

Abstract
Eukaryotic membrane trafficking is a conserved process under tight temporal and spatial regulation in which the fusion of membranes is driven by the formation of the ternary SNARE complex. Syntaxin 1a, a core component of the exocytic SNARE1 complex in neurones and neuroendocrine cells, is regulated directly by munc18-1, its cognate SM (Sec1p/Munc18) protein. SM proteins show remarkable structural conservation throughout evolution indicating a common binding mechanism and function. However, SM proteins possess disparate binding mechanisms and regulatory effects, with munc18-1, the major brain isoform, classed as atypical in both its binding specificity and mode. We now show that munc18-1 interacts with syntaxin 1a through two mechanistically distinct modes of binding, both in vitro and in living cells, in contrast to current models. Furthermore these functionally divergent interactions occur at distinct cellular locations. These findings provide a molecular explanation for the multiple, spatially distinct roles of munc18-1.
In neuronal and neuroendocrine cells, exocytosis is mediated by the plasma membrane proteins (t-SNAREs) syntaxin and SNAP-25 (synaptosome-associated protein 25 kDa) and the vesicular protein synaptobrevin (v-SNARE)(1,2). The cytoplasmic regions of these three proteins interact to form a trimeric, four-helical complex, the generation of which drives fusion of the two opposing bilayers(3). This process is regulated by a conserved set of accessory proteins which operate throughout the trafficking pathway. The Sec1p/Munc18 (SM) protein family represents one such set of modulators, with SM protein mutations characterised by a severe disruption of general secretion or neurotransmitter release(4-7). The mammalian SM protein munc18-1 was originally isolated as a syntaxin 1 binding protein which binds to the monomeric form of syntaxin 1, rendering the t-SNARE unable to form the SDS-resistant ternary SNARE complex(8,9). Syntaxin 1 can act as a molecular switch, adopting two structurally distinct forms(10). In the open form, the SNARE helix does not interact with the N-terminal three helical regulatory domain (termed Habc) and has been shown to not interact with munc18-1(10). In contrast the closed form of syntaxin 1, in which the N-terminal Habc domain interacts with the SNARE helix, exhibits a high affinity for munc18-1. Association of syntaxin 1 with its SNARE partners to form the ternary SNARE complex, which prevents syntaxin adopting the closed conformation, has also been shown to preclude munc18-1 binding(11). However, a recent finding by Zilly et al using lysed cellular membrane sheets provided evidence that munc18-1 may interact with syntaxin 1 when in the binary SNARE complex (a heterodimer of syntaxin and SNAP-25)(12).
The interaction of munc18-1 with its cognate syntaxin is in sharp contrast to the specificity of its yeast homologue Sec1p, which binds its cognate syntaxin, Sso1p, in the ternary SNARE complex and not in the monomeric state(13). This binding specificity has, however, since been questioned(14). Another yeast SM protein, Vps45p, binds to its cognate Golgi syntaxin, Tlg2p, both in the monomeric state and in the ternary SNARE complex(15,16). This interaction can occur through an N-terminal region of Tlg2p, with a similar interaction observed for its mammalian Golgi homologue, syntaxin 16(17). A similar ability to bind syntaxin in both monomeric and complexed forms has been observed for the SM protein Sly1p and its cognate syntaxin Sed5p(18,19) This apparent discrepancy in binding mode suggests differences in binding sites and recognition motifs of SM proteins and their syntaxin partners.
To date, the crystal structures of two SM proteins, munc18-1 and Sly1p (from S. cerevisiae), in complex with their cognate SNAREs, have been solved(20,21). Despite exhibiting high structural homology, munc18-1 and Sly1p appear to interact with their cognate syntaxins, syntaxin 1a and Sed5p, respectively, through independent sites. Munc18-1 binds to the closed conformation of syntaxin 1a with the N-terminal Habc domain folded back on to the helix involved in SNARE complex formation(10,22), whereas Sly1p binds the N-terminus of Sed5p through an independent binding site. The disparate modes of interaction between SM proteins and their cognate syntaxins, observed in different species and for different membrane compartments, are at odds with the conserved role for SM proteins in membrane fusion indicated by genetic experiments(2,23). Importantly, effects exerted by munc18-1 on different stages in the syntaxin lifecycle, from trafficking(24), through vesicle docking(25) to the final fusion event itself(26), cannot be explained using the current model of the munc18-1 - syntaxin 1a interaction. In this study we analysed in detail the syntaxin 1 munc18-1 interaction using mutations shown to lock syntaxin 1 in the open conformation(10). Here we show that munc18-1 binds to an evolutionarily conserved motif at the N-terminus of syntaxin 1 in addition to the closed form binding mode. Using quantitative colocalisation and fluorescence lifetime imaging microscopy (FLIM) we show that these two binding modes of syntaxin1 and munc18-1 are spatially segregated in living cells. Furthermore while the closed form binding mode prevents progress through the SNARE assembly pathway, syntaxin 1 in the open form can progress to the ternary SNARE complex with munc18-1 remaining associated. This is the first demonstration that the major neuronal and neuroendocrine SNARE, syntaxin 1, and its cognate SM protein, munc18-1, can interact through dual modes of binding.
Experimental Procedures
Vectors and cell culture
Plasmids encoding glutathione-S-transferase (GST) fusion proteins with syntaxin 1a (a.a. 1-261, cytoplasmic domain), SNAP-25 (a.a. 1-206) and synaptobrevin (a.a. 1-96) were described previously(27,28). A plasmid encoding a poly histidine tagged munc18-1 (a.a. 1-594) was as previously described(29). Generation of N-terminal truncations of syntaxin 1a was performed by PCR and subsequent ligation into HindIII/KpnI and HindIII/XbaI sites of pmCerulean-C1 and PGEX-KG respectively or into pTargeT (Promega). Distance between the fusion tag and syntaxin 1a was maintained in all constructs. The [L165A, E166A] mutation was generated by site directed mutagenesis using a QuikChange II XL kit (Stratagene). An EYFP-munc18-1 fusion was generated using similar standard techniques. Neuroblastoma 2a (N2a) cells were grown in DMEM supplemented with 10% foetal bovine serum, 10 mM L-glutamine, 50 units penicillin, 50 μg/ml streptomycin and maintained at 37°C in 5% (v/v) CO2, 95% (v/v) air. All transfections were performed using ExGen 500 (Fermentas).
Protein Biochemistry
Recombinant GST fusion proteins were expressed and purified as previously described(30). For in vitro binding reactions, 2 μg GST-syntaxin 1a or SNAP-25 was immobilised on glutathione Sepharose beads (GE Healthcare) and incubated in a total volume of 100 μl of 20 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.1% TX-100 (Buffer A) with 3 μg of protein to be tested for binding for 1 hour at 4°C. Beads were washed by low speed centrifugation and bound protein eluted in SDS-containing sample buffer followed by SDS PAGE and Coomassie staining. For binding reactions involving munc18-1, GST fused syntaxin 1a was incubated with either freshly prepared detergent rat brain extract, as previously described(30), or with freshly prepared detergent bacterial extract containing expressed recombinant His6-munc18-1. Circular dichroism experiments were performed as previously described(31). For the measurement of syntaxin 1a affinity for munc18-1, 0.2 pmol of GST fused syntaxin 1a was immobilised on glutathione Sepharose beads and incubated with increasing concentrations of munc18-1 in detergent bacterial extract in a reaction volume of 0.5 ml at 21°C. The beads were washed three times in buffer A and bound protein analysed by Western immunoblotting using a monoclonal anti-munc18-1 antibody (BD Biosciences) and West Dura enhanced chemiluminescence kit (Pierce). The chemiluminescent signal was imaged using a cooled 16-bit CCD and quantified using ImageJ (http://rsb.info.nih.gov/ij/). The concentration of munc18-1 in the detergent bacterial extract was measured by purification of munc18-1 and in-gel quantification using Sypro Red (Invitrogen). The calculated intensity volumes were fitted with a variable slope dose response relationship using Prism (GraphPad). For binding assays using a purified syntaxin 1a-munc18-1 complex, bacterial detergent extracts containing expressed GST-Syx1-261 (or variant) and His6-munc18-1 were mixed. The resulting protein complex was purified sequentially on glutathione Sepharose resin and IMAC resin (Bio-Rad). Purity was assessed by Sypro Red in-gel staining to confirm an equimolar stoichiometric ratio(30).
Confocal laser scanning microscopy and image analysi
All imaging experiments were performed using a Zeiss LSM 510 Axiovert confocal laser scanning microscope, equipped with a pulsed excitation source (MIRA 900 Ti:Sapphire femtosecond pulsed laser, with a coupled VERDI 10W pump laser (Coherent). Data were acquired using a 1024 × 1024 pixel image size, using a Zeiss Plan NeoFLUAR 1.4 NA 63x oil immersion lens, or a Zeiss C-Apochromat 1.2 NA 63x water corrected immersion objective lens. All imaging was performed using living cells, maintained at 37°C in 5% (v/v) CO2, 95 % (v/v) air. Image data, acquired at Nyquist sampling rates, were deconvolved using Huygens software (Scientific Volume Imaging) and the resulting 3-D models were analysed using ImageJ software (http://rsb.info.nih.gov/ij/). Residual maps were generated by calculating the residual of each voxel from the linear regression fit to the intensities of each channel within each voxel. The resulting residuals are displayed on a colour scale from -1 to 1 (with zero residual coloured cyan) with brightness corresponding to the combined intensity of the two channels. Cell peripheries were determined using transmitted light imaging combined with CLSM data.
TCSPC-FLIM acquisition and analysis
Time-correlated single photon counting (TCSPC) measurements were made under 800-820 nm TPE (two-photon excitation), which efficiently excited cerulean, without any detectable direct excitation or emission from EYFP, using a non-descanned detector (R3809U-50) multichannel plate-photomultiplier tube (MCP-PMT), or a fast photomultiplier tube (H7422; both Hamamatsu Photonics UK) coupled directly to the rear port of the Axiovert microscope. TCSPC recordings were acquired for between 10 s and 60 s, mean photon counts were between 105 - 106 counts per second. Images were recorded at 256 × 256 pixels from a 1024 × 1024 image scan with 256 time bins over a 12 ns period(32). Off-line FLIM data analysis used pixel-based fitting software (SPCImage, Becker & Hickl). The fluorescence was assumed to follow a multi-exponential decay. In addition, an adaptive offset-correction was performed. A constant offset takes into consideration the time-independent baseline due to dark noise of the detector and the background caused by room light, calculated from the average number of photons per channel preceding the rising part of the fluorescence trace. To fit the parameters of the multi-exponential decay to the fluorescence decay trace measured by the system, a convolution with the instrumental response function was carried out. The optimisation of the fit parameters was performed by using the Levenberg-Marquardt algorithm, minimising the weighted chi-square quantity. This approach can be used to separate the interacting from the non-interacting donor fraction in our FRET systems. The long lifetime component τ2 was determined by control assays with cerulean alone, or expressed with (non-interacting) EYFP as described above. This value was subsequently used as a fixed τ2 lifetime for all other experiments. As controls for non-specific FRET, or FRET between GFPs that may form dimers spontaneously when over-expressed in cells, we determined the fluorescence lifetimes of cerulean-Syx1-288 alone, cerulean alone, or cerulean-Syx1-288 co-transfected with EYFP (not shown). No FRET was detected in any of these experiments. Likewise, experiments using ceruleanA206K-fused Syx1-288 revealed no self-dimerisation between fluorescent proteins
RESULTS
We analysed the conservation of amino acids in syntaxin 1 homologues from evolutionarily distant species along with Sed5p from S. cerevisiae (Fig 1A). There is a high degree of similarity between the syntaxin homologues throughout the sequence corresponding to the SNARE helix and head domain of syntaxin. Of note is the additional spike in similarity in the first 15 amino acids. Analysis of the sequence contained in this region demonstrated a high degree of conservation of amino acids with a lysine-aspartate-arginine (KDR) motif absolutely conserved in both Sed5p and the syntaxin homologues. A structural alignment of munc18-1 and Sly1p demonstrates the highly conserved topology of these two proteins despite an overall sequence identity of 18% (Fig. 1B). Highlighted on the bound Sed5p peptide (Fig. 1B) are the residues absolutely conserved in the sequence alignment in Fig. 1A. The conservation of these residues suggested that the amino-terminal of syntaxin 1a may be capable of interacting with munc18-1 in a similar manner and that an “open” mutation of syntaxin 1a(10), could still support an interaction with munc18-1.
Fig. 1.
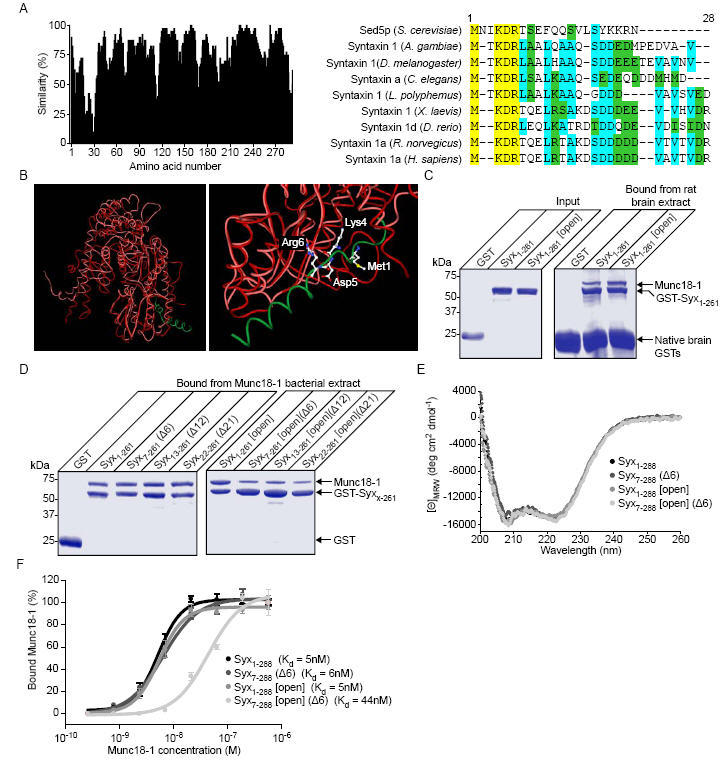
Syntaxin 1a can interact with munc18-1 through its N-terminus in vitro. A, Sequence alignment of syntaxin 1 homologues and Sed5p with similarity scored at each position (left panel). The highly conserved N-terminal region (a.a. 1- 28, right panel) is shown on a colour coded scale (yellow - identical, cyan-conserved, green - similar). B, Structural alignment of Sly1p (red) bound to an N-terminal peptide of Sed5p (green) (PDB: 1MQS(20)) and munc18-1 (pink) (PDB: 1EPU(39)). Highlighted on an enlarged view (right panel) are the absolutely conserved residues from panel a. C, Both Syx1-261 and Syx1-261 [open], immobilised on Sepharose beads, readily bind munc18-1. Native brain GSTs bound directly to the Sepharose resin. D, Truncation of the N-terminus of Syx1-261 [open] reduces its ability to bind munc18-1. N-terminal truncations of GST-Syx1-261 and GST-Syx1-261 [open] were immobilised on glutathione Sepharose beads and incubated with His6-munc18-1 containing bacterial lysate. Bound material was analysed by SDS PAGE and Coomassie staining. E, Circular dichroism spectra of Syx1-261, Syx1-261 (Δ6), Syx1-261 [open] and Syx1-261 [open] (Δ6). F, Measurement of the equilibrium dissociation constant for the syntaxin 1a-munc18-1 complex. GST-Syx1-261, or a mutant form, immobilised on Sepharose beads, was incubated in the presence of varying concentrations of munc18-1. Bound material was analysed by Western immunoblotting. Error bars represent S.E.M. (n=3).
We therefore determined the ability of syntaxin 1a (Syx1-261) and an open mutant of syntaxin [L165A, E166A](10) (Syx 1-261 [Open]) to bind to munc18-1 in rat brain detergent extracts. Both forms of syntaxin readily bound munc18-1 (Fig. 1C). As the crystal structure of the syntaxin 1a - munc18-1 complex (notably excluding the first 26 amino acids of syntaxin) indicates that only closed syntaxin can bind to munc18-1(21), these findings suggest that an additional mechanism and site of interaction exists. To determine whether the conserved amino acids found at the syntaxin N-terminus are involved in a direct interaction with munc18-1, we generated a series of truncations of syntaxin in both the wild type and open mutant forms. These purified proteins were incubated with a bacterial detergent extract containing recombinant His6-munc18-1 (Fig. 1D). Syx1-261 and Syx 1-261 [Open] both readily bound recombinant munc18-1 from bacterial detergent extracts replicating the observation using detergent brain extracts. The N-terminal truncations of syntaxin 1a had no affect on their ability to bind munc18-1, but importantly, the combination of N-terminal truncation and the open mutations severely diminished munc18-1 interaction. As truncation of the first six amino acids produced a decrease in binding of munc18-1 to open mutants of syntaxin 1a (Fig. 1D), similar to the longer deletions, we concentrated on this short truncation in further experiments. Circular dichroism analysis confirmed that the mutations and truncations incorporated in to syntaxin 1a resulted in no major changes to the secondary structure of the proteins (Fig. 1E). Because binding reactions, as used in Fig. 1D are strongly dependent on the relative concentrations of the interacting partners used, the affinity of munc18-1 for syntaxin, and the mutant forms, was measured. Syx1-261, Syx1-261 [open] and Syx7-261 (Δ6) all had a similar affinity for munc18-1. Only when the open mutation was combined with truncation of the N-terminus (Syx7-261 [open] (Δ6)) was there a significant reduction in the affinity of syntaxin 1a for munc18-1 (Fig. 1F).
These in vitro data suggest that syntaxin 1a can interact with munc18-1 through two distinct binding mechanisms. It is unclear, however, whether these binding modes are employed in a cellular context. To address this question, fluorescent fusions of munc18-1 and syntaxin 1a were visualised in living Neuroblastoma 2A (N2A) cells. A quantitative approach was used to investigate colocalisation of syntaxin 1a and munc18-1. In addition to the merger of the two emission channels, to display areas of coincidence, the intensities of both channels within each voxel was displayed as a frequency histogram and fit by linear regression. From this fit, residuals for each voxel were calculated and displayed as a residual map to highlight areas of covariance (expected if 2 proteins interact with a defined stoichiometry) within an image. Syntaxin 1a and munc18-1 exhibited a high degree of coincidence and of intensity covariance on the plasma membrane and in intracellular membranes (Fig. 2A). Similarly, Syx7-288 (Δ6) and Syx1-288 [open] demonstrated a high degree of colocalisation with munc18-1. Importantly, however, munc18-1 no longer colocalised with syntaxin when both the N-terminal truncation and the open syntaxin mutations were combined (Syx 7-288 (Δ6) [Open]). Munc18-1 is a soluble protein with no localisation signals, and as such its membrane association relies on its interaction with syntaxin 1a(33); when co-expressed with Syx7-288 [open] (Δ6), munc18-1 adopted a cytoplasmic localisation. As a negative control for random colocalisation, unfused munc18-1 and EYFP were co-expressed with cerulean-syntaxin 1a. Pearson’s correlation coefficient analysis demonstrated no significant difference between cerulean-Syx1-288, -Syx1-288 [open] and -Syx7-288 (Δ6) covariance with EYFP-munc18-1 (Fig. 2B). However, the combined N-terminal deletion and open mutation of syntaxin 1a (Syx7-288 [open] (Δ6)) resulted in a large decrease in covariance to a level not significantly different from the negative control.
Fig. 2.
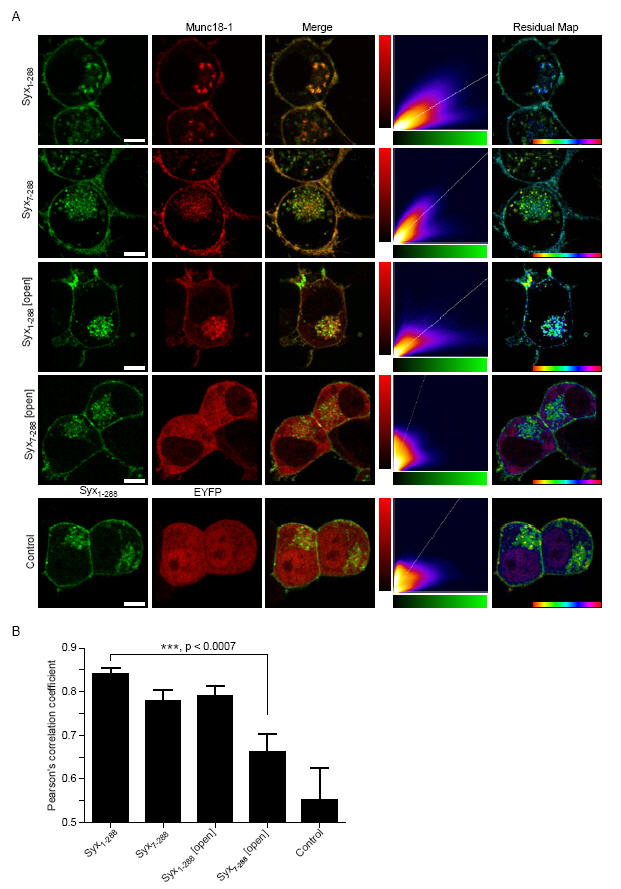
Munc18-1 colocalisation with syntaxin 1a in live cells is dependent on either closed form or N-terminal binding. A, Wild-type or mutant mCer-Syx (green), and EYFP-munc18-1 (red) were expressed in N2a cells and imaged by confocal laser scanning microscopy. The merge image shows areas of coincidence in yellow hues. The 2-D histogram represents the intensity for each channel in each voxel with a colour scale representing frequency. The residual map displays weighted residuals from the line fit to the histogram, thus indicating fluorescence channel covariance. The hue is from -1 to 1 with cyan corresponding to a zero residual. EYFP and unfused munc18-1 were used as a control. Scale bar: 2 μm. B, Combined covariance analysis of cerulean-Syx and EYFP-munc18-1 in N2a cells. Mean Pearson’s coefficients are shown (n>4).
This quantitative analysis of colocalisation between mutant proteins in living cells provided evidence for a role of the N-terminus of syntaxin 1a in binding to munc18-1. However, colocalisation data are limited by the resolution of the microscope (maximally 200 nm) and do not directly indicate interaction. In order to increase our understanding of each mode of syntaxin 1a binding to munc18-1, we employed fluorescence lifetime imaging microscopy (FLIM). FLIM quantifies the fluorescence lifetime of a fluorophore, the duration of which is sensitive to the microenvironment it inhabits(32). Thus, Förster resonance energy transfer (FRET), to an interacting acceptor molecule, shortens dramatically the donor fluorescence lifetime: this effect can be quantified directly in each pixel of an image. Furthermore, the low light levels required for FLIM allowed us to select cells expressing very low levels of exogenous proteins. Using this technique, the fraction of non-interacting and interacting donor fluorescence lifetimes in each pixel can be resolved (Fig. 3A-D)(32).
Fig. 3.
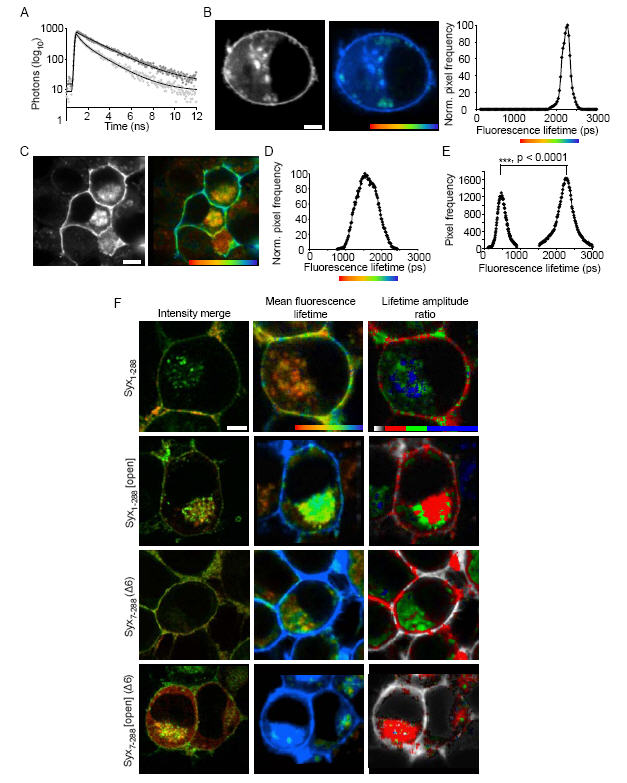
Different binding modes of munc18-1 - syntaxin 1a are spatially distinct. A, The excited state fluorescence decay of cerulean-Syx1-288 in the absence of an energy acceptor followed a mono-exponential decay, as previously described for cerulean(40) (dark circles). When co-expressed with EYFP-munc18-1, the cerulean-Syx1-288 decay data no longer fit to a single exponential, but were well described by a bi-exponential decay function (light grey circles). B, Cerulean-Syx1-288 fluorescence, in the absence of EYFP-munc18-1, exhibited a plasma membrane and intracellular membrane distribution (left panel) The colour scale in the “FLIM map” represents the fluorescence lifetime and brightness represents intensity (centre panel). The fluorescence lifetime values were plotted as a frequency distribution histogram (right panel) with a single fluorescence lifetime of 2288 ± 40 ps (mean ± S.E.M., n=18). C, The intensity localisation of cerulean-Syx1-288, in the presence of EYFP-munc18-1, was unaltered (left panel), but the FLIM map reveals a quenched fluorescence lifetime (right panel). The colours in this map represent the weighted mean of the two lifetimes for each pixel, one identical to the non-FRET lifetime (2288 ps) and one significantly shorter (680 ± 34 ps mean ± S.E.M; p<0.0001, n = 12). These data are plotted as the weighted mean (significantly different to the non-FRET distribution) (D) and as separate fluorescence lifetimes (E), representing the non-FRET and the FRET component contained in each pixel. The amplitudes of these components (which combine to 100%) represent the relative proportion of each lifetime. F, These approaches were applied to each syntaxin 1a mutant, in addition calculating the lifetime amplitude ratio of FRET:non-FRET amplitudes in each pixel. All data treatments were identical. Pixels containing only non-FRET values appear in greyscale. Amplitude ratio scale: zero (no FRET component, greyscale), 0.1-0.66 (red), 0.66-1.33 (green), 1.33-2.5 (blue). For all panels (A-F): scale bars represent 2 μm and fluorescence lifetime color scales 1250 ps (red)-2250 ps (blue).
This approach revealed that fewer Syx1-288 and munc18-1 molecules interact on the plasma membrane compared to intracellular locations (Mann-Whitney, p<0.007, n = 18), and confirmed that Syx1-288 [Open] and munc18-1 interact in cells (Fig. 3F). Surprisingly, FRET (i.e. donor fluorescence lifetime and amplitude ratios) between cerulean-Syx1-288 [Open] and EYFP-munc18-1 was substantially decreased in intracellular membranes compared to Syx1-288 and munc18-1 (p<0.001, n=8; Fig. 3F), with cell surface interactions maintained in evenly distributed puncta. In contrast, cerulean-Syx7-288 (Δ6) showed no decrease in interaction in intracellular membranes, but a large reduction in FRET on the cell surface (p<0.001, n=8). These effects were additive; cerulean-Syx7-288 [Open] (Δ6) exhibited decreased FRET both intracellular and on the cell surface (Fig. 3F). Thus, the two binding modes are spatially distinct, with syntaxin 1a N-terminal deletion affecting plasma membrane interactions, and abolition of syntaxin closed conformation binding affecting predominantly intracellular complexes. Why does deletion of the syntaxin N-terminal amino acids reduce interaction with munc18-1 at the plasma membrane? We have shown that N-terminal binding to munc18-1 is utilised when syntaxin is opened. This being the case, open syntaxin might be able to bind both munc18-1 and the other SNARE(s) simultaneously at the cell surface; loss of N-terminal interaction would thus dissociate munc18-1 in the presence of SNAP-25.
To explore this hypothesis, we used highly purified complexes of syntaxin 1a bound to munc18-1. It was thus possible to probe the effect of N-terminal truncation and the open mutation on progression of syntaxin through the SNARE assembly pathway (Fig. 4A). Incubation of Syx1-261/munc18-1 with immobilised SNAP-25 demonstrated that in this form, syntaxin is unable to engage into either the binary SNARE complex or the ternary SNARE complex (Fig. 4A left panel). The N-terminal truncation of syntaxin did not allow interaction of the Syx7-261 (Δ6)/munc18-1 complex, with no SNAP-25 binding detected (Fig. 4A centre panel). However, incorporation of the open mutation into syntaxin allowed Syx1-261[open]/munc18-1 to interact with SNAP-25 forming both the binary SNARE complex (t-SNARE heterodimer) and the ternary SNARE complex (Fig. 4A right panel). As a control munc18-1 alone was incubated with immobilised SNAP-25 (Fig. 4B). As previously shown(11), munc18-1 was unable to interact directly with SNAP-25. Importantly, munc18-1 remained bound to syntaxin 1a throughout the SNARE assembly pathway. This SNARE complex-munc18-1 interaction cannot include closed form syntaxin, and we conclude therefore that the N-terminal mode of interaction is essential at the cell surface for this reason.
Fig. 4.
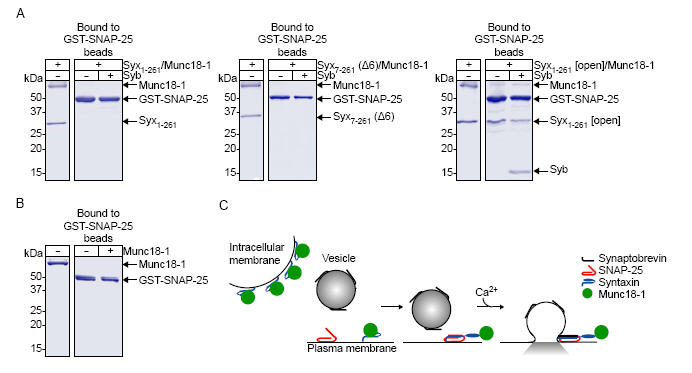
Munc18-1 can remain associated with syntaxin 1a through the SNARE assembly pathway. A, Munc18-1 in complex with Syx1-261 (left panel), Syx7-261 (Δ6) (centre panel) or Syx1-261 [open] (right panel) was incubated with immobilised SNAP-25 in the presence or absence of synaptobrevin (Syb). B, As a control munc18-1 alone was incubated with immobilised SNAP-25. C, Proposed model for munc18-1 interaction with syntaxin 1a. An undefined factor releases syntaxin from its closed state on the plasma membrane permitting binary and ternary SNARE complex formation. Munc18-1 can remain bound to the N-terminus of syntaxin 1a through this process. It was not possible to purify a Syx7-261[open](Δ6)/munc18-1 complex for use in this binding assay because of the decreased affinity of Syx7-261[open](Δ6) for munc18-1.
DISCUSSION
Our data demonstrate that munc18-1 can interact with syntaxin 1a through two discrete modes of binding which are spatially distinct within the cell. The closed form of syntaxin-munc18-1 binding is a structurally constrained state with properties comparable to those previously described for this complex(8,21). This mode of interaction is of high affinity and occurs primarily, but not exclusively, on intracellular membranes (Figs. 1 and 3). We speculate that this conformation of syntaxin 1a is clamped by munc18-1 in order to allow the passage of syntaxin through the endoplasmic reticulum and Golgi apparatus, preventing ectopic intracellular SNARE complex formation. In addition to this form of binding we now show that syntaxin 1a and munc18-1 can interact through an evolutionarily conserved motif at the N-terminus of syntaxin. It is of note that an early clone of syntaxin 1a(22) lacks the conserved N-terminal amino acids identified in Fig. 1 and has since been widely used to examine syntaxin 1a protein-protein interactions. In contrast to a previous report(10) using rat brain extracts (as in Fig. 1D), munc18-1 readily binds to a mutation which prevents syntaxin 1a adopting a closed conformation, exhibiting no reduction in affinity for this interaction (Fig. 1). We are unable to explain this discrepancy, although it should be noted that the mutants used in the NMR studies had a 26-amino-acid N-terminal truncation, as this gave better spectra(10). Importantly the full-length form of this mutant syntaxin rescues the C. elegans unc13 phenotype(34), interpreted as circumventing the requirement for Unc13 to dissociate Unc18 and open syntaxin. However, we now show that this mutation permits syntaxin to progress through the SNARE assembly pathway, all the while still associated with munc18-1 (Fig. 4).
Little or no information is available to describe the intracellular locations of SM-syntaxin interactions. A recent report found that Vps45p, an SM homologue in S. cerevisiae, can interact in vitro with its cognate syntaxin through an undefined second mode of binding in addition to its well characterised N-terminal interaction(35). Additionally, the mammalian SM protein munc18-3 binds its partner syntaxin 4 principally through an N-terminal interaction in vitro(36). However, the authors of this report were unable to exclude that a second mode of binding exists. It is therefore credible that a dual mode of interaction with cognate syntaxins is a conserved feature of the SM protein family.
Why have SM proteins evolved multiple functions and binding modes, and how do they control vesicle fusion? We propose that syntaxin 1a is transported to the plasma membrane in a closed conformation, with the Habc regulatory head domain interacting with the SNARE helix, bound by munc18-1 (Fig. 4C). This is in agreement with a previously suggested chaperone role for munc18-1(24) and would most closely resemble the binding mode observed in the crystal structure of the syntaxin1a/munc18-1 complex(21), although may also include the additional interaction of munc18-1 with the N-terminus of syntaxin 1a. This mode of binding would preclude any aberrant interaction with intracellular or trafficking plasma membrane SNAREs until delivery to the plasma membrane. At the plasma membrane the syntaxin 1a would change conformation from a closed to an open state under the regulation of an as yet undefined plasma membrane factor(30,34,37). In this form munc18-1 binding would occur through the N-terminus of syntaxin 1a through a similar interaction as observed in the Sed5p/Sly1p crystal structure(20). This conformational change of syntaxin 1a would permit the subsequent interaction of syntaxin with SNAP-25 to form the binary t-SNARE heterodimer, whilst retaining the interaction with munc18 through the N-terminal mode of binding. This complex may form a scaffold for vesicle docking, a proposed function for munc18-1(25,38) that cannot be explained adequately using current models. Subsequent triggering of exocytosis would then drive engagement of synaptobrevin with the t-SNARE heterodimer to form the four helical ternary SNARE complex formation driving membrane fusion. The association of munc18-1 with the ternary SNARE complex through the N-terminus of syntaxin 1a can explain the observed effect munc18-1 can exert on the final fusion step (26). It is reasonable to expect that the function of munc18-1 here parallels that of sec1p, which apparently stabilises the yeast exocytic SNARE complex, and is required for vesicle fusion(13). The interaction of munc18-1 with different conformational forms of syntaxin, at spatially distinct sites, can explain the multiple, and somewhat controversial roles munc18-1 has been proposed to play in the cell, and so help unify thinking on SM protein structure and function.
Footnotes
This work was funded by a Wellcome Trust Fellowship award to R.R.D.
- SNARE
- soluble N-ethylmaleimide sensitive factor attachment receptor
- SM
- Sec1p/Munc18
- t-SNARE
- target SNARE
- v-SNARE
- vesicle SNARE
- SNAP-25
- synaptosome-associated protein 25 kDa
- FLIM
- fluorescence lifetime imaging microscopy
- GST
- glutathione-S-transferase
- N2A
- Neuroblastoma 2A
REFERENCES
- 1.Burgoyne RD, Morgan A. Physiol Rev. 2003;83(2):581–632. doi: 10.1152/physrev.00031.2002. [DOI] [PubMed] [Google Scholar]
- 2.Rizo J, Sudhof TC. Nat Rev Neurosci. 2002;3(8):641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- 3.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Nature. 1998;395(6700):347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 4.Brenner S. Genetics. 1974;77(1):71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison SD, Broadie K, van de Goor J, Rubin GM. Neuron. 1994;13(3):555–566. doi: 10.1016/0896-6273(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 6.Novick P, Schekman R. Proc Natl Acad Sci U S A. 1979;76(4):1858–1862. doi: 10.1073/pnas.76.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Science. 2000;287(5454):864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 8.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH. Neuron. 1994;13(2):353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- 9.Hata Y, Slaughter CA, Sudhof TC. Nature. 1993;366(6453):347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- 10.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J. EMBO J. 1999;18(16):4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang B, Steegmaier M, Gonzalez LC, Jr., Scheller RH. J Cell Biol. 2000;148(2):247–252. doi: 10.1083/jcb.148.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zilly FE, Sorensen JB, Jahn R, Lang T. PLoS Biol. 2006;4(10) doi: 10.1371/journal.pbio.0040330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr CM, Grote E, Munson M, Hughson FM, Novick PJ. J Cell Biol. 1999;146(2):333–344. doi: 10.1083/jcb.146.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott BL, Van Komen JS, Irshad H, Liu S, Wilson KA, McNew JA. J Cell Biol. 2004;167(1):75–85. doi: 10.1083/jcb.200405018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryant NJ, James DE. EMBO J. 2001;20(13):3380–3388. doi: 10.1093/emboj/20.13.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nichols BJ, Holthuis JC, Pelham HR. Eur J Cell Biol. 1998;77(4):263–268. doi: 10.1016/s0171-9335(98)80084-8. [DOI] [PubMed] [Google Scholar]
- 17.Dulubova I, Yamaguchi T, Gao Y, Min SW, Huryeva I, Sudhof TC, Rizo J. EMBO J. 2002;21(14):3620–3631. doi: 10.1093/emboj/cdf381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabowski R, Gallwitz D. FEBS Lett. 1997;411(23):169–172. doi: 10.1016/s0014-5793(97)00720-5. [DOI] [PubMed] [Google Scholar]
- 19.Peng R, Gallwitz D. J Cell Biol. 2002;157(4):645–655. doi: 10.1083/jcb.200202006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bracher A, Weissenhorn W. EMBO J. 2002;21(22):6114–6124. doi: 10.1093/emboj/cdf608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Misura KM, Scheller RH, Weis WI. Nature. 2000;404(6776):355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- 22.Calakos N, Bennett MK, Peterson KE, Scheller RH. Science. 1994;263(5150):1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- 23.Toonen RF, Verhage M. Trends Cell Biol. 2003;13(4):177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 24.Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P. J Cell Sci. 1999;112(Pt 12):1865–1877. doi: 10.1242/jcs.112.12.1865. [DOI] [PubMed] [Google Scholar]
- 25.Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Neuron. 2001;31(4):581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- 26.Fisher RJ, Pevsner J, Burgoyne RD. Science. 2001;291(5505):875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]
- 27.Hu K, Carroll J, Fedorovich S, Rickman C, Sukhodub A, Davletov B. Nature. 2002;415(6872):646–650. doi: 10.1038/415646a. [DOI] [PubMed] [Google Scholar]
- 28.Kweon DH, Chen Y, Zhang F, Poirier M, Kim CS, Shin YK. Biochemistry. 2002;41(17):5449–5452. doi: 10.1021/bi0256476. [DOI] [PubMed] [Google Scholar]
- 29.Graham ME, Sudlow AW, Burgoyne RD. J Neurochem. 1997;69(6):2369–2377. doi: 10.1046/j.1471-4159.1997.69062369.x. [DOI] [PubMed] [Google Scholar]
- 30.Rickman C, Davletov B. Chem Biol. 2005;12(5):545–553. doi: 10.1016/j.chembiol.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Rickman C, Jimenez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B. Mol Biol Cell. 2006;17(1):283–294. doi: 10.1091/mbc.E05-07-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duncan RR, Bergmann A, Cousin MA, Apps DK, Shipston MJ. J Microsc. 2004;215(Pt 1):1–12. doi: 10.1111/j.0022-2720.2004.01343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schutz D, Zilly F, Lang T, Jahn R, Bruns D. Eur J Neurosci. 2005;21(9):2419–2432. doi: 10.1111/j.1460-9568.2005.04095.x. [DOI] [PubMed] [Google Scholar]
- 34.Richmond JE, Weimer RM, Jorgensen EM. Nature. 2001;412(6844):338–341. doi: 10.1038/35085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. J Cell Biol. 2006;173(6):927–936. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Latham CF, Lopez JA, Hu SH, Gee CL, Westbury E, Blair DH, Armishaw CJ, Alewood PF, Bryant NJ, James DE, Martin JL. Traffic. 2006;7(10):1408–1419. doi: 10.1111/j.1600-0854.2006.00474.x. [DOI] [PubMed] [Google Scholar]
- 37.Barclay JW, Craig TJ, Fisher RJ, Ciufo LF, Evans GJ, Morgan A, Burgoyne RD. J Biol Chem. 2003;278(12):10538–10545. doi: 10.1074/jbc.M211114200. [DOI] [PubMed] [Google Scholar]
- 38.Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sorensen JB, Klingauf J, Verhage M. EMBO J. 2006;25(16):3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bracher A, Perrakis A, Dresbach T, Betz H, Weissenhorn W. Structure Fold Des. 2000;8(7):685–694. doi: 10.1016/s0969-2126(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 40.Rizzo MA, Springer GH, Granada B, Piston DW. Nat Biotechnol. 2004;22(4):445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]