNA replication system. Cancer Res. 1993;53:5908–14. [PubMed] [Google Scholar]
- 27.Vance JR, Wilson TE. Yeast tdp1 and rad1-rad10 function as redundant pathways for repairing top1 replicative damage. Proc Natl Acad Sci U S A. 2002;99:13669–74. doi: 10.1073/pnas.202242599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El-Khamisy SF, Caldecott KW. Tdp1-dependent DNA single-strand break repair and neurodegeneration. Mutagenesis. 2006;21:219–24. doi: 10.1093/mutage/gel024. [DOI] [PubMed] [Google Scholar]
- 29.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, et al. Mutation of tdp1, encoding a topoisomerase i-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–72. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 30.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–13. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 31.Plo I, Liao ZY, Barcelo JM, Kohlhagen G, Caldecott KW, Weinfeld M, et al. Association of xrcc1 and tyrosyl DNA phosphodiesterase (tdp1) for the repair of topoisomerase i-mediated DNA lesions. DNA Repair (Amst) 2003;2:1087–100. doi: 10.1016/s1568-7864(03)00116-2. [DOI] [PubMed] [Google Scholar]
- 32.Paz-Elizur T, Krupsky M, Blumenstein S, Elinger D, Schechtman E, Livneh Z. DNA repair activity for oxidative damage and risk of lung cancer. J Natl Cancer Inst. 2003;95:1312–9. doi: 10.1093/jnci/djg033. [DOI] [PubMed] [Google Scholar]
- 33.Popanda O, Schattenberg T, Phong CT, Butkiewicz D, Risch A, Edler L, et al. Specific combinations of DNA repair gene variants and increased risk for non-small cell lung cancer. Carcinogenesis. 2004;25:2433–41. doi: 10.1093/carcin/bgh264. [DOI] [PubMed] [Google Scholar]
- 34.Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, Povirk LF. Deficiency in 3′-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (tdp1) Nucleic Acids Res. 2005;33:289–97. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nitiss KC, Malik M, He X, White SW, Nitiss JL. Tyrosyl-DNA phosphodiesterase (tdp1) participates in the repair of top2-mediated DNA damage. Proc Natl Acad Sci U S A. 2006;103:8953–8. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]