

Abstract
Adenovirus E1A mediates its effects on cellular transformation and transcription by interacting with critical cellular proteins involved in cell growth and differentiation. The amino terminus of E1A binds to CBP/p300 and associated histone acetyltransferases such as P/CAF. The carboxyl terminus binds to the carboxyl-terminal binding protein (CtBP), which associates with histone deacetylases. We show that 12S E1A can be acetylated by p300 and P/CAF and map one of the acetylation sites to Lys-239. This Lys residue is adjacent to the consensus CtBP binding motif, PXDLS. Mutation of Lys-239 to Gln or Ala blocks CtBP binding in vitro and disrupts the E1A–CtBP interaction in vivo. Peptide competition assays demonstrated that the interaction of E1A with CtBP is also blocked by Lys-239 acetylation. Supporting a functional role for Lys-239 in CtBP binding, mutation of this residue to Ala decreases the ability of E1A to block cAMP-regulated enhancer (CRE)-binding protein (CREB)-stimulated gene expression. Finally, we demonstrate that Lys-239 is acetylated in cells by using an antibody directed against an acetyl-Lys-239 E1A peptide. CtBP interacts with a wide variety of other transcriptional repressors through the PXDLS motif, and, in many instances, this motif is followed by a Lys residue. We suggest that acetylation of this residue by histone acetyltransferases, and the consequent disruption of repressor complexes, might be a general mechanism for gene activation.
Like other viral transforming proteins, adenovirus E1A mediates its effects by interacting with a variety of cellular factors involved in growth and differentiation (for review, see ref. 1). One class of interacting factors, the retinoblastoma proteins, mediates their transcriptional effects, at least in part, by associating with histone deacetylases (HDACs) (2, 3). Two other classes of proteins associate with domains at the opposing ends of the E1A molecule. CREB-binding protein (CBP) and its homologue p300 bind to sequences at the amino terminus of E1A (4–6). Although differences in CBP and p300 function have been reported, these proteins are generally believed to have highly overlapping activities. CBP/p300 have a central role in transcriptional regulation, bridging a wide variety of DNA-binding transcription factors to components of the general transcriptional machinery (for review, see refs. 7 and 8). Interactions with TFIIB, TBP, and the RNA polymerase II holoenzyme have been proposed to contribute to CBP/p300-mediated transcriptional activation (9–12). In addition, CBP/p300 have been shown to have intrinsic and associated histone acetyltransferase (HAT) activities (13–15). These activities have been proposed to modify the amino-terminal tails of the core histone proteins in a manner that may allow for some, as yet uncharacterized, modification of nucleosome structure. E1A has been reported to inhibit the HAT activity and therefore the transcriptional activation potential of CBP/p300 (16, 17). Although these HAT activities have been shown to be required for transcription of transfected genes in vivo and for chromatinized templates in vitro (18, 19), it is not certain, at least in mammalian cells, that histones are necessarily the most important physiological targets. Several transcription factors are known to be modified by acetylation, and, in some instances, these modifications have been shown to affect various aspects of their transcriptional functions, including nuclear localization, DNA binding, and gene activation (20–23). E1A mutants that fail to interact with CBP/p300 are generally incompetent for transformation (24).
The carboxyl terminus of E1A binds to a protein called CtBP whose properties appear to be the opposite of CBP/p300 (25). CtBP blocks transcription when fused to a heterologous DNA binding domain and thus appears to function as a transcriptional corepressor (26). Like other corepressors, CtBP has been shown to associate with HDACs, although other mechanisms of repression have also been proposed (27, 28). Although the CBP/p300 interaction motifs in E1A and other proteins remain somewhat poorly characterized, the CtBP binding motif, PXDLS, appears to be highly conserved (29). Mutation of this motif in E1A increases the level of E1A-mediated cell transformation (25, 29). Thus, the cellular CtBP and CBP/p300 proteins exert opposing effects on E1A function. Although E1A, CBP/p300, and CtBP are known to be phosphoproteins, there is no evidence that the binding of either protein to E1A is regulated by phosphorylation or by any other mechanism.
As in the case of CBP/p300, CtBP has important functions in cellular regulation aside from its interactions with E1A. These functions have perhaps been demonstrated most definitively in Drosophila, where its contributions to specific developmental processes have been analyzed using genetic techniques (30, 31). These studies have shown that CtBP is essential for “short-range repression,” a process involved in the establishment of localized stripes, bands, and tissue-specific gene expression in the syncytial Drosophila embryo (32). Examples of CtBP-binding transcriptional repressors include knirps, snail, zhf-1, and kruppel (30, 31, 33). Each of these proteins binds to CtBP through a PXDLS motif; in each instance, mutation of this binding motif abrogates repression. Similar processes appear to be used by other organisms. For example, in Xenopus, red cell development is regulated by FOG (friend of GATA), which represses transcription by binding to CtBP (34). BKLF, ZEB, Ikaros, and Net provide examples of mammalian transcriptional repressors that have similarly been shown to mediate their effects through CtBP (35–38). Thus, binding to CtBP appears to represent a common mechanism of transcriptional repression.
Whereas most of the CtBP-interacting proteins identified to date contain the PXDLS consensus motif, a different sequence, PLSLV, has been identified in Hairy, a long-range repressor that also binds to Groucho, and xTcf-3 (39, 40). Moreover, the actions of CtBP are multifaceted and context-dependent. For example, to support proper Hairy function, dCtBP has been shown to interfere with corepressor–HDAC complexes, thereby attenuating transcriptional repression (41). In addition, other proteins, including Groucho, no doubt also play an important role in mediating the functions of many repressors that interact with CtBP. In this report, we describe a regulatory role for CtBP in inhibiting adenovirus E1A-mediated transcriptional repression.
We suggest that CBP/p300 and the associated HAT, P/CAF, block transcriptional repression by disrupting E1A–CtBP complexes. We show that E1A can be acetylated by CBP/p300 as well as by P/CAF. Reminiscent of the differences in their histone acetylation properties, the acetyltransferase specificity of P/CAF for E1A was more restricted than that of CBP/p300. The predominant P/CAF site, also modified by CBP/p300, was localized to a Lys residue immediately adjacent to the consensus CtBP binding site. Remarkably, this Lys residue was frequently conserved adjacent to the CtBP binding sites of other repressor proteins, suggesting that the modification of this residue by acetylation could regulate CtBP binding in a general manner. Acetylation of this conserved Lys blocked CtBP binding in vitro, and mutation of the Lys to Gln (to mimic acetylation) or Ala both blocked CtBP binding in vitro and in vivo. Mutation of Lys-239 in E1A to Ala decreased the ability of E1A to block CREB-stimulated gene expression, supporting the idea that acetylation of E1A leads to the loss of CtBP-mediated repression. We suggest that HATs such as CBP/p300 and P/CAF may activate transcription by altering the binding site for the CtBP corepressor.
Materials and Methods
Plasmids.
Mutations of 12S E1A Lys-239 to Arg, Gln, or Ala were generated from pRcRSV-12S E1A by Quikchange (Novagen) and PCR cloned into a pGEXKG (Amersham Pharmacia) vector. pRcCMV-CtBP and pGST-E1ACter were provided by Dr. G. Chinnadurai (St. Louis University Health Sciences Center). CtBP was subcloned into pET28a (Novagen). pcDNA3-Gal4, pcDNA3-VP16, pRcRSV-cPKA (catalytic subunit of protein kinase A), and vector expressing CRE-luciferase were kindly provided by Dr. R. Maurer (Oregon Health Sciences University, Portland, OR). All of the E1A clones were subsequently cloned into BamHI and ApaI sites of pcDNA3-Gal4 vector or into HindIII and NotI sites of pRcRSV vector (Invitrogen). VP16-CtBP was constructed by cloning human CtBP into AscI and XbaI sites of pcDNA3-VP16 vector. E1A and CtBP were fused to carboxyl termini of the Gal4 DNA binding domain and VP16 activation domain, respectively. pRcRSV-CREB and pRcRSV-CBP have been described previously (10, 42). P/CAF-expressing vector was from Dr. Y. Nakatani (National Institutes of Health, Bethesda, MD).
Proteins.
Glutathione S-transferase (GST)-E1ACter and GST-E1A, both wild-type and mutated forms, were expressed in bacteria and purified by glutathione-Sepharose affinity (Sigma). His-tagged CtBP was expressed in BL21(DE3) and purified by Ni-NTA affinity (Qiagen). 12S E1A was from Dr. J. Lundblad (5). Flag-tagged p300 was expressed in baculovirus-infected SF9 cells and purified using an M2 Flag affinity matrix (Sigma). Core histone octamers were isolated from chicken blood (43). Recombinant yGcn5 and hP/CAF catalytic domains were gifts from Dr. J. Denu (Oregon Health Sciences University, Portland, OR).
Antibodies.
An acetylated E1A peptide spanning Lys-239 (PLDLSC(Ac)KRPRP) was synthesized to immunize rabbits. This antibody was used at 1:104 dilution in Western blotting. Anti-His-tag antibody was from Qiagen, and anti-E1A antibody M73 was from Santa Cruz Biotechnology. Anti-GST antibody was raised in our lab.
Histone Acetylation Assays.
Full-length p300 or catalytic domains of either yGcn5 or hP/CAF were incubated with purified chicken core histones or 12S E1A in a 30-μl reaction buffer containing 10 mM Tris-HCl, pH 8.0/10% glycerol/0.1 mM EDTA/10 mM sodium butyrate, and 3H-acetyl coenzyme (AcCoA) (Amersham Pharmacia) and incubated for 30 min at 30°C. The reactions were subjected to SDS/PAGE analysis and fluorography.
GST Pull-Down Assays.
GST, GST-E1A (both wild-type and mutations), and GST-E1ACter (acetylated and unacetylated) were coupled to glutathione-Sepharose beads (Amersham Pharmacia) and blocked with BSA. Equimolar amounts of GST or GST-fusion proteins were used in the pull-down assays. Recombinant His-tagged CtBP was added to the binding buffer HEG300 (10 mM Hepes, pH 7.4/10% glycerol/300 mM NaCl/0.5 mM EDTA/0.1% Nonidet P-40/1 mM DTT/10 μM NaF/10 μM Na3VO4) plus protease inhibitors (Complete, Boehringer Mannheim) and incubated for 1 h at 4°C. The beads were washed three times with HEG300, boiled in 15 μl of 5 × SDS loading buffer, and electrophoresed on an SDS-polyacrylamide gel. After transfer to a PVDF membrane, the bound fraction was detected by Western blotting using an anti-His-tag antibody.
Peptide Competition Assays.
Ninety-six-well plates were coated with purified CtBP (0.5 μg/well) by incubation overnight at 4°C. Wild-type (EPGQPLDLSCKRPR) and mutated (EPGQPLDLSC(Ac)KRPR, EPGQPLDLSCQRPR, EPGQPLDLSCARPR) E1A peptides, serially diluted from 1–280 μM, were mixed with GST-E1ACter (1.5 μg/ml) and added to each well. After incubation for 1 h at 37°C, the plates were washed four times with TBS containing 0.1% Tween 20. GST-E1ACter bound to CtBP on the plate was determined by ELISA using antibody against GST.
Transfection and Luciferase Assays.
COS 7 cells were maintained in DMEM plus 10% FCS; 50% confluent COS 7 cells were seeded onto 6-cm plates the day before transfection. Transfections of COS 7 cells were performed using the calcium–phosphate method (GIBCO-BRL) for luciferase assays or the Fugene reagent (Roche Molecular Biochemicals) for in vivo acetylation detection per the instructions of the manufacturer. In all experiments, total DNA transfected was kept constant with addition of pcDNA3 or pRcRSV. The maintenance and transfection of F9 cells have been described previously (10). Luciferase assays were performed as described with the modification that 1 mM Na+ pyrophosphate was added (44).
Results
E1A Is Acetylated by CBP/p300, P/CAF, and Gcn5.
Previous studies examining the relationship between E1A and the HATs, CBP/p300 and P/CAF, have focused on the ability of the adenoviral protein to inhibit various coactivator functions (5, 6). In an earlier report by Chakravarti et al. (16), it appeared that 13S E1A might serve as a substrate for these acetyltransferases. We examined this issue further using 12S E1A, which is better characterized as a transcriptional repressor.
Full-length recombinant 12S E1A was expressed in bacteria, purified, and incubated in the presence of 3H-AcCoA with baculovirus-expressed full-length mouse p300, or fragments of human P/CAF or yeast Gcn5 representing the catalytic domains. As shown in Fig. 1a, 12S E1A was acetylated by both p300 and P/CAF. The level of E1A acetylation induced by p300 was reproducibly less than that seen in the presence of P/CAF. Additionally, p300 acetylated itself somewhat more effectively than it acetylated E1A.
Figure 1.

E1A is acetylated by p300, P/CAF, and Gcn5. (a) Recombinant E1A was incubated with full-length mouse p300 expressed in baculovirus-infected SF9 cells or human P/CAF catalytic domain expressed in bacteria in the presence of 3H-AcCoA for 30 min at 30°C. The reaction was subjected to SDS/PAGE analysis and fluorography. Autoacetylation of p300 was shown at the top of the gel. (b) Comparison of E1A and histone acetylation by different HATs. Recombinant E1A or core histones purified from chicken blood were incubated with full-length mouse p300, yeast Gcn5 catalytic domain, or P/CAF catalytic domain in the presence of 3H-AcCoA for 30 min at 30°C. The reaction was subjected to SDS/PAGE analysis and fluorography.
Although it has been proposed that the acetyltransferase catalytic domains of CBP/p300 and P/CAF/Gcn5 are homologous (18, 45), recent evidence suggests that these enzymes have several distinct properties (46). To compare the relative activities of the CBP/p300 and P/CAF/Gcn5 acetyltransferases, we performed parallel assays in the presence of chicken erythrocyte core histones, which are relatively underacetylated (47). Histones were far preferred over E1A as the substrate for p300 (Fig. 1b Left). Although not apparent due to overexposure, all four of the core histone proteins were acetylated by p300, as reported previously (14). It is highly likely, however, that at least some of the difference between the level of acetylation of E1A and the core histones relates to the fact that the histone proteins are acetylated at multiple sites by p300. In contrast to the results for p300, the levels of acetylation of 12S E1A and core histones by P/CAF were essentially equivalent (Fig. 1b Middle). As expected, P/CAF targeted primarily histone H3, which is known to be acetylated at a single site (Lys-14). Similar results were obtained when the assays were performed in the presence of yeast Gcn5 (Fig. 1b Right), although multiple histone proteins were seen to be acetylated. We conclude from these experiments that 12S E1A is a substrate for the p300, P/CAF, and Gcn5 acetyltransferases.
Mapping of the E1A Acetylation Sites.
Fortuitously, the 243-amino acid 12S E1A protein contains only three Lys residues, which greatly facilitates determination of the sites of acetylation. These lysines are located at positions 162, 207, and 239. To determine which of these lysines were acetylated, we first synthesized GST fusion proteins in which Lys-239 was mutated to Arg, Gln, or Ala. The Arg residue was chosen to maintain the positive charge, Gln to mimic an acetylated Lys, and Ala to introduce an uncharged but unrelated substitution. Fusion proteins or GST alone were incubated with full-length p300 or the P/CAF catalytic domain in the presence of 3H-AcCoA and were analyzed by SDS/PAGE. Mutation of Lys-239 to Arg, Gln, or Ala decreased, but did not eliminate, acetylation of the E1A fusion proteins (Fig. 2 Left). This suggests that Lys-239 is acetylated by p300 but is not the only acetylated residue. Subsequent studies indicated that Lys-207 was acetylated by p300 as well (data not shown). GST alone was not acetylated by p300. In contrast, acetylation of E1A by P/CAF appears to occur exclusively on Lys-239. No acetylation was detected when this residue was mutated to Arg, Gln, or Ala or when GST alone was assayed (Fig. 2 Right). This high degree of specificity of P/CAF, as opposed to p300, is reminiscent of the activities of these enzymes on histones and suggests that they have fundamentally different catalytic mechanisms.
Figure 2.

p300 and P/CAF acetylate E1A at Lys-239. GST-E1A fusion proteins were generated containing Lys-239 mutated to Arg (K239R), Gln (K239Q), or Ala (K239A). Wild-type (WT) and mutated GST-E1A proteins were incubated with full-length mouse p300 (Left) or P/CAF catalytic domain (Right) in the presence of 3H-AcCoA for 30 min at 30°C. Note that the mutations at Lys-239 abrogated the acetylation by P/CAF completely but only decreased the acetylation by p300. Similar sequences around Lys-14 of histone H3 (H3) and Lys-239 of 12S E1A (E1A) are aligned, with identical residues shaded and the other residues of interest denoted by an asterisk.
Role of Lys-239 in CtBP Binding.
The Lys-239 acetylation site is located adjacent to the sequence Pro-Leu-Asp-Leu-Ser (PLDLS), which has been shown to be responsible for the binding of E1A to CtBP (29). Interaction with CtBP reduces the ability of E1A to mediate cellular transformation and enhances E1A-directed transcriptional repression (25). Of interest, a Lys residue in the same position relative to the PLDLS motif is conserved in many other transcriptional repressors that similarly interact with CtBP (Fig. 3). We asked, therefore, whether mutation of Lys-239 in E1A blocked the CtBP interaction. GST-E1A fusion proteins containing either wild-type sequences or various Lys-239 mutations were coupled to glutathione beads and incubated with bacterially expressed CtBP. As shown in Fig. 4a, binding of CtBP was reduced in the presence of the Gln mutant and almost eliminated in the presence of the Ala mutant. Mutation of the Lys to Arg slightly increased CtBP binding. No binding was seen in the presence of GST alone. To assess the effects of the Lys-239 mutations in vivo, we examined the interaction of E1A and CtBP by mammalian two-hybrid assays in COS 7 cells. For these studies, Gal4 fusion proteins containing either the carboxyl-terminal 67 amino acids of E1A (Gal4-E1ACter) or the full-length protein (Gal4-E1A) were cotransfected into COS 7 cells with a vector containing CtBP fused onto the activation domain of VP16. These assays used a 5XGal4-E1b-luciferase reporter, which is only minimally activated by the Gal4-E1A proteins. As indicated in Fig. 4b, the combination of Gal4-E1A (containing either the Cter fragment or full-length E1A) and VP16-CtBP strongly activated the reporter. This activation was prevented by mutating Lys-239 to either Gln or Ala, but not to Arg. These experiments demonstrate that Lys-239 is important for CtBP binding to E1A and raise the possibility that it is an integral component of the PLDLS binding motif found in other transcriptional repressors.
Figure 3.
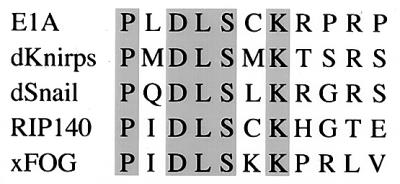
Alignment of PXDLS motifs in other transcriptional repressors. d, Drosophila; x, Xenopus. Identical residues are shaded.
Figure 4.

Effects of E1A Lys-239 mutations on E1A-CtBP binding. (a) E1A Lys-239 mutations abolish the interaction between E1A and CtBP in vitro. GST-E1A fusion proteins, both wild-type (WT) and mutated [Lys-239 mutated to Arg (K239R), Gln (K239Q), or Ala (K239A)], were coupled to glutathione-Sepharose beads, blocked with BSA, and then incubated with His-tagged human CtBP for 1 h at 4°C. Equal amounts of GST and GST-fusion proteins were used in each pull-down assay. The bound material was dissolved in sample buffer and subjected to SDS/PAGE and Western blotting using an antibody against the His-tag. (b) E1A Lys-239 mutations abolish the interaction between E1A and CtBP in vivo. Gal4 fusion proteins (0.2 μg) containing the carboxyl-terminal 67 amino acids of E1A (Gal4-E1ACter Left) or the full-length E1A (Gal4-E1A Right) wild-type (WT) and Lys-239 mutations (K239A, K239Q, K239R) were cotransfected with 0.2 μg of VP16-CtBP and 1 μg of 5XGal-E1b-luciferase as indicated, and the luciferase activity was measured. The basal luciferase activity from 5XGal-E1b-luciferase alone was subtracted from all of the samples. Data are presented as mean value ± SD (n = 3 for Gal4-E1ACter; n = 6 for Gal4-E1A). The differences between the wild-type and K239A/K239Q mutations were significant in both experiments (P < 0.01). The difference between wild-type and K239R was significant (P < 0.01) in the Gal4-E1A experiment.
Role of Lys-239 in CtBP-Mediated Repression.
To assess the functional importance of Lys-239, we examined the abilities of the E1A mutants to block CREB-stimulated gene expression. F9 teratocarcinoma cells [which lack functional protein kinase A (PKA) and CREB] were transfected with a CRE-luciferase reporter gene. In the absence of PKA, CREB and CBP are relatively ineffective in activating the reporter. The combination of CREB, CBP, and PKA stimulated CRE-reporter expression by about 80-fold (Fig. 5). Addition of a vector expressing wild-type 12S E1A reduced expression of the reporter in a dose-dependent manner, as reported previously (5). E1A has been proposed to block gene expression by interfering with the ability of CBP to interact with RNA polymerase II and P/CAF. We reasoned that other effects of E1A might be identified by examining the actions of the E1A mutants at low concentrations. At a low dose of E1A (1 μg), the K239A mutant (which does not bind CtBP) was ineffective in blocking reporter expression. In contrast, the K239R mutant (which retains the ability to bind CtBP) was highly effective at this dose. These data indicate that interaction with CtBP is essential for “low-dose” E1A repression. Interestingly, the wild-type E1A resembled K239A more than K239R. We believe this is because wild-type E1A is largely acetylated in these assays (and thus incapable of interacting with CtBP) because of the coexpression of exogenous CBP, which we showed acetylates the Lys-239 site. Higher concentrations of all E1A mutants blocked reporter expression, probably due to the known effects on CBP.
Figure 5.

Contribution of CtBP-binding to E1A-mediated transcription repression. Four micrograms of CRE-luciferase, 8 μg of pRcRSV-CREB, 10 μg of pRcRSV-CBP, 4 μg of PKA, and various amounts of E1A expression vectors as indicated were transfected into F9 cells. Equal amounts of proteins from each sample were used for luciferase assay. Data are presented as mean value ± SD (n = 3).
Acetylation of Lys-239 in CtBP Binding.
To test whether acetylation of E1A affects its interaction with CtBP, GST-fusion proteins containing the carboxyl-terminal 67 amino acids of E1A (GST-E1ACter) were treated with p300 or the catalytic domain of P/CAF in the presence or absence of AcCoA (Fig. 6a). Binding of CtBP to the acetylated proteins was reduced by about 50%, similar to the reduction seen with the Lys-239 to Gln mutation. In the context of a GST-fusion protein, the stoichiometry of E1A acetylation is relatively low (30% for P/CAF and 23% for p300, data not shown). It is likely, therefore, that the fraction of E1A that is acetylated at Lys-239 is highly deficient in its ability to bind CtBP. To determine the effects of the E1A mutations and acetylation more quantitatively, we used an ELISA (48). Briefly, this method involved mixing a GST-fusion protein containing the carboxyl-terminal 67 amino acids of E1A (GST-E1ACter) with immobilized full-length CtBP in the presence of varying concentrations of wild-type and mutated E1A peptides. Bound GST-E1ACter was measured using an anti-GST antibody. As shown in Fig. 6b, the IC50 of wild-type E1A peptide was 7.26 μM. This value is similar to that measured by Molloy et al. (48). Mutating the Lys-239 residue in the competitor peptide to Gln or Ala increased the IC50 to 11.97 μM and 21.85 μM, respectively. Peptides containing an acetylated Lys had an IC50 of 26.62 μM. Thus, in these more quantitative assays, acetylation of Lys-239 was slightly more effective than mutation of this residue to Ala in disrupting the E1A-CtBP interaction. This suggests that the K239A and K239Q mutants may underestimate the effects of Lys-239 acetylation in vivo.
Figure 6.

Effects of E1A acetylation on CtBP binding. (a) GST-E1ACter fusion proteins were treated with p300 or P/CAF in the presence or absence of 3H-AcCoA for 60 min at 37°C and coupled to glutathione-Sepharose beads. After washing, the beads were blocked with BSA and incubated with His-tagged human CtBP for 1 h at 4oC. The bound material was dissolved in sample buffer and subjected to SDS/PAGE and Western blotting using an antibody against the His-tag. The stoichiometry of GST-E1ACter acetylation is approximately 30% for P/CAF and 23% for p300. (b) Peptide competition assay. CtBP was immobilized onto 96-well plates. Serially diluted wild-type (EPGQPLDLSCKRPR), mutated (EPGQPLDLSCQRPR, EPGQPLDLSCARPR), or acetylated (EPGQPLDLSC(Ac)KRPR) E1A peptides were mixed with GST-E1ACter and added to each well. GST-E1ACter bound to CtBP on the plate was determined by an ELISA using antibody against GST. Results from wild-type and acetylated E1A peptides are plotted in the figure. Data were curve-fitted by the Michaelis–Menton equation. All curves have chi-square values less than 0.05 and R values larger than 0.96. IC50 values for each peptide are presented in the Inset.
Acetylation of E1A on Lys-239 in Vivo.
To assess whether Lys-239 is acetylated in vivo, we generated an antibody directed against an acetylated E1A peptide (Fig. 7a). Commercially available anti-acetyl-Lys antibodies were not capable of recognizing E1A that had been acetylated at this site in vitro (data not shown). The anti-acetyl-Lys-239 antibody was highly specific for acetylated E1A and did not recognize the unacetylated form even at high concentrations (Fig. 7a). Wild-type and mutated forms of E1A were transfected into COS 7 cells, along with a P/CAF expression vector. E1A proteins were immunoprecipitated from the cell lysate and analyzed by Western blotting. All of the samples, except for the vector alone, contained similar E1A protein levels. Only the wild-type E1A was detected by the anti-acetyl-Lys-239 antibody (Fig. 7b). We conclude from these experiments that Lys-239 in E1A can be acetylated in vivo.
Figure 7.

Acetylation of E1A Lys-239 in vivo. (a) Specificity of the anti-acetyl-E1A antibody. Serially diluted unacetylated and P/CAF-acetylated E1A protein was analyzed by Western blotting using the anti-acetyl-E1A antibody (Upper) or anti-E1A antibody (Lower). Arrow indicates E1A protein. (b) In vivo acetylation of E1A at Lys-239. Wild-type (WT, 0.5 μg) or Lys-239-mutated pRcRSV-E1A were transfected into COS 7 cells, along with 0.5 μg of P/CAF expression vector. E1A proteins were immunoprecipitated from the cell lysate and analyzed by Western blotting using the anti-acetyl-E1A antibody (Upper) or anti-E1A antibody (Lower). All of the samples, except for the vector alone, contained similar levels of E1A protein. Only extracts from the cells expressing the wild-type E1A stained positively with the anti-acetyl-Lys-239 antibody. Arrow indicates E1A protein.
Discussion
The correlation of histone acetylation and deacetylation with gene activation and repression has become a central tenet in the field of gene regulation. Nonetheless, it is still not entirely clear whether acetylation of histones modulates nucleosomal structure or how these presumed structural changes in the nucleosome might alter chromatin function. The recognition that many transcriptional activators are directly regulated by acetylation led us to search for repressor complexes that could be disrupted by activities of the prototypical HATs, such as CBP/p300 and P/CAF. We focused on adenovirus E1A because it is known to function as a transcriptional repressor and it associates with both histone acetyltransferases and deacetylases.
The early region of adenovirus gives rise to two proteins, designated 12S and 13S E1A. The amino-terminal domain of E1A interacts with CBP/p300 and is believed to mediate cell immortalization (1, 4). The importance of these amino-terminal interactions is apparent from the numerous studies in tissue culture showing that E1A-mediated transformation is blocked by overexpression of CBP/p300 (49). Additional CBP/p300 associated factors, such as P/CAF, may contribute to these effects as well. For example, overexpression of P/CAF suppresses the cell-cycle progressive effects of E1A (15). The carboxyl terminus of E1A contains the CtBP interaction motif. The association of CtBP with E1A negatively regulates transformation, as deletion of the PLDLS sequence near the E1A carboxyl terminus increases the number of transformants in cells transfected with E1A and ras (25, 29). This effect has been attributed to the ability of CtBP to interact with HDACs, although other mechanisms have been proposed as well (27, 50).
Previous studies of E1A have focused on its ability to inhibit CBP/p300 function. Precisely how this occurs is not clear. E1A binds to the same region of CBP/p300 that interacts with TFIIB and components of the RNA polymerase II holoenzyme (5, 6). Thus, E1A may function in part by preventing coactivators from interacting with proteins in the preinitiation complex. E1A has also been reported to block the association of CBP/p300 with the HAT, P/CAF, presumably by competing for binding to the same region (15). These models were supported by the recent studies of Kraus et al., which indicated that a GST-E1A fusion protein containing only the amino-terminal sequences of E1A blocked p300-directed transcription in the context of reconstituted chromatin templates (19). Additionally, E1A has been reported to block the intrinsic acetyltransferase activities of CBP/p300 and P/CAF directly (16, 17).
Although not addressed explicitly, acetylation of 13S E1A by CBP/p300 and P/CAF was evident in the data of Chakravarti et al. (16). Our studies show that 12S E1A can be acetylated by these enzymes as well. In addition to the apparently greater activity of P/CAF, as compared with CBP/p300, P/CAF appears to be more specific, in that it recognizes only a single Lys of 12S E1A. The increased specificity of P/CAF over CBP/p300 recapitulates the properties of these enzymes in acetylating histones. Whereas CBP/p300 acetylates E1A more weakly than it does histones, the ability of P/CAF to acetylate these two substrates was approximately equal.
Given the specificity of P/CAF for a single Lys residue in histone H3 and E1A, it is interesting to consider whether these two substrates might be related. The catalytic domain of P/CAF is similar to that of Gcn5, whose structure has been solved (51). Crystallographic analysis of the Tetrahymena Gcn5/CoA/histone H3 complex indicated that sequences carboxyl-terminal to the target Lys are particularly critical for enzyme recognition (52). In addition to the target Lys at position 14 in the histone substrate, a Pro residue at position 16 was also thought to be important for binding specificity (52). The KXPR motif in histone H3 is also present in E1A, which may explain their similar sensitivity to the P/CAF acetyltransferase. Although residues amino-terminal to Lys-14 in histone H3 were not proposed to contribute importantly to histone H3 recognition by Gcn5, recent studies have shown that phosphorylation of Ser-10 increases the affinity of histone H3 peptides for Gcn5 by 10-fold (53). E1A contains an Asp at this site (Fig. 2), a large negatively charged residue that could mimic the phosphorylated Ser.
CtBP appears to mediate the actions of many transcriptional repressors, possibly through its ability to associate with HDACs (27). Most interactions involving CtBP occur through the binding motif, PXDLS. Recent studies, however, have suggested that the binding site in E1A for CtBP extends over a slightly larger sequence than originally suggested (48). In many instances, this motif is followed at the carboxyl-terminal end by a Lys residue, but the function of this Lys has never before been addressed. In the case of E1A, acetylation of the Lys by P/CAF or CBP/p300 disrupts the CtBP–E1A interaction, potentially leading to the relief of CtBP-mediated repression. Evidence for this model was obtained by examining the repressive effect of E1A on CREB-mediated transcription. We and others have previously shown that 12S E1A blocks CREB-stimulated gene expression in a dose-dependent manner. This effect has been attributed to the ability of E1A to inhibit CBP/p300 function. We reasoned that the role of CtBP in E1A function might be identified by examining the actions of the E1A mutants at different levels of expression. At low concentrations of E1A, the K239A mutant (which is defective in CtBP binding) was ineffective in blocking reporter expression. In contrast, the K239R mutant (which retains the ability to bind CtBP) was highly effective at this dose. We predict that the same mode of regulation might pertain to other repressor–CtBP complexes. This additional level of control could be important for the regulation of developmental pathways in Drosophila, where the function of CtBP is probably best understood. This model would predict that the generation of repressor complexes responsible for the spatially localized patterns of gene expression might be regulated not only by the gradient of specific transcription factors but also by the active disruption of repressor complexes by transcriptional activators and their associated HATs. Evidence for this model will require additional experimentation, but preliminary studies on another CtBP interacting protein, the ligand-dependent nuclear hormone receptor repressor RIP 140, suggest that this mechanism is a general one (N.V. and R.H.G., unpublished observations). It is likely that the overall biological effects of most repressors are not due to CtBP alone but rather depend on the combinatorial affects of multiple interacting proteins.
By providing an additional example of how HATs can activate gene expression in a histone-independent manner, our studies argue against the idea that the critical competition between acetylases and deacetylases occurs exclusively at the level of the nucleosome. On the other hand, in a larger context, the models are not so different. In both cases, the target of the acetyltransferase activity is a repressor protein whose function is blocked by the acetylation of specific Lys residues. In all likelihood, both mechanisms are likely to contribute to gene regulation.
Acknowledgments
We thank G. Chinnadurai and J. E. Gambee for reagents, K. Tanner and J. Denu for reagents and helpful suggestions, J. Notis for help with many of the experiments, and G. Mandel for comments on the manuscript. This work was supported by grants from the National Institutes of Health and the McKnight Foundation.
Abbreviations
- CtBP
carboxyl-terminal binding protein
- GST
glutathione S-transferase
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- PKA
protein kinase A
- CRE
cAMP-regulated enhancer
- CREB
CRE-binding protein
- CBP
CREB-binding protein
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.011283598.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.011283598
References
- 1.Moran E. Curr Opin Genet Dev. 1993;3:63–70. doi: 10.1016/s0959-437x(05)80342-9. [DOI] [PubMed] [Google Scholar]
- 2.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain J P, Troalen F, Trouche D, Harel-Bellan A. Nature (London) 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 3.Martinez-Balbas M A, Bauer U M, Nielsen S J, Brehm A, Kouzarides T. EMBO J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stein R W, Corrigan M, Yaciuk P, Whelan J, Moran E. J Virol. 1990;64:4421–4427. doi: 10.1128/jvi.64.9.4421-4427.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundblad J R, Kwok R P, Laurance M E, Harter M L, Goodman R H. Nature (London) 1995;374:85–88. doi: 10.1038/374085a0. [DOI] [PubMed] [Google Scholar]
- 6.Eckner R, Ewen M E, Newsome D, Gerdes M, DeCaprio J A, Lawrence J B, Livingston D M. Genes Dev. 1994;8:869–884. doi: 10.1101/gad.8.8.869. [DOI] [PubMed] [Google Scholar]
- 7.Shikama M, Lyon J, Thangue N B. Trends Cell Biol. 1997;7:230–236. [Google Scholar]
- 8.Goodman R H, Smolik S. Genes Dev. 2000;14:1553–1577. [PubMed] [Google Scholar]
- 9.Felzien L K, Farrell S, Betts J C, Mosavin R, Nabel G J. Mol Cell Biol. 1999;19:4241–4246. doi: 10.1128/mcb.19.6.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwok R P, Lundblad J R, Chrivia J C, Richards J P, Bachinger H P, Brennan R G, Roberts S G, Green M R, Goodman R H. Nature (London) 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 11.Nakajima T, Uchida C, Anderson S F, Lee C G, Hurwitz J, Parvin J D, Montminy M. Cell. 1997;90:1107–1112. doi: 10.1016/s0092-8674(00)80376-1. [DOI] [PubMed] [Google Scholar]
- 12.Swope D L, Mueller C L, Chrivia J C. J Biol Chem. 1996;271:28138–28145. doi: 10.1074/jbc.271.45.28138. [DOI] [PubMed] [Google Scholar]
- 13.Bannister A J, Kouzarides T. Nature (London) 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 14.Ogryzko V V, Schiltz R L, Russanova V, Howard B H, Nakatani Y. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 15.Yang X J, Ogryzko V V, Nishikawa J, Howard B H, Nakatani Y. Nature (London) 1996;382:319–324. doi: 10.1038/382319a0. [DOI] [PubMed] [Google Scholar]
- 16.Chakravarti D, Ogryzko V, Kao H Y, Nash A, Chen H, Nakatani Y, Evans R M. Cell. 1999;96:393–403. doi: 10.1016/s0092-8674(00)80552-8. [DOI] [PubMed] [Google Scholar]
- 17.Hamamori Y, Sartorelli V, Ogryzko V, Puri P L, Wu H Y, Wang J Y, Nakatani Y, Kedes L. Cell. 1999;96:405–413. doi: 10.1016/s0092-8674(00)80553-x. [DOI] [PubMed] [Google Scholar]
- 18.Martinez-Balbas M A, Bannister A J, Martin K, Haus-Seuffert P, Meisterernst M, Kouzarides T. EMBO J. 1998;17:2886–2893. doi: 10.1093/emboj/17.10.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraus W L, Manning E T, Kadonaga J T. Mol Cell Biol. 1999;19:8123–8135. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soutoglou E, Katrakili N, Talianidis I. Mol Cell. 2000;5:745–751. doi: 10.1016/s1097-2765(00)80253-1. [DOI] [PubMed] [Google Scholar]
- 21.Gu W, Roeder R G. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Bieker J J. Proc Natl Acad Sci USA. 1998;95:9855–9860. doi: 10.1073/pnas.95.17.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyes J, Byfield P, Nakatani Y, Ogryzko V. Nature (London) 1998;396:594–598. doi: 10.1038/25166. [DOI] [PubMed] [Google Scholar]
- 24.Dyson N, Harlow E. Cancer Surv. 1992;12:161–195. [PubMed] [Google Scholar]
- 25.Boyd J M, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. EMBO J. 1993;12:469–478. doi: 10.1002/j.1460-2075.1993.tb05679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sollerbrant K, Chinnadurai G, Svensson C. Nucleic Acids Res. 1996;24:2578–2584. doi: 10.1093/nar/24.13.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundqvist A, Sollerbrant K, Svensson C. FEBS Lett. 1998;429:183–188. doi: 10.1016/s0014-5793(98)00588-2. [DOI] [PubMed] [Google Scholar]
- 28.Sewalt R G, Gunster M J, van der Vlag J, Satijn D P, Otte A P. Mol Cell Biol. 1999;19:777–787. doi: 10.1128/mcb.19.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaeper U, Boyd J M, Verma S, Uhlmann E, Subramanian T, Chinnadurai G. Proc Natl Acad Sci USA. 1995;92:10467–10471. doi: 10.1073/pnas.92.23.10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nibu Y, Zhang H, Levine M. Science. 1998;280:101–104. doi: 10.1126/science.280.5360.101. [DOI] [PubMed] [Google Scholar]
- 31.Nibu Y, Zhang H, Bajor E, Barolo S, Small S, Levine M. EMBO J. 1998;17:7009–7020. doi: 10.1093/emboj/17.23.7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mannervik M, Nibu Y, Zhang H, Levine M. Science. 1999;284:606–609. doi: 10.1126/science.284.5414.606. [DOI] [PubMed] [Google Scholar]
- 33.Postigo A A, Ward E, Skeath J B, Dean D C. Mol Cell Biol. 1999;19:7255–7263. doi: 10.1128/mcb.19.10.7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deconinck A E, Mead P E, Tevosian S G, Crispino J D, Katz S G, Zon L I, Orkin S H. Development. 2000;127:2031–2040. doi: 10.1242/dev.127.10.2031. [DOI] [PubMed] [Google Scholar]
- 35.Criqui-Filipe P, Ducret C, Maira S M, Wasylyk B. EMBO J. 1999;18:3392–3403. doi: 10.1093/emboj/18.12.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koipally J, Georgopoulos K. J Biol Chem. 2000;275:19594–19602. doi: 10.1074/jbc.M000254200. [DOI] [PubMed] [Google Scholar]
- 37.Postigo A A, Dean D C. Proc Natl Acad Sci USA. 1999;96:6683–6688. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner J, Crossley M. EMBO J. 1998;17:5129–5140. doi: 10.1093/emboj/17.17.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poortinga G, Watanabe M, Parkhurst S M. EMBO J. 1998;17:2067–2078. doi: 10.1093/emboj/17.7.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brannon M, Brown J D, Bates R, Kimelman D, Moon R T. Development. 1999;126:3159–3170. doi: 10.1242/dev.126.14.3159. [DOI] [PubMed] [Google Scholar]
- 41.Phippen, T. M., Sweigart, A. L., Moniwa, M., Krumm, A., Davie, J. R. & Parkhurst, S. M. (2000) J. Biol. Chem., in press. [DOI] [PubMed]
- 42.Loriaux M M, Rehfuss R P, Brennan R G, Goodman R H. Proc Natl Acad Sci USA. 1993;90:9046–9050. doi: 10.1073/pnas.90.19.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng H P, Scherl D S, Widom J. Biochemistry. 1993;32:7824–7831. doi: 10.1021/bi00081a030. [DOI] [PubMed] [Google Scholar]
- 44.Impey S, Obrietan K, Wong S T, Poser S, Yano S, Wayman G, Deloulme J C, Chan G, Storm D R. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 45.Dutnall R N, Tafrov S T, Sternglanz R, Ramakrishnan V. Cell. 1998;94:427–438. doi: 10.1016/s0092-8674(00)81584-6. [DOI] [PubMed] [Google Scholar]
- 46.Lau O D, Kundu T K, Soccio R E, Ait-Si-Ali S, Khalil E M, Vassilev A, Wolffe A P, Nakatani Y, Roeder R G, Cole P A. Mol Cell. 2000;5:589–595. doi: 10.1016/s1097-2765(00)80452-9. [DOI] [PubMed] [Google Scholar]
- 47.Brotherton T W, Covault J, Shires A, Chalkley R. Nucleic Acids Res. 1981;9:5061–5073. doi: 10.1093/nar/9.19.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molloy D P, Milner A E, Yakub I K, Chinnadurai G, Gallimore P H, Grand R J. J Biol Chem. 1998;273:20867–20876. doi: 10.1074/jbc.273.33.20867. [DOI] [PubMed] [Google Scholar]
- 49.Smits P H, de Wit L, van der Eb A J, Zantema A. Oncogene. 1996;12:1529–1535. [PubMed] [Google Scholar]
- 50.Sewalt R G, Gunster M J, van der Vlag J, Satijn D P, Otte A P. Mol Cell Biol. 1999;19:777–787. doi: 10.1128/mcb.19.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trievel R C, Rojas J R, Sterner D E, Venkataramani R N, Wang L, Zhou J, Allis C D, Berger S L, Marmorstein R. Proc Natl Acad Sci USA. 1999;96:8931–8936. doi: 10.1073/pnas.96.16.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rojas J R, Trievel R C, Zhou J, Mo Y, Li X, Berger S L, Allis C D, Marmorstein R. Nature (London) 1999;401:93–98. doi: 10.1038/43487. [DOI] [PubMed] [Google Scholar]
- 53.Cheung P, Tanner K G, Cheung W L, Sassone-Corsi P, Denu J M, Allis C D. Mol Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]