

Abstract
The clinical impact of MLL partial tandem duplication (MLL-PTD) was evaluated in 238 adults aged 18 to 59 years with cytogenetically normal (CN) de novo acute myeloid leukemia (AML) who were treated intensively on similar Cancer and Leukemia Group B protocols 9621 and 19808. Twenty-four (10.1%) patients harbored an MLL-PTD. Of those, 92% achieved complete remission (CR) compared with 83% of patients without MLL-PTD (P = .39). Neither overall survival nor disease-free survival significantly differed between the 2 groups (P = .67 and P = .55, respectively). Thirteen MLL-PTD+ patients relapsed within 1.4 years of achieving CR. MLL-PTD+ patients who relapsed more often had other adverse CN-AML–associated molecular markers. In contrast with previously reported studies, 9 (41%) MLL-PTD+ patients continue in long-term first remission (CR1; range, 2.5-7.7 years). Intensive consolidation therapy that included autologous peripheral stem-cell transplantation during CR1 may have contributed to the better outcome of this historically poor-prognosis group of CN-AML patients with MLL-PTD.
Introduction
The MLL partial tandem duplication (MLL-PTD) produces an in-frame, elongated protein and occurs in approximately 8% of patients with cytogenetically normal (CN) acute myeloid leukemia (AML).1 The MLL-PTD affects only 1 allele in CN-AML, and the second, wild-type (WT), MLL allele is silenced.2 Unlike MLL chimeric fusion proteins arising from translocations involving 11q23, the MLL-PTD protein retains the C-terminal domains including the histone H3 lysine 4 methyltransferase activity.3 In a murine knock-in model, the Mll-PTD acts as a gain-of-function allele, giving rise to aberrations in skeletal development and conferring proliferation and self-renewal advantages to hematopoietic stem/progenitor cells without causing frank leukemia.4
The MLL-PTD was the first adverse prognostic molecular marker identified in CN-AML.5,6 In several subsequent studies, the presence of MLL-PTD was associated with a shorter remission duration, with most patients relapsing within 1 year.7–10 We report herein for the first time that younger adults with MLL-PTD (MLL-PTD+) treated on 2 recent frontline Cancer and Leukemia Group B (CALGB) protocols had a clinical outcome comparable to that of CN-AML patients without MLL-PTD (MLL-PTD−) and that a substantial percentage of MLL-PTD+ patients are disease-free beyond 2.5 years.
Patients, materials, and methods
A combination of real-time reverse transcriptase–polymerase chain reaction (RT-PCR) and nested RT-PCR/sequencing was used to detect MLL-PTD in pretreatment bone marrow (BM) or blood (PB) samples as previously described.2,7 All patients gave informed consent for the research use of their specimens, in accordance with the Declaration of Helsinki. The clinical trials and companion protocols were approved by the Ohio State University Institutional Review Board and the Cancer and Leukemia Group B. Screening was conducted as described previously for additional molecular markers associated with CN-AML (ie, FLT3 internal tandem duplication [FLT3-ITD], FLT3 tyrosine kinase domain [FLT3-TKD] mutation, NPM1 mutation, and high BAALC and ERG expression).11–14 All screening assays were performed at The Ohio State University Comprehensive Cancer Center.
Patients enrolled on CALGB 9621 and 19808 received induction treatment that included cytarabine, etoposide, and daunorubicin with or without PSC-833, a multi-drug resistance protein inhibitor also called valspodar. Patients who achieved complete remission (CR) underwent autologous peripheral blood stem-cell transplantation (auto-PBSCT) phase.15,16 Clinical end points were disease-free survival (DFS) and overall survival (OS), as defined previously.14
Pretreatment clinical features were compared between the MLL-PTD+ and MLL-PTD− groups as well as between MLL-PTD+ patients who did and those who did not relapse using Fisher 2-sided exact and Wilcoxon rank sum tests for categoric and continuous variables, respectively. Estimatedprobabilities of DFS and OS were calculated using the Kaplan-Meier method, and the log-rank test evaluated differences between survival distributions. All analyses were performed by the CALGB Statistical Center.
Results and discussion
Of the 238 CN-AML patients, 24 (10.1%) harbored an MLL-PTD, which represents the largest series of MLL-PTD+ CN-AML patients treated similarly and with outcome reported. MLL-PTD+ patients differed from MLL-PTD− patients with respect to white blood cell (WBC) counts (median, 4.35 vs 25.15 X109/L; P < .001), extramedullary involvement (9% vs 31%; P = .03), and French-American-British (FAB) subgroups (P = .04); a higher percentage of FAB M2 (48% vs 32%) and a lower percentage of FAB M4/M5 (10% vs 35%) was observed in the MLL-PTD+ group (Table 1). Among patients with available tissue, MLL-PTD+ patients more often presented with wild-type NPM1 (NPM1-WT; 76% vs 32%; P < .001) and high BAALC expression (81% vs 46%; P = .009). In contrast, there was no significant association between the MLL-PTD status and the presence of FLT3-ITD (P = .37); 25% and 35% of MLL-PTD+ and MLL-PTD− patients, respectively, had FLT3-ITD (Table 1).
Table 1.
Comparison of CN-AML patients with and without MLL-PTD and comparison of MLL-PTD+ patients who relapsed with MLL-PTD+ patients who did not relapse
| Characteristic |
MLL-PTD |
P |
MLL-PTD+ |
P | ||
|---|---|---|---|---|---|---|
| Negative, n=214 | Positive, n=24 | No relapse, n=9 | Relapse, n=13 | |||
| Age, y | .39 | .62 | ||||
| Median | 46 | 49 | 47 | 51 | ||
| Range | 18-59 | 22-59 | 22-59 | 23-59 | ||
| Sex, no. of males (%) | 100 (47) | 13 (54) | .52 | 5 (56) | 7 (54) | 1.00 |
| Race, n (%) | 1.00 | .16 | ||||
| White | 186 (88) | 22 (92) | 7 (78) | 13 (100) | ||
| Nonwhite | 25 (12) | 2 (8) | 2 (22) | 0 (0) | ||
| Hemoglobin, g/dL | .29 | .07 | ||||
| Median | 9.3 | 8.75 | 9.5 | 8.3 | ||
| Range | 4.6-13.6 | 6.2-11.8 | 7.8-10.3 | 6.2-11.2 | ||
| Platelet count, ×109/L | .78 | .15 | ||||
| Median | 60.5 | 64.5 | 31 | 72 | ||
| Range | 7-466 | 5-395 | 5-395 | 23-120 | ||
| WBC count, ×109/L | <.001 | .76 | ||||
| Median | 25.15 | 4.35 | 4.1 | 6.7 | ||
| Range | 0.8-295.0 | 0.8-64.2 | 0.8-27.8 | 0.9-64.2 | ||
| Blood blasts, % | .40 | .08 | ||||
| Median | 56 | 39.5 | 26 | 54 | ||
| Range | 0-97 | 5-95 | 7-75 | 10-95 | ||
| Bone marrow blasts, % | .17 | .03 | ||||
| Median | 65 | 55 | 40.5 | 66 | ||
| Range | 12-99 | 10-94 | 10-80 | 38-94 | ||
| Centrally reviewed FAB, n (%) | .04 | .83 | ||||
| M0 | 4 (3) | 1 (5) | 0 (0) | 1 (9) | ||
| M1 | 40 (27) | 5 (24) | 2 (25) | 2 (18) | ||
| M2 | 46 (32) | 10 (48) | 4 (50) | 5 (45) | ||
| M4 | 37 (25) | 2 (10) | 0 (0) | 2 (18) | ||
| M5 | 14 (10) | 0 (0) | 0 (0) | 0 (0) | ||
| M6 | 1 (1) | 2 (10) | 1 (13) | 1 (9) | ||
| Unclassified | 4 (3) | 1 (5) | 1 (13) | 0 (0) | ||
| Extramedullary involvement, n (%)* | .03 | 1.00 | ||||
| No | 146 (69) | 21 (91) | 8 (89) | 11 (92) | ||
| Yes | 65 (31) | 2 (9) | 1 (11) | 1 (8) | ||
| Lymphadenopathy, n (%) | .05 | NA | ||||
| No | 184 (86) | 24 (100) | 9 (100) | 13 (100) | ||
| Yes | 29 (14) | 0 (0) | 0 (0) | 0 (0) | ||
| FLT3-ITD, n (%) | .37 | .33 | ||||
| Absent | 139 (65) | 18 (75) | 8 (89) | 8 (62) | ||
| Present | 75 (35) | 6 (25) | 1 (11) | 5 (38) | ||
| FLT3-TKD, n (%) | 1.00 | 1.00 | ||||
| Absent | 180 (91) | 18 (95) | 6 (100) | 10 (91) | ||
| Present | 18 (9) | 1 (5) | 0 (0) | 1 (9) | ||
| NPM1, n (%) | <.001 | .12 | ||||
| Wild-type | 63 (32) | 13 (76) | 3 (50) | 9 (90) | ||
| Mutated | 136 (68) | 4 (24) | 3 (50) | 1 (10) | ||
| BAALC expression, n (%)† | .009 | .03 | ||||
| Low | 83 (54) | 3 (19) | 3 (60) | 0 (0) | ||
| High | 72 (46) | 13 (81) | 2 (40) | 9 (100) | ||
| ERG expression, n (%)‡ | .27 | .23 | ||||
| Low | 101 (64) | 12 (80) | 5 (100) | 5 (63) | ||
| High | 57 (36) | 3 (20) | 0 (0) | 3 (38) | ||
| Complete remission rate, n (%) | 177 (83) | 22 (92) | .39 | NA | NA | NA |
| Relapse rate, n (%) | 96 (54) | 13 (59) | .82 | NA | NA | NA |
| Overall survival/ | .67 | NA | NA | NA | ||
| Median, y | 3.7 | NR | NA | NA | ||
| Alive at 3 y (95% CI), % | 52 (45-59) | 50 (29-68) | NA | NA | ||
| Alive at 5 y (95% CI), % | 45 (38-52) | 50 (29-68) | NA | NA | ||
| Disease-free survival | .55 | NA | NA | NA | ||
| Median, y | 2.5 | 1.0 | NA | NA | ||
| Disease-free at 3 y (95% CI), % | 48 (40-55) | 41 (21-60) | NA | NA | ||
| Disease-free at 5 y (95% CI), % | 43 (35-51) | 41 (21-60) | NA | NA | ||
The median follow-up for the 116 of 238 patients alive is 4.7 years, ranging from 1.2 to 8.9 years. For the 199 patients who achieved CR, median disease-free survival duration for the 90 who have not relapsed is 5.4 years, ranging from 1.5 to 8.8 years.
MLL-PTD indicates partial tandem duplication of the MLL gene; WBC, white blood cell; FAB, French-American-British; FLT3-ITD, internal tandem duplication of the FLT3 gene; FLT3-TKD, tyrosine kinase domain mutation of the FLT3 gene; CI, confidence interval; NA, not applicable; and NR, not reached.
Includes involvement of the central nervous system, hepatomegaly, splenomegaly, lymphadenopathy, skin infiltrates, gum hypertrophy, and/or a mediastinal mass.
For patients on protocol 9621, cut point same as in Baldus et al.13 For patients on protocol 19808, median BAALC expression value used for cut point.
For patients on protocol 9621, cut point same as in Marcucci et al.14 For patients on protocol 19808, median ERG expression value used for cut point.
CR rates were not significantly different between the MLL-PTD+ and MLL-PTD− patients (92% vs 83%; P = .39; Table 1). The CR rate for MLL-PTD+ patients was similar to that reported in a German study (Döhner et al9) that administered idarubicin, cytarabine, and etoposide–based intensive double-induction therapy, and higher than CR rates observed in other studies (Table S1, available on the Blood website; see the Supplemental Materials link at the top of the online article).7,8,17 Younger age (< 60 years) and/or inclusion of etoposide may have contributed to the high CR rates in the CALGB and German studies.
With a median follow-up of 4.7 years, no significant differences in DFS (P = .55) or OS (P = .67) between MLL-PTD+ and MLL-PTD− patients were observed (Table 1). The estimated 3-year DFS and OS rates for MLL-PTD+ patients were, respectively, 41% and 50% compared with, respectively, 48% and 52% for MLL-PTD− patients (Table 1). Most striking was that 50% of MLL-PTD+ patients were alive at last follow-up and 9 (41%) of 22 were still in first CR (CR1), ranging from 2.2 to 7.7 years (Figure 1). This is markedly different from data previously reported, where few adult MLL-PTD+ patients maintained a remission beyond 2 years (Table S1). In the Döhner et al9 study, CN-AML patients aged 16 to 60 years in CR1 were treated with a high-dose cytarabine and mitoxantrone-based (HAM) regimen for the first consolidation therapy and yet 11 of 16 MLL-PTD+ patients relapsed within 2 years, and the 5 patients remaining in CR1 were censored with short follow-ups, from only a few months to about a year after achieving remission. Two patients in the Döhner et al9 study, who achieved a second CR and received allogeneic transplantation, were alive with approximately 3 and a half to 4 years of follow-up. In the current study, 18 of 22 MLL-PTD+ patients in CR1 underwent auto-PBSCT, which may have contributed to the reduced number of early relapses in our patients with MLL-PTD+ CN-AML. Three of the other 4 received multiple courses of high-dose cytarabine and 1 received 1 course of high-dose cytarabine before refusing further treatment.
Figure 1.
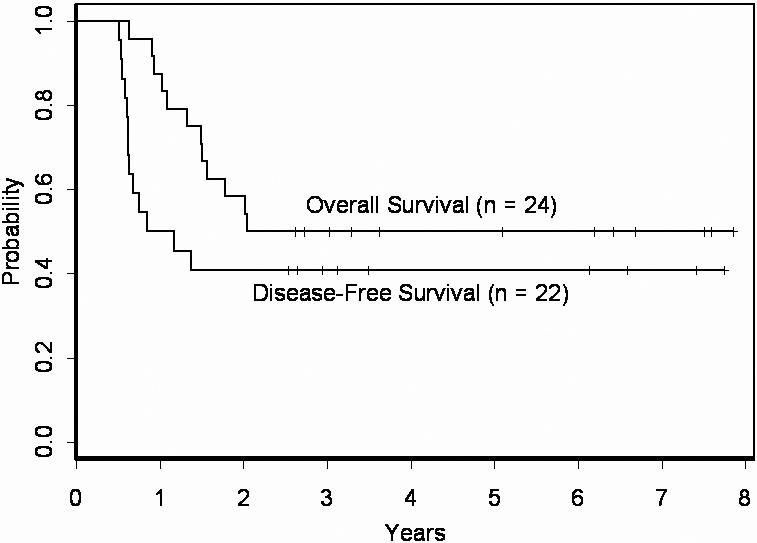
Overall survival and disease-free survival of CN-AML patients with MLL-PTD evaluated in this study. Fifty percent of the patients are alive and 41% remain disease-free in CR1 beyond 2.5 years.
Although a considerable fraction of MLL-PTD+ patients were alive and relapse-free, the majority relapsed within the first 1.4 years of remission (Figure 1). Thus, we analyzed the MLL-PTD+ group for pretreatment and/or molecular characteristics that might explain some of the differences in outcome in this subgroup. Relapsed patients had a higher percentage of BM blasts at diagnosis (median, 66% vs 40.5%; P = .03) and more often were high BAALC expressers (P = .03; Table 1). Nine of 11 MLL-PTD+ patients with high BAALC relapsed, whereas all 3 patients with low BAALC remain in remission (Figure S1A). While not statistically significant, the presence of other adverse molecular prognostic markers was also more frequent among relapsed MLL-PTD+ patients compared with MLL-PTD+ patients who remain in remission. Thirty-eight percent of the relapsed patients had FLT3-ITD compared with only 11% of those still in CR1. Similar trends were observed for NPM1-WT and high ERG expression. Among relapsed MLL-PTD+ patients evaluated for NPM1 mutations and ERG expression, 9 of 10 had only NPM1-WT alleles and 3 of 8 were high ERG expressers. In contrast, among MLL-PTD+ patients remaining in remission, 3 of 6 carried NPM1 mutations and 0 of 5 were high ERG expressers (Table 1; Figure S1B-D).
In conclusion, we have observed for the first time a relatively good outcome for MLL-PTD+ patients who achieved CR, with 41% of such patients remaining in first remission beyond 2.5 years. This contrasts with previous studies reporting only a few patients who achieved long-lasting remissions. Besides the multitude of other potential factors, we postulate that treatment advances for younger adults may have contributed to the better outcome observed for the MLL-PTD+ patients reported in this study. Although these are encouraging findings, it is clear that the majority of MLL-PTD+ patients still relapse early. Our data suggest that the presence of additional, negative prognostic markers may be a contributing factor. Larger studies are necessary to confirm our results and to elucidate the underlying mechanisms of leukemogenesis, which could lead to the development of molecularly targeted curative therapies.
Supplementary Material
Acknowledgments
We thank Ms Donna Bucci of the CALGB Leukemia Tissue Bank and the Ohio State University Comprehensive Cancer Center's Nucleic Acid Shared Resource for technical support. We thank CALGB-participating institutions, medical professionals, and AML patients for their valuable involvement with this study.
This work was supported in part by National Institutes of Health (NIH) grants CA101140, CA77658, CA16058, CA102031, CA096887, CA089341, CA41287, and CA098933 and the Coleman Leukemia Research Foundation.
Footnotes
The online version of this manuscript contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.P.W., A.S.R., G.M., K.M., and C. D. Bloomfield contributed to the design and analysis of this study. S.P.W., A.S.R., G.M., K.M., P.P., and C. D. Bloomfield contributed to the writing of this manuscript and all authors agreed on the final version. S.P.W., P.P., C.L., C. D. Baldus, J.W., and T.V. carried out laboratory-based research. A.S.R. performed statistical analyses. B.L.P., A.J.C., J.E.K., R.A.L., M.A.C., G.M., and C. D. Bloomfield were involved directly or indirectly in care of patients and/or sample procurement.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the CALGB study group can be found in Document S1, available on the Blood website; see the Supplemental Materials link at the top of the online article.
Correspondence: Susan P. Whitman, Comprehensive Cancer Center and James Cancer Hospital and Solove Research Institute, The Ohio State University, 2001 Polaris Parkway, Columbus, OH 43240; e-mail: susan.whitman@osumc.edu.
References
- 1.Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007;109:431–448. doi: 10.1182/blood-2006-06-001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitman SP, Liu S, Vukosavljevic T, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood. 2005;106:345–352. doi: 10.1182/blood-2005-01-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol. 2006;135:438–449. doi: 10.1111/j.1365-2141.2006.06301.x. [DOI] [PubMed] [Google Scholar]
- 4.Dorrance AM, Liu S, Yuan W, et al. Mll partial tandem duplication induces aberrant Hox expression in vivo via specific epigenetic alterations. J Clin Invest. 2006;116:2707–2716. doi: 10.1172/JCI25546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caligiuri MA, Schichman SA, Strout MP, et al. Molecular rearrangement of the ALL-1 gene in acute myeloid leukemia without cytogenetic evidence of 11q23 chromosomal translocations. Cancer Res. 1994;54:370–373. [PubMed] [Google Scholar]
- 6.Schichman SA, Caligiuri MA, Gu Y, et al. ALL-1 partial duplication in acute leukemia. Proc Natl Acad Sci U S A. 1994;91:6236–6239. doi: 10.1073/pnas.91.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caligiuri MA, Strout MP, Lawrence D, et al. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 1998;58:55–59. [PubMed] [Google Scholar]
- 8.Schnittger S, Kinkelin U, Schoch C, et al. Screening for MLL tandem duplication in 387 unselected patients with AML identify a prognostically unfavorable subset of AML. Leukemia. 2000;14:796–804. doi: 10.1038/sj.leu.2401773. [DOI] [PubMed] [Google Scholar]
- 9.Döhner K, Tobis K, Ulrich R, et al. Prognostic significance of partial tandem duplications of the MLL gene in adult patients 16 to 60 years old with acute myeloid leukemia and normal cytogenetics: a study of the Acute Myeloid Leukemia Study Group Ulm. J Clin Oncol. 2002;20:3254–3261. doi: 10.1200/JCO.2002.09.088. [DOI] [PubMed] [Google Scholar]
- 10.Muñoz L, Nomdedéu JF, Villamor N, et al. Acute myeloid leukemia with MLL rearrangements: clinicobiological features, prognostic impact and value of flow cytometry in the detection of residual leukemic cells. Leukemia. 2003;17:76–82. doi: 10.1038/sj.leu.2402708. [DOI] [PubMed] [Google Scholar]
- 11.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 12.Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 13.Baldus CD, Tanner SM, Ruppert AS, et al. BAALC expression predicts clinical outcome of de novo acute myeloid leukemia patients with normal cytogenetics: a Cancer and Leukemia Group B study. Blood. 2003;102:1613–1618. doi: 10.1182/blood-2003-02-0359. [DOI] [PubMed] [Google Scholar]
- 14.Marcucci G, Baldus CD, Ruppert AS, et al. Overexpression of the ETS-related gene, ERG, predicts a worse outcome in acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:9234–9242. doi: 10.1200/JCO.2005.03.6137. [DOI] [PubMed] [Google Scholar]
- 15.Kolitz JE, George SL, Dodge RK, et al. Dose escalation studies of cytarabine, daunorubicin, and etoposide with and without multidrug resistance modulation with PSC-833 in untreated adults with acute myeloid leukemia younger than 60 years: final induction results of Cancer and Leukemia Group B study 9621. J Clin Oncol. 2004;22:4290–4301. doi: 10.1200/JCO.2004.11.106. [DOI] [PubMed] [Google Scholar]
- 16.Kolitz JE, George SL, Marcucci G, et al. A randomized comparison of induction therapy for untreated acute myeloid leukemia (AML) in patients < 60 years using P-glycoprotein (Pgp) modulation with Valspodar (PSC833): preliminary results of Cancer and Leukemia Group B study 19808 [abstract]. Blood. 2005;106:122a–123a. Abstract 407. [Google Scholar]
- 17.Shiah H-S, Kuo Y-Y, Tang J-L, et al. Clinical and biological implications of partial tandem duplication of the MLL gene in acute myeloid leukemia without chromosomal abnormalities at 11q23. Leukemia. 2002;16:196–202. doi: 10.1038/sj.leu.2402352. AQ1: “respectively” inserted. Please confirm that your meaning has not been changed. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.