

Abstract
Airway inflammation is orchestrated by cell-cell interactions involving soluble mediators and cell adhesion molecules. Alterations in the coordination of the multicellular process of inflammation may play a major role in the chronic lung disease state of cystic fibrosis (CF). The aim of this study was to determine whether direct cell-cell interactions via gap junctional communication is affected during the inflammatory response of the airway epithelium. We have examined the strength of intercellular communication and the activation of nuclear factor-κB (NF-κB) in normal (non-CF) and CF human airway cell lines stimulated with tumor necrosis factor-α (TNF-α). TNF-α induced maximal translocation of NF-κB into the nucleus of non-CF as well as CF airway cells within 20 minutes. In non-CF cells, TNF-α progressively decreased the extent of intercellular communication. In contrast, gap junctional communication between CF cells exposed to TNF-α remained unaltered. CF results from mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Interestingly, transfer of wild-type CFTR into CF cells by adenovirus-mediated infection was associated with the recovery of TNF-α-induced uncoupling. These results suggest that expression of functional CFTR is necessary for regulation of gap junctional communication by TNF-α. Gap junction channels close during the inflammatory response, therefore limiting the intercellular diffusion of signaling molecules, and thereby the recruitment of neighboring cells. Defects in this mechanism may contribute to the excessive inflammatory response of CF airway epithelium.
Chronic airway inflammation is a hallmark of cystic fibrosis (CF). CF is an autosomal recessive disease resulting from mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. 1 CFTR acts as a cAMP-activated Cl− channel as well as a regulator of the activity of other ion channels and transporters in epithelial cells. 2 Despite considerable progress in understanding the structure-function relationship of CFTR, the link between the absence or dysfunctions of CFTR and the early chronic inflammation of airways from CF patients remains unclear.
Airway inflammation is the result of a network of events involving complex cell-cell interactions via paracrine factors and cell adhesion molecules. 3-5 In CF, the release of a variety of cytokines and chemokines by lung epithelial cells and macrophages, such as tumor necrosis factor-α (TNF-α) and interleukin-8 (IL-8), along with an excessive neutrophil influx into the airways lead to a progressively destructive inflammatory reaction. Lung injury and progressive loss of pulmonary function follow the release of cytotoxic neutrophil products into the airways. 6 The reason for persistent neutrophil infiltrates into the airways is uncertain. Some authors have proposed that chronic inflammation is maintained by increased adherence, 7 decreased clearance, 8 or decreased killing 9 of CF-specific pathogens. Other studies suggest that the early inflammation in the CF lung is associated with abnormalities in the production of pro-inflammatory cytokines, even in the absence of infectious stimuli. 5,10-12 Altogether, these data suggest that a primary dysregulation in the coordination of the multicellular process of inflammation occurs in CF airways.
Specialized cell junctions are particularly important in the function of epithelia. Direct cell-cell interactions via gap junctional communication provide a low resistance pathway to coordinate multicellular activity by mediating the intercellular diffusion of ions, second messengers, and small metabolites. Gap junctional communication is thought to contribute to the maintenance of cell differentiation and homeostasis. 13-15 Conversely, abnormal gap junctional communication has been associated with a number of pathologies, 16,17 including CF. 18 However, little is known about the role of gap junctional communication in the inflammatory process. Some studies have documented alteration in gap junction protein connexin (Cx) expression and intercellular communication by pro-inflammatory mediators in endothelial, 19,20 hepatic, 21,22 cardiac, 23 and Schwann cells. 24 Whether gap junctional communication is affected in human airway cells during the inflammatory process remains to be established.
To address this question, we have studied the effects of TNF-α on the inflammatory response and gap junctional communication of normal (non-CF) and CF human airway cell lines. We show that TNF-α concurrently induced the activation of the nuclear factor-κB (NF-κB), a transcriptional activator of immunomodulatory genes, and closure of gap junction channels in non-CF cells. Although TNF-α also elicited inflammatory responses in CF cells, gap junctional communication was not affected. This effect was related to the absence of a functional CFTR, because adenovirus-mediated transfer of wild-type CFTR into CF cells was associated with the recovery of TNF-α-induced uncoupling.
Materials and Methods
Cell Culture
The human bronchial Beas2B cell line was purchased from the American Type Culture Collection (Rockville, MD); the human nasal CF15 cell line, which was derived from a patient homozygous for the ΔF508 mutation of CFTR, was previously characterized by Jefferson and colleagues 25 ; the human bronchial IB3-1 cell line, 26 which was derived from a patient with CF (ΔF508/W1282X), was kindly provided by Dr. P. L. Zeitlin (Johns Hopkins University School of Medicine, Baltimore, MD). Beas2B cells were maintained in Dulbecco’s modified Eagle’s medium; CF15 cells were cultured on surfaces coated with 50 μg/ml of human placental collagen IV (Sigma Chemical Co., St. Louis, MO) and maintained in 3:1 (vol/vol) Dulbecco’s modified Eagle’s medium/F12 supplemented with growth factors; 25 IB3-1 cells were cultured on surfaces coated with collagen IV and 10 μg/ml bovine plasma fibronectin (Life Technologies AG, Basel, Switzerland), and maintained in bronchial epithelial cell growth medium (Promocell, Heidelberg, Germany). All media were supplemented with 10% fetal calf serum (SeraTech, Griesbach, Switzerland), 30 U/ml of penicillin, and 30 μg/ml of streptomycin (GibcoBRL, Basel, Switzerland).
IL-8 Production
Subconfluent monolayers of non-CF and CF cells were rinsed three times with phosphate-buffered saline (PBS), and incubated in their respective culture medium without fetal calf serum and with 0.4% bovine serum albumin for 30 minutes. Cells were then refreshed with 500 μl of media containing 100 U/ml of TNF-α (Bachem AG, Bubendorf, Switzerland) and incubated for 2 hours. After this period, the supernatant was removed and stored at −20°C before further analysis. Cells were lysed with 500 μl of distilled water and total protein content was determined by a Bio-Rad protein assay (Biorad Laboratories GmBH, München, Germany). IL-8 was measured in supernatants using an enzyme-linked immunosorbent assay kit (CLB, Amsterdam, The Netherlands). Only assays having standard curves with a calculated regression line value >0.95 were accepted for analysis. IL-8 production was normalized to total protein content. Data are expressed as mean ± SEM and compared using unpaired t-tests.
NF-κB Activity
NF-κB translocation was determined by electrophoretic mobility-shift assays (EMSA) and IκBα degradation by Western blot analysis, as previously described. 27 Subconfluent monolayers of cells, which had been treated with 100 U/ml TNF-α or control medium for the appropriate time, were washed with ice-cold PBS, harvested by scraping into 1 ml of PBS, and pelleted in a 1.5-ml microfuge tube at 6000 rpm for 5 minutes. The pellet was washed twice in ice-cold PBS, pelleted, and then suspended in a lysis buffer containing 10 mmol/L Hepes (pH 7.9), 10 mmol/L KCl, 0.1 mmol/L ethylenediaminetetraacetic acid (EDTA), 1.5 mmol/L MgCl2, 0.25% Nonidet P-40, 1 mmol/L dithiothreitol, and 0.1 mmol/L phenylmethyl sulfonyl fluoride (PMSF). After a 5-minute incubation on ice, the nuclear pellet was isolated by centrifugation. The supernatant, which represents the cytoplasmic extract, was ulteriorly used for Western blot analysis of IκBα. The nuclear pellet was then resuspended in an ice-cold solution containing 20 mmol/L Hepes (pH 7.9), 420 mmol/L NaCl, 0.1 mmol/L EDTA, 1.5 mmol/L MgCl2, 25% (vol:vol) glycerol, 1 mmol/L dithiothreitol, and 0.5 mmol/L PMSF for 5 minutes. The debris was removed by centrifugation and nuclear extracts were stored at −70°C before use.
For EMSA, equivalent quantities of nuclear protein were incubated on ice for 10 minutes in a buffer containing 12 mmol/L Hepes (pH 7.9), 4 mmol/L Tris-HCl (pH 7.9), 25 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 50 ng/ml poly[d(I-C)], and 0.2 mmol/L PMSF. NF-κB probes for EMSA were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The probes were end-labeled with 32P and added to the reaction mixture for 10 minutes on ice. Bound and free probes were resolved through nondenaturing polyacrylamide gel electrophoresis. To test for specificity in NF-κB binding activity, competition assay was performed by adding an excess of cold NF-κB and AP-1 probe (100×). For Western blot analysis of IκBα degradation, 50 μg of cytoplasmic protein were boiled for 3 minutes. The samples were then subjected to electrophoresis on a 10% Tris-glycine gel (Novex, San Diego, CA) at 140 V for 1.5 hours. The protein was transferred to nitrocellulose membranes and nonspecific binding sites were blocked with 5% milk in Tris-buffered saline supplemented with 0.05% Tween 20. The membranes were then probed with an antibody raised against IκBα (c21, Santa Cruz Biotechnology). After washing the membranes in Tris-buffered saline supplemented with 0.05% Tween 20, the membranes were probed with a goat anti-rabbit antibody conjugated to horseradish peroxidase (Calbiochem, La Jolla, CA). Immunoreactivity was detected using an ECL chemiluminescent detection kit, according to the manufacturer’s instructions (Amersham, Arlington Heights, IL).
Cx Expression
For reverse transcriptase-polymerase chain reaction, cellular mRNA was isolated from subconfluent cells using oligo-dT columns (Pharmacia Biotech, Dübendorf, Switzerland), according to the manufacturer’s instructions. Reverse transcription was performed using random hexamers and the resulting cDNAs were amplified by polymerase chain reaction using primer pairs specific for human Cx43: sense 5′-GCAACATGGGTGACTGGAGCG and antisense 5′-GCCAGGTACAAGAGTGTGGGT (predicted size, 285 bp). After a 5-minute incubation at 94°C, amplification of cDNA was performed for 35 cycles, each comprising 1 minute at 94°C, 1 minute at 55°C, and 1 minute at 72°C, using an UNOII polymerase chain reaction cycler (Biometra GmBH, Göttingen, Germany). After the last cycle, an elongation step of 5 minutes at 72°C was performed. Amplified DNA fragments were separated in a 1.5% agarose gel and visualized by exposure to UV after ethidium bromide staining. No products were amplified in the absence of reverse transcriptase (not shown).
For Western blots, subconfluent monolayers of cells were rinsed with PBS and scraped into an ice-cold solubilization buffer containing 50 mmol/L Tris-HCl (pH 7.4), 150 mmol/L NaCl, 1% Nonidet P-40, 1 mmol/L PMSF, and a cocktail of protease inhibitors (Boehringer Mannheim, Mannheim, Germany). After a 30-minute incubation, the samples were centrifuged at 4°C for 10 minutes at 50,000 × g. Supernatants were recovered and total amounts of protein were determined by a bicinchoninic acid quantification assay (Sigma Chemical Co.). Fifteen μg of protein were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and electrotransferred onto Immobilon-P polyvinylidene difluoride membranes (Millipore AG, Volketswill, Switzerland). Membranes were then soaked overnight at 4°C in a 2% defatted milk saturation buffer containing 10 mmol/L Tris-HCl (pH 7.4), 2 mmol/L EDTA, 133 mmol/L NaCl, 0.05% Triton X-100, and 0.2% sodium azide. Blotted proteins were then incubated for 1 hour at room temperature with mouse Cx43 (1:500 dilution) antibodies (Chemicon International Inc., Temecula, CA). This step was followed by a 1-hour incubation with goat anti-mouse secondary antibodies conjugated to peroxidase (Jackson Laboratories, West Grove, PA). Immunoreactivity was detected through the Super Signal West Pico kit (Pierce, Rockford, IL).
Dye Coupling
Subconfluent monolayers of cells were treated with 100 U/ml TNF-α or control medium for the appropriate time at 37°C. For dye coupling studies, the medium was changed to a solution containing 136 mmol/L NaCl, 4 mmol/L KCl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 2.5 mmol/L glucose, 10 mmol/L Hepes (pH 7.4), supplemented with or without TNF-α. Single cells were impaled with microelectrodes backfilled with a 4% lucifer yellow solution prepared in 150 mmol/L LiCl (buffered to pH 7.2 with 10 mmol/L Hepes). The fluorescent tracer was allowed to fill the cells by simple diffusion for 3 minutes. After the injection period, the electrode was removed and the number of fluorescent cells was counted. Cells were visualized using epifluorescent illumination provided by a 100 W mercury lamp and the appropriate filters. To test for the specificity of the dye coupling experiments, two gap junction blockers were also used. Heptanol (1.5 mmol/L) or 18-α-glycyrrethinic acid (10 μmol/L) inhibited dye coupling within minutes so that data obtained with both gap junction blockers were pooled. Results are expressed as mean ± SEM and compared using unpaired t-tests.
Recombinant Adenovirus and Viral Infection
The replication-defective adenovirus were derived from the human adenovirus serotype 5, and contained either the CFTR cDNA controlled by the RSV promoter (AdTG6429) or the CMV promoter (AdTG6418), the CMV promoter-driven eGFP (enhanced green fluorescent protein) gene (AdTG6297), or the RSV promoter-driven lacZ gene. All vectors, in which E1 and E3 were deleted, were constructed as infectious plasmids by homologous recombination in Escherichia coli, as previously described. 28 The transgenes are incorporated in place of the viral E1 gene. Viral stocks were stored at −80°C in 1 mol/L sucrose, 10 mmol/L Tris-HCl (pH 8.5), 1 mmol/L MgCl2, 150 mmol/L NaCl, and 0.005% Tween 80.
Subconfluent monolayers of CF15 cells were infected for 16 hours with adenovirus at a multiplicity of infection (MOI) of 25 to 500 (for RSV-based viruses) and of 1 to 200 (for CMV-based viruses), where a MOI of 1 represents 1 infectious unit/cell (∼1/2 plaque-forming unit/cells). The cells were then rinsed and cultured for an additional 24 hours in normal medium before the experiment, as previously described. 29 To visualize the expression of β-galactosidase, infected cells were rinsed with PBS, fixed with 0.5% glutaraldehyde for 10 minutes, and incubated for 6 hours at 37°C in PBS supplemented with 1 mg/ml X-Gal, 5 mmol/L K+ ferricyanide, 5 mmol/L K+ ferrocyanide, and 1 mmol/L MgCl2. β-galactosidase activity was detected by light microscopy as nuclear-localized blue staining.
Results
NF-κB Activation in Non-CF and CF Airway Cells
The production of IL-8 after NF-κB translocation to the nucleus is an indication of the inflammatory response. We first established a dose-response curve for TNF-α stimulation of IL-8 production. For all airway cell lines, maximal release of IL-8 after a 2 hour-exposure period was observed for 80 to 120 U/ml TNF-α. Therefore, a dose of 100 U/ml was used throughout the study. Basal and stimulated values of IL-8 production between non-CF and CF cells are shown in Table 1 ▶ .
Table 1.
Production of IL-8 (ng/μg of Total Protein) by Non-CF and CF Airway Epithelial Cells
| Airway cell line | Basal* | TNF-α | n† | P‡ |
|---|---|---|---|---|
| Beas2B | 0.35 ± 0.09 | 13.20 ± 3.3 | 6 | <0.003 |
| CF15 | 0.50 ± 0.06 | 1.30 ± 0.3 | 6 | <0.04 |
| IB3-1 | 0.03 ± 0.015 | 1.90 ± 0.4 | 3 | <0.01 |
*Values are expressed as mean ± SEM.
†n is the number of experiments.
‡P is the degree of significance. The production of IL-8 in response to 100 U/ml TNF-α is enhanced 38-fold in Beas2B cells, 2.6-fold in CF15 cells, and 63-fold in IB3-1 cells.
To investigate the TNF-α-induced inflammatory response in non-CF and CF cells, the activation of NF-κB was measured by EMSA and Western blots. Nuclear and cytoplasmic extracts were prepared from subconfluent monolayers of cells exposed to TNF-α for increasing amounts of time. As shown in Figure 1A ▶ , EMSA analysis on non-CF Beas2B cells treated with TNF-α revealed an increase in NF-κB binding activity in the nuclear extracts, reaching a steady-state after 20 minutes (representative example of three experiments). Nonspecific binding of the NF-κB probe can also be seen. Cold competition assays confirmed that the band detected in Figure 1A ▶ is specific for NF-κB (data not shown). In parallel experiments, Western blots analysis of cytoplasmic extracts showed that addition of TNF-α to the non-CF cells induced a rapid loss of IκBα (Figure 1B) ▶ . IκBα degradation followed a time course that was similar to that detected for the translocation of NF-κB to the nucleus. Similar results were obtained for both CF (CF15 and IB3-1) airway cell lines (Figure 2) ▶ . TNF-α-induced NF-κB translocation and IκBα degradation was maximal within 20 minutes in non-CF and CF lines and remain unchanged for longer stimulation periods of 30 and 60 minutes (not shown). Although CF airway cells seem to exhibit larger amount of translocated NF-κB under basal conditions, the effect of TNF-α on NF-κB translocation and IκBα degradation is indicative of an inflammatory response in all non-CF and CF lines.
Figure 1.

Time-course of NF-κB translocation and IκBα degradation in non-CF Beas2B cells exposed to TNF-α. A: EMSA analysis of NF-κB binding activity in nuclear extracts from Beas2B cells treated for the indicated time with 100 U/ml TNF-α. TNF-α induced the increase in NF-κB binding activity in nuclear extracts in a time-dependent manner. Maximal nuclear translocation was reached after 20 minutes in the presence of TNF-α. The single arrow corresponds to binding of NF-κB to the probe, and the double arrow indicates the free probe. Nonspecific binding of the NF-κB probe can also be seen on the EMSAs (asterisk). Competition with 100× excess of cold competitors (see Materials and Methods) abolished binding of NF-κB to the probe but not the nonspecific binding. B: Detection of IκBα by Western blot analysis of cytoplasmic extracts from Beas2B cells exposed to TNF-α. TNF-α rapidly induced the degradation of IκBα, in parallel to NF-κB translocation. The arrow indicates the IκBα band at ∼36 kd.
Figure 2.

A: NF-κB translocation and IκBα degradation in non-CF and CF airway cells exposed to TNF-α. In all cell lines, EMSA analysis shows that TNF-α evoked maximal translocation of NF-κB within 20 minutes. Note nonspecific binding of the NF-κB probe in EMSAs on Beas2B cells (asterisk). B: The parallel degradation of IκBα elicited by TNF-α in Beas2B, IB3-1, and CF15 cells was detected by Western blot experiments after 20 minutes of cell exposure to TNF-α. The arrow indicates the IκBα band at ∼36 kd.
Expression of Connexins in Non-CF and CF Airway Cells
The identification of the Cxs, expressed in non-CF and CF airway cells, was first evaluated by reverse transcriptase-polymerase chain reaction. Thus, mRNA was extracted from all cell types and reverse-transcribed into cDNA. The cDNAs were amplified by polymerase chain reaction using specific primer pairs for human Cx26, Cx32, Cx37, Cx40, Cx43, or Cx45. As shown in Figure 3A ▶ , amplification products corresponding to Cx43 mRNA were detected in all cell lines. In addition to Cx43, mRNA for Cx45 in Beas2B and IB3-1 cells, and mRNA for Cx32 in IB3-1 cells were also detected (not shown).
Figure 3.

Expression of Cx43 in non-CF and CF airway cells. A: Reverse transcriptase-polymerase chain reaction was performed on mRNA isolated from Beas2B (lane 2), CF15 (lane 3), and IB3-1 (lane 4) cells using primer pairs specific for human Cx43. Amplification products of the expected sizes for Cx43 (285 bp) were detected in all non-CF and CF cell lines. Molecular markers are shown in lanes 1 and 5. B: Western blot analysis of Cx43 expression in non-CF and CF airway cells. The anti-Cx43 antibody revealed one band at ∼41 kd in cytosolic protein extracts from Beas2B (lane 2), CF15 (lane 3), and IB3-1 (lane 4) cells. Lane 1 corresponds to rat atrium samples that were used as positive controls. Several bands at 41 to 46 kd could be detected, corresponding to various phosphorylated forms of Cx43. The lower band likely corresponds to a degradation product of Cx43. Lane 5 corresponds to SKHep1 cells, which do not express Cx43, that were used as negative controls.
The expression of Cx43 protein was confirmed by Western blots in all non-CF and CF airway cells. Total protein preparations were isolated from subconfluent monolayers of each cell type, and equal amounts of protein were blotted and probed with anti-Cx43 antibodies. One major isoform with an apparent molecular weight of ∼41 kd was detected by the Cx43 antibodies in all airway cell lines (Figure 3B) ▶ .
Extent of Dye Coupling in Non-CF and CF Airway Cells
The strength of intercellular communication was evaluated in non-CF and CF airway cells by injection of lucifer yellow. In all airway cell lines, lucifer yellow rapidly spread from the injected cell to several neighboring cells. Interestingly, the extent of lucifer diffusion was markedly reduced in the presence of TNF-α in non-CF cells (Figure 4 ▶ , Table 2 ▶ ). The time course of TNF-α-induced uncoupling is shown in Figure 5 ▶ . TNF-α progressively decreased the extent of intercellular communication between Beas2B cells, a steady-state level being reached after 20 minutes of exposure. Quantitative analysis revealed that, on average, TNF-α decreased (P < 0.002) intercellular communication between non-CF cells by 2.3-fold (Figure 6A) ▶ and increased the proportion of uncoupled cells (Table 2) ▶ . The uncoupling effect was observed for up to 90 minutes after washing out TNF-α (not shown). In contrast, TNF-α had no effect on the extent of dye coupling between CF15 cells (Figure 6B ▶ and Table 2 ▶ ). Dye coupling between CF15 cells was not affected by increasing the concentration of TNF-α up to 1000 U/ml (not shown). Similarly, TNF-α did not change dye coupling between IB3-1 cells, another CF cell line (Figure 6C ▶ and Table 2 ▶ ). In all cell lines, gap junction channel blockers inhibited cell-to-cell diffusion of lucifer yellow (Figure 6) ▶ .
Figure 4.

Effects of TNF-α on the extent of dye coupling between non-CF Beas2B cells. Under control conditions, lucifer yellow (LY) diffused from the microinjected cell to five to six neighbors (A). In contrast, the extent of LY diffusion was markedly decreased in the presence of 100 U/ml TNF-α (B). Scale bar, 15 μm.
Table 2.
Extent of Dye Coupling Between Non-CF and CF Airway Epithelial Cells
| Number of labeled cells | Beas2B | CF15 | IB3-1 | |||
|---|---|---|---|---|---|---|
| Control* | TNF-α† | Control | TNF-α | Control | TNF-α | |
| 1–2 | 7 (20) | 17 (63) | 2 (7) | 3 (16) | 5 (20) | 1 (7) |
| 3–4 | 7 (20) | 9 (33) | 8 (30) | 1 (5) | 6 (24) | 3 (23) |
| > 5 | 21 (60) | 1 (4) | 17 (63) | 15 (79) | 14 (56) | 9 (70) |
*Values indicate the number of microinjections. Percent values are given in parentheses.
†TNF-α (100 U/ml) decreased the proportion of dye coupled Beas2B (non-CF) cells but not that of CF (CF15 and IB3-1) cells.
Figure 5.

Time course of TNF-α-induced dye uncoupling between non-CF cells. In the absence of TNF-α (time 0), the number of cells labeled with lucifer yellow (LY) averaged 5.3 ± 0.65 cells (n = 13 microinjections). Exposure of Beas2B cells to TNF-α was associated with a time-dependent decrease in the number of LY-labeled cells. Maximal uncoupling was reached within 20 minutes. Bars indicate the mean number of LY-labeled cells.
Figure 6.
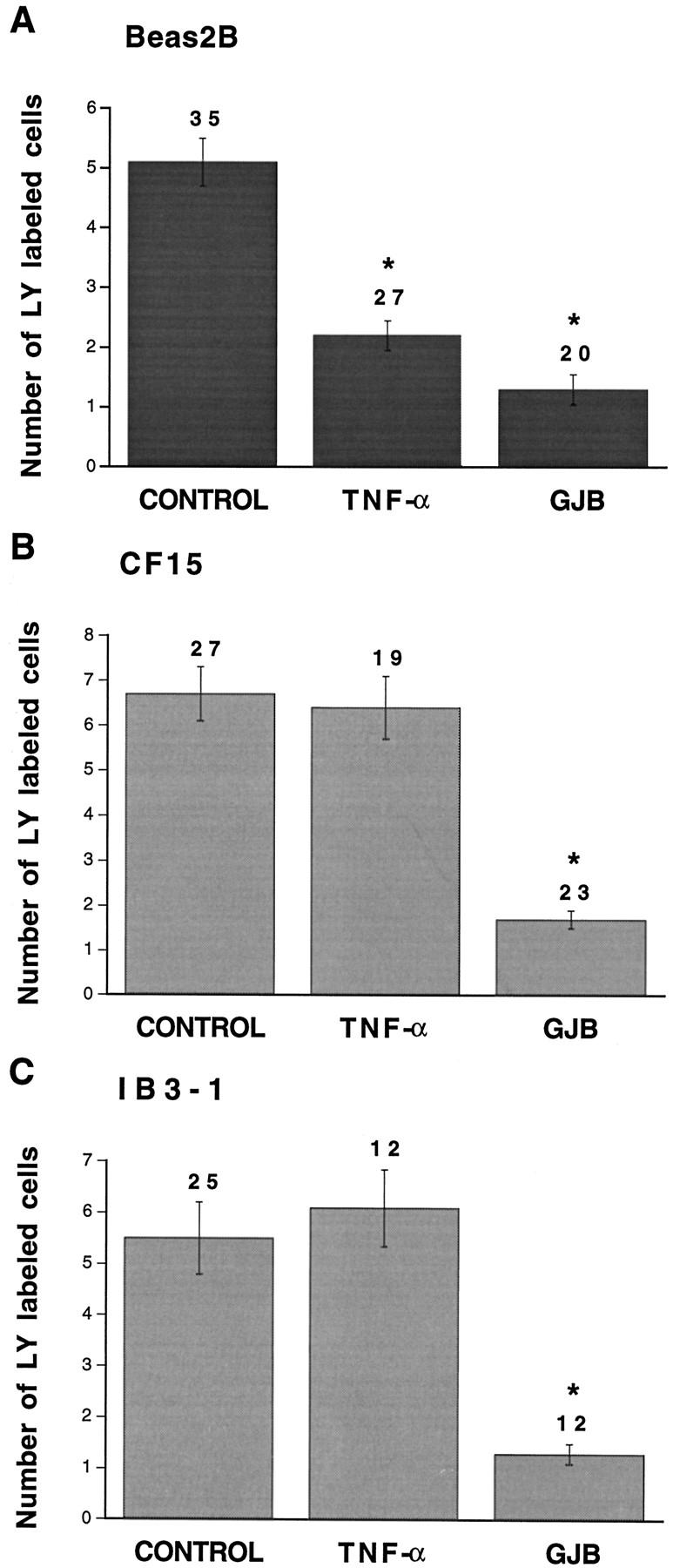
Quantitative evaluation of dye coupling in non-CF and CF airway cells exposed to TNF-α. Under control conditions, transfer of lucifer yellow (LY) was detected in Beas2B (A), CF15 (B), and IB3-1 (C) cells. Whereas TNF-α significantly decreased dye transfer in non-CF Beas2B cells, the pro-inflammatory mediator had no effect on intercellular communication between CF (CF15 and IB3-1) cells. In all cells lines, gap junction channel blockers (GJB) inhibited dye coupling. Asterisks indicate differences at P < 0.002 levels.
CF15 and IB3-1 cells are cultured in the presence of various growth factors, which may affect the sensitivity of gap junction channels to TNF-α. To test for this possibility, Beas2B cells were maintained for 3 to 4 days in the culture medium used for CF cells and subjected again to dye coupling. Under these conditions, the strength of intercellular communication between Beas2B cells exposed to CF culture medium did not change, averaging 6.1 ± 1 cells (n = 18). Exposure of the cells to TNF-α was still associated (P < 0.003) with cell uncoupling (2.8 ± 0.5 cells, n = 21). These results suggest that TNF-α directly modulates intercellular communication between non-CF but not CF airway cells.
Extent of Dye Coupling in Corrected CF Airway Cells
To evaluate whether CFTR contributes to the modulation of intercellular communication by TNF-α, CF15 cells were infected with increasing concentrations of recombinant adenovirus containing wild-type CFTR cDNA. The expression of CFTR was controlled either by the RSV promoter (AdRSV CFTR) or the CMV promoter (AdCMV CFTR). The efficiency of transgene expression was evaluated by using the RSV promoter-driven lacZ (AdRSV βgal) gene or the CMV promoter-driven eGFP (AdCMV GFP) gene. As previously reported, 29 increasing the MOI of AdRSV βgal from 25 to 500 was associated with an increase in the number of CF15 cells positive for X-Gal. Similarly, the number of fluorescent cells as well as the intensity of the fluorescent signal increased with increasing MOIs (ranging from 1 to 200) of AdCMV GFP (not shown).
The strength of intercellular communication in CF15 cells infected with AdCFTR was determined. As shown in Figure 7A ▶ , low and intermediate MOIs (25 to 100) of AdRSV CFTR had no effect on dye coupling between CF15 cells. However, dye coupling was strongly reduced at higher MOIs (250 to 500). A similar dye-coupling pattern was observed for CF15 infected with AdRSV βgal. Although 25 to 100 MOI of AdRSV βgal did not alter the extent of dye coupling between CF15 cells (7 ± 1 labeled cells, n = 8 versus 6.7 ± 0.6 cells, n = 27 in controls), the diffusion of lucifer yellow was decreased for higher MOIs (1.8 ± 0.5 labeled cells, n = 4). This effect was not restricted to adenovirus using the RSV promoter because high MOIs (100 to 200) of AdCMV CFTR also reduced dye coupling between CF15 cells (Figure 7B) ▶ . These results indicate that high MOIs of recombinant adenovirus exert toxic effects on gap junctional communication.
Figure 7.
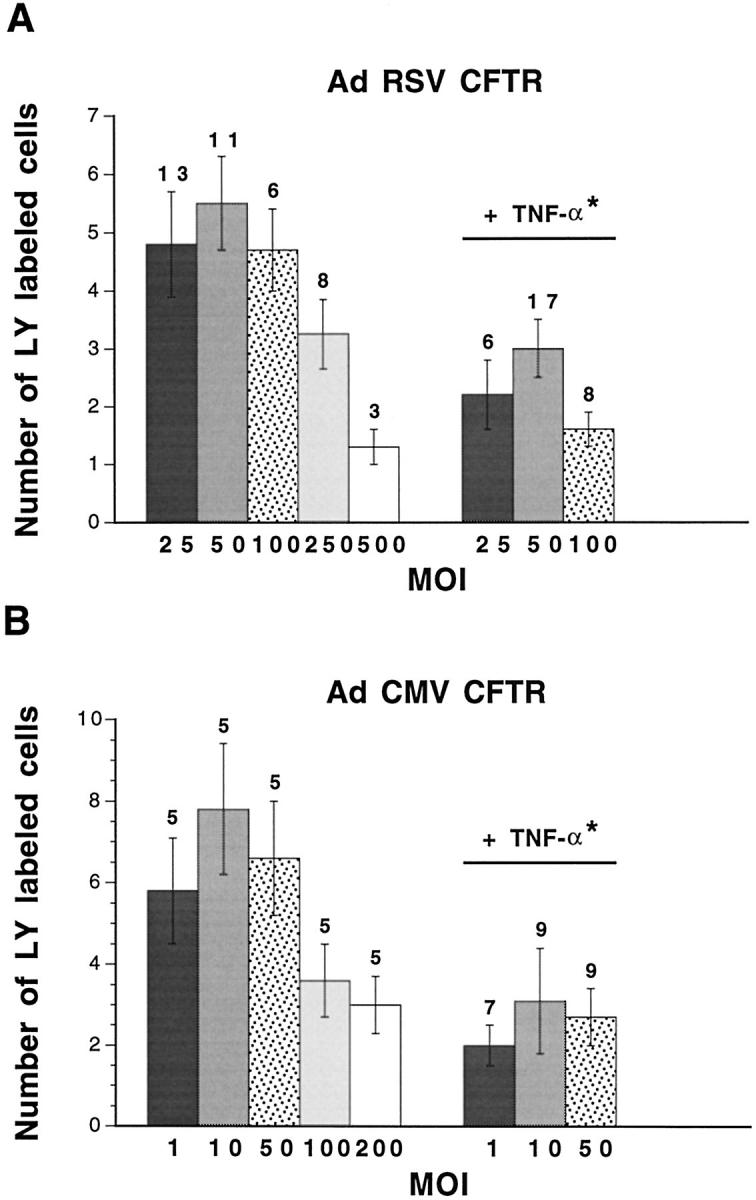
Dye coupling in CF15 cells infected with AdRSV CFTR or AdCMV CFTR. High MOIs of AdRSV CFTR (A) and of AdCMV CFTR (B) decreased lucifer yellow (LY) diffusion between CF15 cells. Lower MOIs of AdRSV CFTR (25 to 100) and of AdCMV CFTR (1 to 50) did not affect the normal extent of intercellular communication between CF15 cells. For the latter range of concentrations, exposure of the cells to TNF-α was associated with a significant reduction of gap junctional communication. Thus, difference at P < 0.001 (asterisk) was calculated for the pooled values obtained from 25 to 100 MOIs of AdRSV CFTR- or 1 to 50 MOIs of AdCMV CFTR-infected cells between control and TNF-α conditions.
Because of these limitations, only CF15 cells infected with low or intermediate MOIs of AdRSV CFTR (25 to 100 MOI) or AdCMV CFTR (1 to 50 MOI), and therefore exhibiting unaltered strength of intercellular communication, were exposed to TNF-α. Under these conditions, exposure of AdRSV CFTR-infected (Figure 7A) ▶ and AdCMV CFTR-infected (Figure 7B) ▶ CF15 cells to TNF-α resulted in a marked reduction of intercellular communication. This was caused by an enhanced proportion of cells that did not allow the passage of the dye from 17 to 68% (Table 3) ▶ . In contrast, TNF-α had no effect on dye coupling of CF15 cells infected with 25 to 100 MOI AdRSV βgal (7.6 ± 1 labeled cells, n = 10). These results indicate that expression of a functional CFTR is necessary for TNF-α-dependent regulation of gap junctional communication in airway epithelial cells.
Table 3.
Extent of Dye Coupling in CF15 Cells Infected with AdRSV CFTR
| Number of labeled cells | CF15 + AdRSV CFTR* | CF15 + AdRSV CFTR + TNF-α† |
|---|---|---|
| 1–2 | 5 (16) | 21 (68) |
| 3–4 | 8 (27) | 4 (13) |
| > 5 | 17 (57) | 6 (19) |
*Values indicate the number of microinjections. Percent values are given in parentheses.
†TNF-α (100 U/ml) decreased the proportion of dye coupled CF15 cells that have been infected with 25 to 100 MOI of AdRSV CFTR.
Discussion
Our results describe the temporal relationship between changes in NF-κB translocation into the nucleus and gap junctional communication of non-CF and CF human airway cells exposed to TNF-α. Although TNF-α induced NF-κB translocation in both cell types, the pro-inflammatory cytokine reduced the strength of intercellular communication only between CFTR-expressing airway cells.
The transcription factor NF-κB, by controlling the activation of numerous immunomodulatory genes in response to pathogens and pro-inflammatory cytokines, is essential in the development of acute and chronic inflammation. 30,31 In this regard, it has been reported that NF-κB translocation was increased in response to Pseudomonas aeruginosa in respiratory cells expressing wild-type CFTR, whereas in CF cells NF-κB activity appeared to be already elevated in unstimulated cells. 32 More recently, primary CF bronchial gland cells were shown to produce abnormally high levels of IL-8 through constitutively activated NF-κB. 33 NF-κB is present in the cytosol of most cell types as an inactive heterodimer that is bound to an inhibitor subunit, IκBα. Pro-inflammatory cytokines activate NF-κB by stimulating the activity of protein kinases that phosphorylate IκBα, allowing its ubiquitination and then rapid proteasomal degradation. This allows nuclear translocation of the active NF-κB and DNA binding. 30,34 TNF-α is an essential effector cytokine for immune response and inflammation. We observed that TNF-α stimulated maximal translocation of NF-κB into the nucleus and IκBα degradation within 20 minutes of Beas2B, CF15, and IB3-1 cells. The fact that unstimulated CF airway cell lines exhibit a larger amount of translocated NF-κB might be a key component of the airway inflammation in CF but also be explained by the variability between cell lines. This fact, however, does not affect the time course of the response to TNF-α, which evoked an inflammatory response in all airway cells used in this study irrespective of their CF or non-CF origins.
Whereas all airway cell lines developed an inflammatory response, TNF-α rapidly reduced the extent of intercellular communication in non-CF but not CF cells. Each gap junction channel type is characterized by intrinsic properties and differential sensitivities to modulation by intracellular signaling pathways that are dictated by their Cx composition. 13-15 Changes in Cx expression during inflammation have been shown in various cell systems after long-term exposure to pro-inflammatory cytokines. 20-24 So far, only one study reported a short-term (within 30 minutes) modulation of intercellular communication by TNF-α in human myoendothelial junctions. 19 Three types of Cxs were detected in non-CF and CF cells, Cx45, Cx32, and Cx43. Cx43 is most likely the Cx involved in the TNF-α-dependent regulation of gap junctional communication. Although mRNA for Cx45 was found in Beas2B and IB3-1 cells, Cx45 has been previously shown not to transfer lucifer yellow. 35 mRNA for Cx32 was detected only in IB3-1 cells, therefore this Cx cannot be responsible for the difference in modulation of intercellular coupling between non-CF and CF cells. In contrast to Cx45 and Cx32, Cx43 mRNA and protein were expressed in all cell lines. Cx43 is the gap junction phosphoprotein that has received most attention so far. Short-term modulation of Cx43 permeability and/or single-channel conductance has been demonstrated in response to various phosphorylating treatments, including activation of protein kinase C, 35,36-38 mitogen-activated protein kinase, 39,40 and c-Src tyrosine kinases. 41 Interestingly, these signaling pathways have also been found to mediate the cellular responses of TNF-α in various cell types 42,43 including airway epithelial cells. 44 In preliminary studies, exposure of Beas2B cells to TNF-α for up to 90 minutes had no apparent effect on the level of expression of Cx43 (Marc Chanson, unpublished observations). Regardless of the specific mechanism, our results demonstrate that intercellular communication is suppressed during the inflammatory response of normal human airway cells. This down-regulation, however, is defective in CF airway cells. Whether abnormal posttranslational modifications of Cx43 are responsible for the differential modulation of gap junctional coupling between non-CF and CF cells remains to be investigated.
It is now clearly established that CFTR, in addition to functioning as a Cl− channel, plays an important role in conferring regulatory properties on other ion channels of the plasma membrane. 2 Recent observations also suggested a role for CFTR in the control of gap junctional coupling between pancreatic duct cells. 18 Gap junctional communication was therefore examined in CF15 airway cells before and after correction of their phenotype by transfer of wild-type CFTR. The transduction of functional CFTR into CF15 cells with AdRSV CFTR or AdCMV CFTR has been previously characterized. 29 The Cl− secretion defect of CF15 cells was indeed corrected by CFTR-containing adenovirus. We report now that TNF-α-induced down-regulation of intercellular communication was restored in cells infected with AdCFTR but not with adenovirus encoding a reporter gene. These results, therefore, lead to propose that expression of CFTR is necessary for the regulation of gap junction-mediated intercellular communication by TNF-α in airway cells.
The links between the expression of a normal CFTR protein and the modulation of gap junction channels are not known. It is conceivable that abnormal cell functions in CF cells may be a consequence of cell stress caused by trafficking defects of mutant CFTR proteins. 32 CFTR might also influence activities and regulation of other transport pathways by direct or indirect protein-protein interactions. Thus, the NH2-terminus of CFTR binds syntaxin 1A 45 and the CFTR tail binds the α1 subunit of AMP-activated protein kinase. 46 CFTR has also been shown to associate with submembranous scaffolding proteins via PDZ-binding domains. 47 PDZ-binding domain proteins are involved in the clustering of transmembrane ion channels and in connecting intracellular signaling pathways. Consequently, it has been suggested that protein-protein interactions may be required for CFTR-mediated regulation of other ion channels. 48 In this context, it is noteworthy that Cx43 co-localizes and specifically interacts via PDZ-binding domains with the zonula occludens-1 protein. 49,50 Future studies might provide insights into the mechanisms that couple CFTR-dependent functions to gap junctional communication. Our data, however, support the view that absence of functional CFTR is associated with defective regulation of intercellular communication during the inflammatory response evoked by TNF-α in CF airway epithelial cells.
Cell-specific expression of Cxs and differential modulation of gap junctional permeability to signaling molecules are thought to coordinate the appropriate response of groups of cells to external stimuli. In this context, altering the level of intercellular communication by manipulating Cx expression has been shown to be associated with modulation of glycogen metabolism, 51 digestive enzyme secretion, 52 gene expression, 53 and Ca2+ signaling by controlling ATP release. 54 One challenge for future studies will be to explore the pathophysiological consequences of a defective regulation of airway epithelial cell-to-cell communication on the inflammatory process. The closure of gap junction channels during sustained inflammation may restrict the intercellular diffusion of signaling molecules, thereby preventing the recruitment of bystander cells into inflammatory responses. Defects in this mechanism may decrease the capacity to localize the inflammatory reaction to the areas stimulated by invading pathogens, and thus contribute to the widespread inflammatory response of the CF airway epithelium.
Acknowledgments
We thank Brenda Kwak, Marc A. Thomas, and Thomas M. Boat for critical reading of the manuscript; and Melete Solomon for excellent technical assistance.
Footnotes
Address reprint requests to Dr. Marc Chanson, Laboratory of Clinical Investigation III, Department of Pediatrics, HUG-PO Box 14, Micheli-du-Crest, 24, 1211 Geneva 14, Switzerland. E-mail: marc_chanson@hotmail.com.
Supported by grants from the Swiss National Science Foundation no. 32-55745.98 (to M. C.), l’Association Française de Lutte contre la Mucoviscidose (to M. C.), and the Swiss National Science Foundation (to P.-Y. B.).
References
- 1.Welsh MJ, Tsui L-C, Boat TM, Beaudet AL: Cystic fibrosis. ed 7 Scriver CR Beaudet AL Sly WS Valle D eds. The Metabolic and Molecular Basis of Inherited Disease, 1995, :pp 3799-3876 McGraw-Hill, New York [Google Scholar]
- 2.Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB: CFTR is a conductance regulator as well as a chloride channel. Physiol Rev 1999, 79:S145-S166 [DOI] [PubMed] [Google Scholar]
- 3.Tosi MF, Stark JM, Smith CW, Hamedani A, Gruenert DC, Infeld MD: Induction of ICAM-1 expression on human airway epithelial cells by inflammatory cytokines: effects on neutrophil-epithelial cell adhesion. Am J Respir Cell Mol Biol 1992, 7:214-221 [DOI] [PubMed] [Google Scholar]
- 4.Bonfield TM, Panuska JR, Konstan MW, Hilliard KA, Hilliard JB, Ghnaim H, Berger M: Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med 1995, 152:2111-2118 [DOI] [PubMed] [Google Scholar]
- 5.Noah TL, Black HR, Cheng PW, Wood RE, Leigh MW: Nasal and bronchoalveolar lavage fluid cytokines in early cystic fibrosis. J Infect Dis 1997, 175:638-647 [DOI] [PubMed] [Google Scholar]
- 6.Döring G: The role of neutrophil elastase in chronic inflammation. Am J Respir Crit Care Med 1995, 152:163-168 [DOI] [PubMed] [Google Scholar]
- 7.Saiman L, Prince A: Pseudomonas aeruginosa pili bind to asialoGM1 which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest 1993, 92:1875-1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pier GB, Grout M, Zaidi TS: Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci USA 1997, 94:12088-12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith JJ, Travis SM, Greenberg EP, Welsh MJ: Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 1996, 85:229-236 [DOI] [PubMed] [Google Scholar]
- 10.Balough K, McCubbin M, Weinberger M, Smits W, Ahrens R, Fick R: The relationship between infection and inflammation in the early stages of lung disease from cystic fibrosis. Pediatr Pulmonol 1995, 20:63-70 [DOI] [PubMed] [Google Scholar]
- 11.Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DWH: Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med 1995, 151:1075-1082 [DOI] [PubMed] [Google Scholar]
- 12.Tabary O, Zahm JM, Hinnrasky J, Couetil JP, Cornillet P, Guenounou M, Gaillard D, Puchelle E, Jacquot J: Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am J Pathol 1998, 153:921-930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar NM, Gilula NB: The gap junction communication channel. Cell 1996, 84:381-388 [DOI] [PubMed] [Google Scholar]
- 14.Goodenough DA, Goliger JA, Paul DL: Connexins, connexons, and intercellular communication. Annu Rev Biochem 1996, 65:475-502 [DOI] [PubMed] [Google Scholar]
- 15.Spray DC: Molecular physiology of gap junction channels. Clin Exp Pharmacol Physiol 1996, 23:1038-1040 [DOI] [PubMed] [Google Scholar]
- 16.Yamasaki H, Naus CC: Role of connexin genes in growth control. Carcinogenesis 1996, 17:1199-1213 [DOI] [PubMed] [Google Scholar]
- 17.White TW, Paul DL: Genetic diseases and gene knockouts reveal diverse connexin functions. Annu Rev Physiol 1999, 61:283-310 [DOI] [PubMed] [Google Scholar]
- 18.Chanson M, Scerri I, Suter S: Defective regulation of gap junctional coupling in cystic fibrosis pancreatic duct cells. J Clin Invest 1999, 103:1677-1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Cotgreave IA: Differential regulation of gap junctions by proinflammatory mediators in vitro. J Clin Invest 1997, 99:2312-2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Rijen HV, van Kempen MJ, Postma S, Jongsma HJ: Tumor necrosis factor alpha alters the expression of connexin43, connexin40, and connexin37 in human umbilical vein endothelial cells. Cytokine 1998, 10:258-264 [DOI] [PubMed] [Google Scholar]
- 21.Gingalewski C, Wang K, Clemens MG, De Maio A: Posttranscriptional regulation of connexin32 expression in liver during acute inflammation. J Cell Physiol 1996, 166:461-467 [DOI] [PubMed] [Google Scholar]
- 22.Temme A, Traub O, Willecke K: Downregulation of Connexin32 protein and gap-junctional intercellular communication by cytokine-mediated acute-phase response in immortalized mouse hepatocytes. Cell Tissue Res 1998, 294:345-350 [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Cobo M, Gingalewsky C, Drujan D, De Maio A: Downregulation of connexin43 gene expression in rat heart during inflammation. The role of tumor necrosis factor. Cytokine 1999, 11:216-224 [DOI] [PubMed] [Google Scholar]
- 24.Chandross KJ, Spray DC, Cohen RI, Kumar NM, Kremer M, Dermietzel R, Kessler JA: TNF alpha inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol Cell Neurosci 1996, 7:479-500 [DOI] [PubMed] [Google Scholar]
- 25.Jefferson DM, Valentich JD, Marini FC, Grubman SA, Iannuzzi MC, Dorkin HL, Li M, Klinger KW, Welsh MJ: Expression of normal and cystic fibrosis phenotypes by continuous airway epithelial cell lines. Am J Physiol 1990, 259:L496-L505 [DOI] [PubMed] [Google Scholar]
- 26.Zeitlin PL, Lu L, Rhim J, Cutting G, Stetten G, Kieffer KA, Craig R, Guggino WB: A cystic fibrosis bronchial epithelial cell line: immortalization by adeno-12-SV40 infection. Am J Respir Cell Mol Biol 1991, 4:313-319 [DOI] [PubMed] [Google Scholar]
- 27.Fiedler MA, Wernke-Dollries K, Stark JM: Inhibition of TNF-α-induced NF-κB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol 1998, 19:259-268 [DOI] [PubMed] [Google Scholar]
- 28.Lusky M, Christ M, Rittner K, Dieterlé A, Dreyer D, Mourot B, Schultz H, Stoeckel F, Pavirani A, Mehtali M: In vitro and in vivo biology of recombinant adenovirus vectors with E1, E1/E2A, or E1/E4 deleted. J Virol 1998, 72:2022-2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pizurki L, Morris MA, Chanson M, Solomon M, Pavirani A, Bouchardy I, Suter S: Cystic fibrosis transmembrane conductance regulator does not affect neutrophil migration across cystic fibrosis airway epithelial monolayers. Am J Pathol 2000, 156:1407-1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baeuerle PA, Baltimore D: NF-κB: ten years after. Cell 1996, 87:13-20 [DOI] [PubMed] [Google Scholar]
- 31.Barnes PJ, Karin M: Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med 1997, 336:1066-1071 [DOI] [PubMed] [Google Scholar]
- 32.DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A: Activation of NF-κB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest 1998, 101:2598-2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tabary O, Escotte S, Couetil JP, Hubert D, Dusser D, Puchelle E, Jacquot J: High susceptibility for cystic fibrosis human airway gland cells to produce IL-8 through the I kappaB kinase alpha pathway in response to extracellular NaCl content. J Immunol 2000, 164:3377-3384 [DOI] [PubMed] [Google Scholar]
- 34.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M: The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 1997, 91:243-252 [DOI] [PubMed] [Google Scholar]
- 35.Kwak BR, Hermans MMP, De Jonge HR, Lohmann SM, Jongsma HJ, Chanson M: Differential regulation of distinct types of gap junction channels by similar phosphorylating conditions. Mol Biol Cell 1995, 6:1707-1719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musil LS, Cunningham BA, Edelman GM, Goodenough DA: Differential phosphorylation of the gap junction protein connexin43 in junctional communication-competent and -deficient cell lines. J Cell Biol 1990, 111:2077-2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreno AP, Saez JC, Fishman GI, Spray DC: Human connexin43 gap junction channels: regulation of unitary conductances by phosphorylation. Circ Res 1994, 74:1050-1057 [DOI] [PubMed] [Google Scholar]
- 38.Lampe PD, TenBroek EM, Burt JM, Kurata WE, Johnson RG, Lau AF: Phosphorylation of connexin43 on serine 368 by protein kinase C regulates gap junctional communication. J Cell Biol 2000, 149:1503-1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanemitsu MY, Lau AF: Epidermal growth factor stimulates the disruption of gap junctional communication and connexin43 phosphorylation independent of 12-O-tetradecanoylphorbol 13-acetate-sensitive protein kinase C: the possible involvement of mitogen-activated protein kinase. Mol Biol Cell 1993, 4:837-848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warn-Cramer BJ, Cottrell GT, Burt JM, Lau AF: Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J Biol Chem 1998, 273:9188-9196 [DOI] [PubMed] [Google Scholar]
- 41.Postma FR, Hengeveld T, Alblas J, Giepmans BB, Zondag GC, Jalink K, Moolenaar WH: Acute loss of cell-cell communication caused by G protein-coupled receptors: a critical role for c-Src. J Cell Biol 1998, 140:1199-1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eder J: Tumor necrosis factor α and interleukin 1 signalling: do MAPKK kinases connect it all? TIPS 1997, 18:310-322 [DOI] [PubMed] [Google Scholar]
- 43.Abu-Am Y, Ross FP, McHugh KP, Livolsi A, Peyron J-F, Teitelbaum SL: Tumor necrosis factor-α activation of nuclear transcription factor-κB in marrow macrophages is mediated by c-Src tyrosine phosphorylation of IκBα. J Biol Chem 1998, 273:29417-29423 [DOI] [PubMed] [Google Scholar]
- 44.Krunkosky TM, Fischer BM, Martin LD, Jones N, Akley NJ, Adler KB: Effects of TNF-α on expression of ICAM-1 in human airway epithelial cells in vitro. Signaling pathways controlling surface and gene expression. Am J Respir Cell Mol Biol 2000, 22:685-692 [DOI] [PubMed] [Google Scholar]
- 45.Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL: Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature 1997, 390:302-305 [DOI] [PubMed] [Google Scholar]
- 46.Hallows KR, Raghuram V, Kemp BE, Witters LA, Foskett JK: Inhibition of cystic fibrosis transmembrane conductance regulator by novel interaction with the metabolic sensor AMP-activated protein kinase. J Clin Invest 2000, 105:1711-1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Short DB, Trotter KW, Reczek D, Kreda SM, Bretscher A, Boucher RC, Stutts MJ, Milgram SL: An apical PDZ protein anchors the cystic fibrosis transmembrane conductance regulator to the cytoskeleton. J Biol Chem 1998, 273:19797-19801 [DOI] [PubMed] [Google Scholar]
- 48.Mohler PJ, Kreda SM, Boucher RC, Sudol M, Stutts MJ, Milgram SL: Yes-associated protein 65 localizes p62c-Yes to the apical compartment of airway epithelia by association with EPB50. J Cell Biol 1999, 147:879-890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M: Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem 1998, 273:12725-12731 [DOI] [PubMed] [Google Scholar]
- 50.Giepmans BNG, Moolenaar WH: The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol 1998, 8:931-934 [DOI] [PubMed] [Google Scholar]
- 51.Nelles E, Bützler C, Jung D, Temme A, Gabriel H-D, Dahl U, Traub O, Stümpel F, Jungermann K, Zielasek J, Toyka KV, Dermietzel R, Willecke K: Defective propagation of signals generated by sympathetic nerve stimulation in the liver of connexin32-deficient mice. Proc Natl Acad Sci USA 1996, 93:9565-9570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chanson M, Fanjul M, Bosco D, Nelles E, Suter S, Willecke K, Meda P: Enhanced secretion of amylase from exocrine pancreas of connexin32-deficient mice. J Cell Biol 1998, 141:1267-1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lecanda F, Towler DA, Ziambaras K, Cheng SL, Koval M, Steinberg TH, Civitelli R: Gap junctional communication modulates gene expression in osteoblastic cells. Mol Biol Cell 1998, 9:2249-2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M: Connexins regulate calcium signalling by controlling ATP release. Proc Natl Acad Sci USA 1998, 95:15735-15740 [DOI] [PMC free article] [PubMed] [Google Scholar]