

Abstract
The predisposition of the testis and ovary to primarily synthesize testosterone and estradiol, respectively, is due to gonadal-specific cell types that differentially express the various hydroxysteroid (17β) dehydrogenase (HSD17B) isoforms. In testes, Leydig cells rely on LH stimulation to maintain expression of the type 3 (HSD17B3) isoform, which specifically converts androstenedione to testosterone. In ovaries, thecal-interstitial cells also rely on LH to induce androgen synthesis but lack HSD17B3 and therefore secrete androgens of low biological activity. Therefore, thecal cells may possess a mechanism to repress the Leydig cell phenotype and HSD17B3 expression. Estradiol is known to inhibit experimentally Leydig cell function and proliferation. In the current study, we provide evidence that estradiol prevents the development of functional Leydig-like cells in the murine ovary; and that this action is mediated by estrogen receptor-α (ERα). ERα-null (αERKO) female mice exhibit testis-like levels of Hsd17b3 expression in the ovaries and male-like levels of plasma testosterone. Herein, we demonstrate that a) Hsd17b3 expression in αERKO ovaries is a primary effect of the loss of intraovarian ERα actions, b) αERKO ovarian cells produce substantial levels of testosterone in vitro and this is blocked by a HSD17B3 specific inhibitor, c) Hsd17b3 expression in αERKO ovaries is LH regulated and localized to the secondary/thecal interstitial cells, and d) αERKO secondary/thecal interstitial cells possess Leydig-like ultrastructural features. These data indicate that intraovarian ERα actions are required to repress Hsd17b3 expression in the ovary and may be important to maintaining a female phenotype in secondary/thecal interstitial cells.
Keywords: 17β-hydroxysteroid dehydrogenase, steroidogenesis, thecal cells
INTRODUCTION
The well-conserved sexually dimorphic pattern of gonadal steroid hormone secretion in mammals is vital to the proper development and function of reproductive tissues and behaviors. The predisposition of the testis and ovary to primarily produce testosterone and estradiol, respectively, is due primarily to the development of gonadal-specific cell types that differentially express enzymes involved in the final stages of steroid hormone synthesis, namely isoforms of the hydroxysteroid (17β) dehydrogenase (HSD17B) family (1–3). The gonads of both sexes possess the enzymatic capacity to convert C27 sterols (e.g., cholesterol) to C19 steroids, primarily dehydroepiandrosterone and androstenedione, which have low biological activity but are precursors for conversion to the more biologically active steroids, testosterone and estradiol. In mammalian males, the Leydig cells of the testes are especially proficient at reducing androstenedione to testosterone because they specifically express the androgenic or type 3 (HSD17B3) isoform (2, 4–7). Furthermore, because testes possess a relatively modest level of aromatase (CYP19A1), which converts androstenedione and testosterone to estrone and estradiol, respectively, the latter androgen is allowed to accumulate and be secreted. In contrast, granulosa cells in adult female ovaries possess substantially high levels of CYP19A1 but lack HSD17B3 (1, 2, 8), thereby favoring aromatization of androstenedione to estrone rather than reduction to testosterone. The accumulating estrone is then reduced to estradiol by the estrogenic or type 1 (HSD17B1) isoform, which is highly expressed in the granulosa cells of growing follicles, as well as in the uterus, adrenals and mammary gland (1).
The importance of sexual dimorphic expression of HSD17B1 and HSD17B3 in female and male gonads, respectively, is well appreciated yet poorly understood. This is especially true of the mechanisms that restrict HSD17B3 expression to testicular Leydig cells. Leydig cells constitutively express the LH-receptor and rely on LH stimulation to maintain HSD17B3 expression and testosterone synthesis (6, 9, 10). Thecal-interstitial cells of the ovary, however, also constitutively possess LH-receptor and rely on LH stimulation to maintain steroidogenesis (11, 12) but are limited to the secretion of C19 steroids of low biological activity because they lack HSD17B3 expression (13). This divergence between Leydig and thecal-interstitial cells, which are thought to arise from a common primordial cell during gonadal differentiation, suggests that the latter cell type either lack an LH-stimulated transcription factor(s) specific to the induction of HSD17B3 expression or possess a mechanism(s) to actively repress HSD17B3 expression. Estradiol has long been considered a leading candidate to fulfill this postulated inhibitory mechanism since it exists at significantly higher levels in ovaries than testes and inhibits Leydig cell function and proliferation in experimental studies (14, 15), but has received little investigative attention in this regard. However, our recent studies provide the first definitive evidence of estradiol-mediated repression of Hsd17b3 expression in the ovary and suggest that receptor-mediated actions of estradiol may play an important physiological role during ovarian development and function. We have previously shown that the ovaries of mice lacking functional estrogen receptor-α (αERKO) exhibit testis-like levels of Hsd17b3 expression and male-like levels of plasma testosterone (16). These findings are corroborated by a report that female mice null for CYP19A1 and therefore lacking endogenous estradiol synthesis exhibit comparable ovarian Hsd17b3 expression that is abolished by exogenous estradiol treatments (17). These data indicate that ligand-dependent ERα actions are fundamental to the repression of Hsd17b3 expression in the ovary. In the current study, we expand our description of this unique αERKO ovarian phenotype by demonstrating that a) ectopic Hsd17b3 expression is fundamentally due to the loss of intraovarian ERα actions and not a secondary effect of the hypergonadotropism that follows the neuroendocrine loss of ERα functions, b) ovarian HSD17B3 activity in αERKO females is responsible for their characteristic male-like testosterone burden, c) Hsd17b3 expression in αERKO ovaries exhibits a pattern of LH regulation comparable to that of Leydig cells in the testes, d) ectopic Hsd17b3 expression in αERKO ovaries is localized to the secondary/thecal interstitial cells, and e) these same cells exhibit ultrastructural features that are generally considered unique to Leydig cells.
MATERIALS AND METHODS
Animals
The Animal Care and Use Committee of the NIEHS preapproved all procedures involving animals. Animals were maintained in plastic cages in a temperature-controlled room (21–22ºC) under a 12-h light:12-h dark schedule and given NIH 31 mouse chow and fresh water ad libitum. Wild type (Esr1+/+) and αERKO (Esr1−/−) mice were of C57BL/6 strain and obtained from our colony at Taconic Farms, Germantown, NY. LH-CTP transgenic mice were a mixed background strain (C57BL/6 x CF-1) and obtained from our in-house colony established with breeders from the original colony at Case Western Reserve University, Cleveland, OH (as a gift from Dr. John H. Nilson). Compound hypogonadal Gnrhhpg/Esr1+/+ (wild typehpg) or Gnrhhpg/Esr1−/−(αERKOhpg) mice were obtained from our colony that was established by crossing heterozygous Gnrhhpg males (C3H/HeH x 101/H; Jackson Laboratories, Bar Harbor, ME) with Esr1+/− females (C57BL/6), and maintained at Charles River Laboratories (Wilmington, MA). All animals were genotyped by PCR on DNA extracted from tail biopsies using the Wizard SV 96 Genomic DNA extraction kit (Promega, Madison, WI). The procedures for genotyping Esr1−/− (16) and LH-CTP (18) mice have been previously described. All potential Gnrhhpg mice were genotyped using a 3-primer PCR scheme based on the original description of the mutant Gnrhhpg allele (19). This PCR method used the following common forward primer and two reverse primers to differentiate the wild type and mutant Gnrh alleles: forward 5′-ATGATGCTGCCCACAATCG (bp 2446–2464) to reverse 5′-TCCCAGACAGGAGTGAAGTGC (bp 2847–2827), these primers flank the mutant breakpoint at bp 2700 and produce a 402 bp amplimer from a wild type Gnrh allele and no amplimer from a mutant Gnrh allele since sequences homologous to the reverse primer are deleted; and the above forward primer paired with the following reverse primer 5′-TTTCTACTCTCTTGAAACAGGCAAATT, which is homologous to sequences 47 bp beyond the mutant breakpoint and therefore produces a 301 bp amplimer that is unique to the mutant Gnrhhpg allele. All PCR results were evaluated by agarose gel electrophoresis.
Animal treatments and tissue collection
All animals were 2–5 mo of age at the time of use. Animals were killed via CO2 asphyxiation, whole blood was immediately drawn from the inferior vena cava, mixed with heparin and the plasma later separated by centrifugation; gonads were promptly removed, trimmed of surrounding tissue and processed according to the intended downstream analysis (as described below). Two experiments required the treatment of animals with gonadotropins prior to tissue collection. In the first (described in Fig. 3), animals were implanted (s.c.) with an Alzet® osmotic pump (Durect Corporation, Cupertino, CA) rated for drug delivery of 0.5 μl/h for 7 days. Pumps were filled with either a) vehicle (0.85% saline), b) purified human LH (hLH) at 0.83 I.U./μl or c) recombinant human FSH (hFSH) at 0.83 I.U./μl to provide a calculated delivery of 10 I.U. gonadotropin per day. Gonadal tissues were collected as described above on the morning of the 7th day. Human LH and hFSH (hFSH) preparations were purchased from A.F. Parlow via the National Hormone and Peptide Program (Torrance, CA). In the second experiments involving gonadotropin treatment (described in Figs. 5 and 6), animals were injected (s.c.) twice daily for 3 consecutive days with vehicle (0.85% saline), 5 I.U. pregnant mares’ serum gonadotropin (PMSG) (Sigma, St. Louis, MO) or 5 I.U. human choriogonadotropin (hCG) (Sigma). Gonadal tissues were then collected as described above on the morning of the 6th day, approximately 1 h after the final treatment.
FIG. 3.
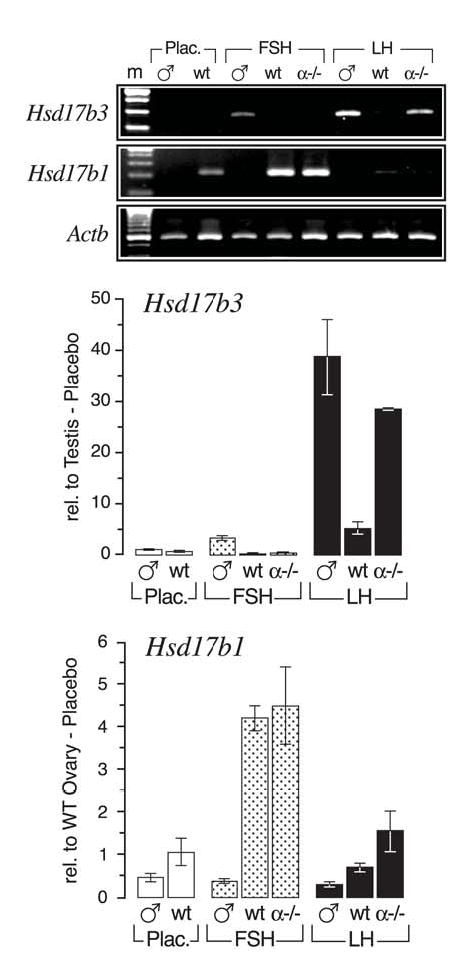
Gonadotropin induction of Hsd17b3 in hypogonadal αERKO ovaries. Adult wild-typehpg (WThpg) males and females, and αERKOhpg females were treated for 1 week with purified human LH or recombinant human FSH (see Materials and Methods). (top) Shown is a representative agarose gel of semi-quantitative RT-PCR for Hsd17b3 and Hsd17b1 transcripts in the gonads of each genotype and treatment. Hsd17b3 expression is relatively unique to testes and maximally induced by hLH, while Hsd17b1 is relatively unique to ovaries and maximally induced by hFSH. Also apparent is the testis-like induction of Hsd17b3 expression in the ovaries of αERKOhpg females. A small amount of Hsd17b3 expression is detectable by RT-PCR in the ovaries of LH-treated WThpg females. (bottom) Shown are the results of real-time RT-PCR quantitative assays (average ± SEM) for Hsd17b3 and Hsd17b1 transcripts in the gonads of each genotype and treatment. Sample numbers (n) for each group were: WThpg males (all groups), 4 per group; WThpg-none, 2 pools (3–4 ovary pairs per pool); WThpg-hFSH, 6; WThpg-hLH, 3; αERKOhpg (all groups), 2 per group.
FIG. 5.
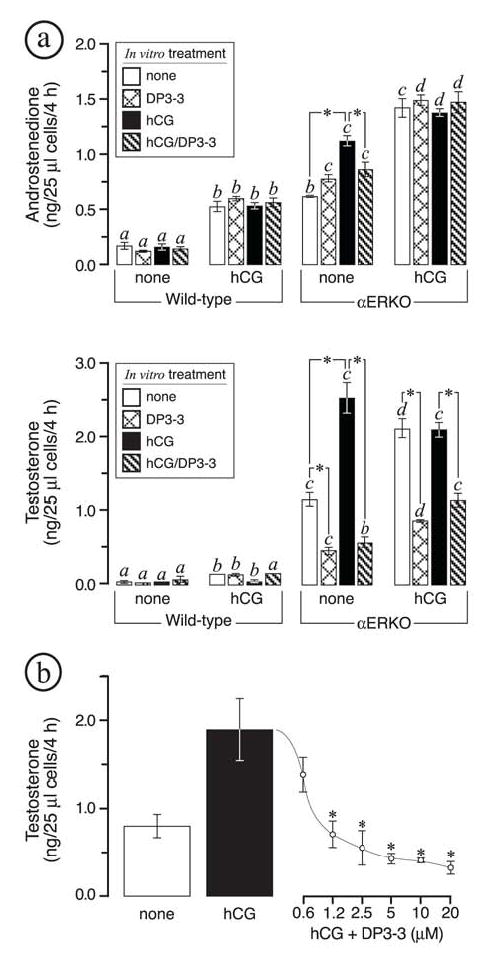
In vitro evaluation of HSD17B3 activity in dispersed αERKO ovarian cells. All data are from in vitro acute steroidogenic assays on dispersed ovarian cells (see Materials and Methods). These experiments were repeated three times and found to yield comparable results, shown are the results of one independent trial. Wild type (WT) and αERKO females were first treated twice daily for 3 days with vehicle or human choriogonadotropin (hCG) as indicated along the x-axis; dispersed ovarian cells were then prepared from each genotype/treatment group and incubated for 4 h in the indicated in vitro treatments. (a) In vitro androstenedione (top) and testosterone (bottom) synthesis indicates that in vivo hCG treatments led to increased androstenedione production in both WT and αERKO ovarian cells but increased testosterone synthesis in αERKO cells only. In vitro testosterone synthesis in dispersed αERKO ovarian cells was inhibited by the HSD17B3 inhibitor (DP3-3) at 10 μM. (b) Shown is a dose-response curve illustrating the effect of the HSD17B3 inhibitor (DP3-3) on testosterone synthesis in dispersed αERKO ovarian cells. All data are from dispersed ovarian cells from untreated αERKO females that were incubated in medium alone, medium with hCG (10 I.U./ml), or medium with hCG (10 I.U./ml) plus increasing concentrations of DP3-3. Each bar or point represents the average of 3 replicates, each of which was assayed in duplicate for each steroid. Bars that do not share the same letter are significantly different (P < 0.05). Significant differences (P < 0.05) within each genotype/treatment group are indicated by asterisks.
FIG. 6.
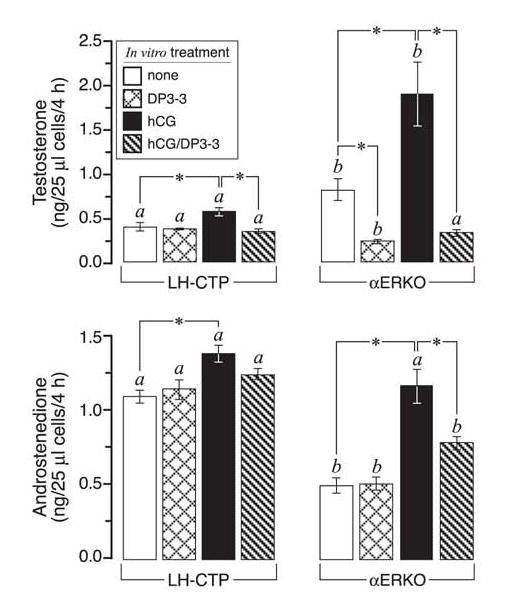
In vitro evaluation of HSD17B3 activity in dispersed LH-CTP and αERKO ovarian cells. All data are from in vitro acute steroidogenic assays on dispersed ovarian cells (see Materials and Methods). Dispersed ovarian cells were prepared from untreated adult LH-CTP and αERKO females and incubated for 4 h in the indicated in vitro treatments. These experiments were repeated two times and found to yield comparable results, shown are the results of one independent trial. Although LH-CTP ovarian cells produce increased amounts of androstenedione relative to αERKO ovarian cells, they produce much less testosterone due to the absence of HSD17B3 activity. Bars that do not share the same letter are significantly different (P < 0.05). Significant differences (P < 0.05) within each genotype/treatment group are indicated by asterisks. Each bar represents the average of 4 (αERKO) or 3 (LH-CTP) replicates, each of which was assayed in duplicate for each steroid.
Granulosa – thecal cell isolation
Ovaries were immediately removed from adult wild type and αERKO females upon death by CO2 asphyxiation and pooled according to genotype in 100 mm cell culture dishes containing ice-cold M199 medium (Invitrogen, Carlsbad, CA). Granulosa cells were then expressed into the medium by manual puncture of the ovaries with a 25-gauge needle and by applying slight pressure with a sterile spatula. The suspended granulosa cells in the medium were then concentrated via centrifugation at 250 x g for 5 min at 4°C and washed two times in M199 medium; the pellets were stored at –70ºC for later RNA isolation. The remaining ovarian fragments were considered to represent an enriched stromal/thecal-interstitial cell fraction and as such were washed several times in M199 medium and concentrated by centrifugation; the pellets were stored at –70ºC for later RNA isolation.
RNA isolation and gene expression assays
Total RNA was isolated from snap-frozen tissues or cell pellets using TRIZOL reagent (Invitrogen) according to the manufacturer’s protocol. The concentration and integrity of all final preparations was calculated via an A260 reading using a Molecular Devices Spectramax (Sunnyvale, CA) spectrophotometer and agarose gel electrophoresis of a 1 μg aliquot.
Northern blots were generated from 20 μg total RNA per sample using NorthernMax formaldehyde based reagents and BrightStar (Ambion, Austin, TX) positively charged nylon membrane according to the manufacturer’s protocol. Blots were sequentially probed, stripped and reprobed for serial gene expression analyses using StripEZ (Ambion) generated riboprobes and stripping reagents. Radiolabeled anti-sense riboprobes for Hsd17b3 (bp 363–729 of U66827) and Cyp17a1 (bp 522–932 of M4863) mRNAs were generated from previously described cDNA clones (16) using the StripEZ transcription reagents (Ambion) and [32P]α-UTP (Amersham Biosciences, Piscataway, NJ). Hybridizations were carried out in ULTRAhyb hybridization solution (Ambion) with approximately 1 x 107 cpm riboprobe per ml in a 68ºC rotisserie oven (Thermo-Hybaid, Franklin, MA) and then washed according to the manufacturer’s protocol. Blots were exposed to a phosphorimager screen and the data analyzed with a Storm 860 and accompanying ImageQuant Software (Molecular Dynamics, Sunnyvale, CA).
RT-PCR was carried out on total RNA that was first rid of contaminating DNA by use of the DNA-free® reagents (Ambion) according to the manufacturer’s protocol. For each sample, cDNA was generated from 1 μg RNA in a 25 μl reaction using random hexamers and the Superscript cDNA synthesis system (Invitrogen) according to the manufacturer’s protocol. Traditional PCR reactions were then prepared from the equivalent of 1 μl cDNA per 15 μl reaction for each respective primer set using PCR reagents and Platinum Taq Polymerase (Invitrogen) as previously described. PCR was carried out in a Thermo Hybaid Multiblock System (Thermo-Hybaid) with the following cycling conditions: 95°C/30 sec (1X); 95°C/30 sec, 58°C/45 sec, 72°C/30 sec (28–35X); 72°C/7 min. The primer sets for assessing Hsd17b3, Hsd17b1 and Actb mRNAs have been previously described (16). All samples were electrophoresed on an agarose gel (2% NuSieve/0.7% SeaKem, BMA Bioproducts, Rockland, ME) in 1X Trisborate-EDTA buffer, stained with ethidium bromide and photographed using an EC3 Imaging System (UVP, Upland, CA).
For real-time RT-PCR assessment of gene expression, Applied Biosystems Primer Express (Applied Biosystems, Foster City, CA) software was used to select primers specific for the amplification of murine Hsd17b1, Hsd17b3, Cyp17a1 and Cyp19a1 cDNAs (Table I). All primer sets were a) designed to lie in separate exons to avoid erroneous amplification of contaminating genomic DNA, b) confirmed to amplify a single product via dissociation analysis and gel electrophoresis, and c) confirmed to amplify the expected sequences by restriction enzyme mapping of the amplified product. Each sample was assayed in duplicate using the equivalent of 0.01 μl cDNA (prepared as described above), 10 pmoles of each primer, and 1X SYBR Green Master Mix (Applied Biosystems) in a total reaction volume of 25 μl. For normalization purposes, an identical set of reactions were prepared using primers specific for ribosomal 18S RNA (Rn18s) (Table I). Amplification was carried out in an ABI PRISM 7700 Sequence Detection System (Applied Biosystems) as follows: 50°C/2 min, 95°C/10 min (1X); 95°C/15 sec, 60°C/30 sec (40X). Quantitative differences in the cDNA target between samples were determined using the mathematical model of Pfaffl (20) in which an expression ratio was determined for each sample by calculating (Etarget) ΔCt(target)/(ERn18s) ΔCt(Rn18s), where E is the efficiency of the primer set and ΔCt = Ct(normalization cDNA)-Ct(experimental cDNA). The amplification efficiency of each primer set was calculated from the slope of a standard amplification curve of log μl cDNA/reaction vs. Ct value over at least 4 orders of magnitude (E = 10−(1/slope)); Hsd17b3 primers, E = 1.97 (vs. testis cDNA); Hsd17b1 primers, E = 2.02 (vs. ovary cDNA); Cyp17a1 primers, E = 2.16 (vs. ovary cDNA); Cyp19a1 primers, E = 2.13 (vs. ovary cDNA).
TABLE I.
Primers used for RT-PCR
| Gene | Accession # | Amplifed Sequences (bp) | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|---|
| Cyp17a1 | NM 007809 | 1356–1436 | GATCGGTTTATGCCTGAGCG | TCCGAAGGGCAAATAACTGG |
| Cyp19a1 | D00659 | 135–210 | TGATCATGGGCCTCCTTCTC | CCCAGACAGTAGCCAGGACCT |
| Hsd17b1 | NM 010475 | 689–765 | CTGCGTGGTTATGAGCAAGC | CGCATTGCAGTCAAGAAGAGC |
| Hsd17b3 | NM 008291 | 560–635 | ATGGAGTCAAGGAGGAAAGGC | GGCTGTAAAGAGGCCAGGG |
| Rn18s | X56974 | 1271–1351 | GAAACTGCGAATGGCTCATTAA | GAATTACCACAGTTATCCAAGTAGGA |
In situ hybridization
Phylogeny Inc. (Columbus, OH) conducted all in situ hybridization experiments under paid contract. Gonadal tissues were collected from adult wild type and αERKO animals immediately upon death, fixed overnight in Phylogeny’s proprietary fixative, then dehydrated and infiltrated with paraffin. Serial sections of 5–8 μm were mounted on gelatin-coated slides, deparaffinized in xylene, and rehydrated through a series of graded ethanol baths in 1X phosphate-buffered saline, pH 7.5 (PBS). The sections were treated with proteinase K, then triethanolamine/acetic anhydride, washed and dehydrated. Radiolabeled sense and anti-sense riboprobes for Hsd17b3 mRNA (bp 363–729 of U66827) were generated from a previously described cDNA clone (16) using Maxiscript reagents (Ambion) and 35S-αUTP (>1000 Ci/mmol; Amersham) according to the manufacturer’s protocol. Sections were hybridized overnight at 55°C in a solution of 50% deionized formamide, 0.3 M NaCl, 20 mM Tris-HCl pH 7.4, 5 mM EDTA, 10 nM NaPO4, 10% dextran sulfate, 1X Denhardt’s, 50 μg/ml total yeast RNA, and radiolabeled riboprobe at 5–8 x 105 cpm/μl. The sections were then subjected to stringent washing at 65°C in a solution of 50% formamide, 2X sodium chloride/sodium citrate (SSC) buffer with 10 mM dithiothreitol; washed in 1X PBS; then treated with 20 μg/ml ribonuclease A at 37°C for 30 min. The slides were washed in 2X SSC, then 0.1X SSC for 10 min per wash at 37°C; dehydrated in a series of graded ethanol baths; dipped in Kodak NTB-2 nuclear track emulsion; exposed for 21 days in light-tight boxes with desiccant at 4°C. All slides were then developed in Kodak D-19, lightly counterstained with hematoxylin and eosin (H & E), and viewed and photographed under both light- and dark-field microscopy.
In vitro acute steroidogenic assays
The acute steroidogenic capacity of dispersed ovarian cells from wild type, αERKO and LH-CTP animals was assessed in vitro using a method first described by Magoffin and Erickson (11, 21), with some modifications. In brief, ovaries were collected from 5–6 untreated or hCG-treated adult females of each genotype and pooled according to genotype and in vivo treatment in Medium 199 with 100 mg/l L-glutamine and 25 mM HEPES (M199; Invitrogen) on ice. The pooled ovaries were minced into 8–10 pieces each and the fragments gently washed in M199. The ovarian fragments were incubated in M199 supplemented with 4 mg/ml collagenase (Sigma), 10 mg/ml deoxyribonuclease I (Sigma) and 10 mg/ml bovine serum albumin (Sigma) at 37ºC for 90 min in an ambient atmosphere. Several times during this incubation, the fragments were flushed through sterile pipettes with successively smaller orifices to obtain a preparation of dispersed cells. Each cell preparation was washed 3 times in M199 and the final cell pellet suspended in 0.16 ml M199 per pair of ovaries in the original pool.
Acute steroidogenic assays were conducted in sterile 12 x 75 polystyrene culture tubes (BD Falcon, Bedford, MA). Each assay consisted of 25 μl dispersed ovarian cells in a 500 μl final volume of M199 supplemented with 10 I.U./ml hCG or an equivalent volume of vehicle (0.85% saline), and/or a HSD17B3 inhibitor (DP3-3) or equivalent volume of vehicle (100% ethanol). The generation and characterization of DP3-3 (3β-((N-cyclohexylmethyl-N-cyclopropylcarbonyl)aminomethyl)-3α-hydroxy-5α-androstan-17-one)) has been previously described (22) as compound 213 and was provided for these studies by D.P. All assays were prepared at a minimum of duplicate per genotype, per in vivo treatment, per in vitro treatment. Immediately following preparation, all assays were incubated for 4 h in a 37ºC water bath in an ambient atmosphere. The cells were then pelletted by centrifugation at 8,000 x g for 5 min and the medium transferred to a fresh tube and stored at –70ºC for later analyses of androstenedione and testosterone content by radioimmunoassay (RIA). These experiments were repeated in 3 independent trials.
Hormone radioimmunoassays (RIA)
Plasma and media androgen levels were assessed using the Active Androstenedione RIA and Active Testosterone RIA kits (Diagnostic Systems Laboratories, Webster, TX) according to the manufacturer’s protocol. All plasma hormone assays were performed in duplicate (when sample volume allowed) on samples collected from individual animals. All media hormone assays were performed on individual experimental samples and always in duplicate. Final assay samples were quantified using an Apex Automation Gamma Counter (Micromedic Systems, Seattle, WA) and accompanying software. For the androstenedione RIA, the least detectable concentration was 0.03 ng/ l, the average intra-assay coefficient of variation was 2.5%, and the inter-assay coefficient of variation was 9.2%. For the testosterone RIA, the least detectable concentration was 0.08 ng/ l, the average intra-assay coefficient of variation was 3.0%, and the inter-assay coefficient of variation was 11.6%. RIAs on an equivalent volume of M199 supplemented with 10 I.U./ml hCG or an equivalent volume of vehicle (0.85% saline), and/or 10 μM HSD17B3 inhibitor (DP3-3) or equivalent volume of vehicle (100% ethanol) indicated that the androstenedione and testosterone content was below the level of detection for each steroid.
Transmission electron microscopy
Adult animals were administered a sufficient dose of pentobarbital to induce deep anesthetization and then promptly perfused (whole-body) with a modified Karnovsky’s fixative containing 2% paraformaldehyde and 2.5% glutaraldehyde, buffered to pH 7.4 in 0.1 M sodium cacodylate (Electron Microscopy Sciences, Hatfield, PA). Gonads were excised from the animals, placed whole in the above fixative for approximately 2 h, cut into 1–2 mm cubes and then stored in the above fixative at 4ºC until further processing. The tissues were then washed in 0.1 M sodium cacodylate, postfixed in 0.1 M sodium cacodylate-buffered 1% OsO4 (Electron Microscopy Sciences), rinsed in distilled water, dehydrated through a series of graded ethanol baths followed by propylene oxide, and embedded in Polybed epoxy resin (Polysciences, Warrington, PA). Ultrathin (90 nm) sections were prepared and stained on-grid with 5% uranyl acetate and Reynold’s lead citrate. All sections were examined in a FEI Tecnai 12 (Hillsboro, OR) transmission electron microscope operated at 80 kV and equipped with Microsoft Windows 2000 (Redmond, WA) and the MegaView III Soft Imaging System (Lakewood, CO). Several electron micrographs were taken for each sample to give an adequate sampling and representative overview.
Statistical analysis
All data were analyzed for statistical significance (P < 0.05) using JMP software (SAS Institute, Cary, NC). Data sets were first tested for homoscedasticity of variance using the Levene’s test and if failed were log-transformed prior to further statistical analysis. All data sets were then evaluated by a one-way ANOVA followed by the Tukey-Kramer HSD post-hoc test when applicable.
The testosterone/androstenedione (T/A4) ratios were calculated for each individual animal or in vitro assay sample using the formula first described by Weibe and Morris (23) to evaluate the endocrine profiles of women diagnosed with ovarian androgen excess. This value more effectively compares the degree of elevated testosterone above normal (vs. a simple ratio) and employs the following formula: (plasma T ÷ 95% upper limit normal T)/(plasma A4 ÷ 95% upper limit normal A4). For determining the in vivo T/A4 ratios (Fig. 1), the individual plasma levels from a group of adult wild type female mice (n = 24) were used to determine the 95% upper limit for T and A4 as 0.10 ng/ml and 0.25 ng/ml, respectively. For determining the in vitro T/A4 ratios (Figs. 5 and 6), the acute in vitro steroid synthesis by dispersed cells from untreated wild type ovaries (n = 3 replicates) were used to determine the 95% upper limit for T and A4 as 0.20 ng/ml and 0.60 ng/ml, respectively.
FIG. 1.
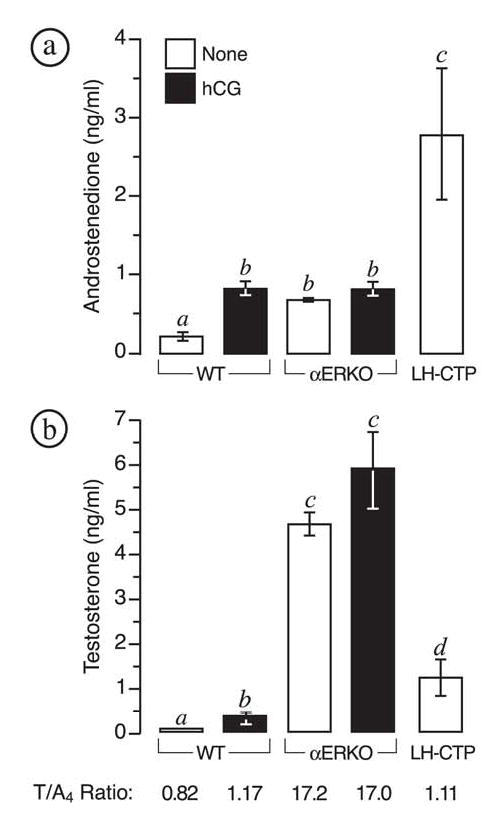
αERKO females exhibit elevated plasma androgens. Shown are the average (± SEM) plasma levels for (a) androstenedione and (b) testosterone in untreated adult wild type, αERKO and LH-CTP females; and wild type (WT) and αERKO females treated twice daily with 5 I.U. human choriogonadotropin (hCG) for 5 consecutive days (see Materials and Methods). Sample numbers (n) for each group were: wild type, 24; wild type + hCG, 22; αERKO, 28; αERKO + hCG, 8; LH-CTP, 8. Bars that do not share the same letter are significantly different (P < 0.05). (bottom) Shown is the average testosterone to androstenedione (T/A4) ratio for each genotype and treatment group as calculated using the formula of Weibe and Morris (23).
RESULTS
αERKO females exhibit abnormally elevated plasma androgens
Adult αERKO females possess levels of circulating androstenedione (A4) and testosterone (T) that are more than 3- and 50-fold that of wild type females, respectively (Fig. 1). Wild type females treated with hCG twice daily for 3 consecutive days exhibited significantly increased plasma androgen levels, but only androstenedione and not testosterone approached the levels characteristic of αERKO females (Fig. 1). Similar treatment of αERKO females had no additive effect on plasma androgen levels. Female LH-CTP mice, a transgenic line that possess chronically elevated LH levels that are more comparable to those of αERKO females (16, 24), also exhibited significantly increased plasma androstenedione levels relative to wild type and even αERKO females, but much lower testosterone levels compared to the latter genotype (Fig. 1). Therefore, αERKO females largely differ from other models of ovarian LH-hyperstimulation in their unique capacity to produce much greater levels of testosterone. This male-like capacity of αERKO ovaries to synthesize testosterone is especially revealed when T/A4 ratios among the different genotypes are compared (Fig. 1). Wild type females exhibited a T/A4 ratio of 0.82, which was significantly increased to 1.17 (P < 0.05) following 3 days of hCG treatment. Remarkably, LH-CTP females exhibited a similar T/A4 ratio (1.11) despite a life-long elevation in circulating LH. In contrast, αERKO females exhibited a striking T/A4 ratio of 17.2 and showed no further increase following hCG treatment (17.0). These data indicate that chronic, excessive LH stimulation alone does not lead to male-like efficiency of testosterone synthesis in murine ovaries; but instead indicate that this phenotype is a primary effect of the loss of intraovarian ERα-mediated actions.
αERKO ovaries exhibit testis-like expression and regulation of the Hsd17b3 gene
Comparative Northern blot analyses of gonadal RNAs indicated that αERKO ovaries repeatedly possess levels of Hsd17b3 expression that are equal to or even greater than those observed in the testes of normal adult males, while levels in wild type ovaries were below the level of detection (Fig. 2a). These data were confirmed by quantitative real-time RT-PCR, which indicated the level of Hsd17b3 expression in αERKO ovaries was 2-fold that of adult testes (Fig. 2c). Similar analyses of LH-CTP ovaries also indicated a lack of Hsd17b3 expression but increased Cyp17a1 expression (Figs. 2b and c), the latter being a known ovarian marker of LH stimulation. αERKO ovaries also exhibited increased levels of Cyp17a1 expression that were 6- and 3-fold higher than those of wild type and LH-CTP ovaries, respectively (Fig. 2c).
FIG. 2.
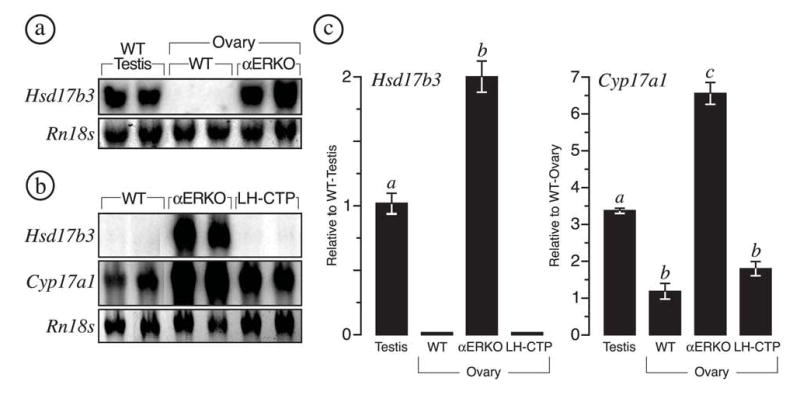
Ectopic Hsd17b3 expression is unique to αERKO ovaries. (a) Northern blot analysis for Hsd17b3 transcripts in adult wild type (WT) testes, WT ovaries, and αERKO ovaries. The level of Hsd17b3 expression in adult αERKO ovaries is comparable to that of WT testes; but undetectable in WT ovaries. (b) Northern blot analysis for Hsd17b3 transcripts in adult WT, αERKO and LH-CTP ovaries indicates that ectopic expression of Hsd17b3 does not occur in the ovaries of hypergonadotropic LH-CTP females. The same Northern blot was reprobed for Cyp17a1 transcripts, a known LH-regulated gene in the ovary. All Northern blots were probed for ribosomal Rn18s (18S rRNA) to demonstrate equal loading of total RNA among samples. (c) Data from real-time RT-PCR (average ± SEM) for Hsd17b3 and Cyp17a1 expression in adult WT testes, WT ovaries, αERKO ovaries, and LH-CTP ovaries. Hsd17b3 expression in αERKO ovaries is 2-fold that of WT testes. αERKO ovaries also exhibit significantly increased Cyp17a1 expression relative to WT testes and ovaries. Sample numbers (n) for each group were as follows: WT testes, 3 males; wild type ovaries, 5 females; αERKO, 3 females; LH-CTP, 3 females. Bars that do not share the same letter are significantly different (P < 0.05).
Because LH is the principle hormone for the induction and maintenance of Hsd17b3 expression in Leydig cells of adult testes (25–28), we sought to determine if Hsd17b3 expression in αERKO ovaries exhibits a similar, testis-like pattern of gonadotropin regulation. These experiments were conducted by treating hypogonadal (hpg) wild type (wild typehpg) and αERKO (αERKOhpg) females, which lack circulating endogenous gonadotropins, with a continuous infusion of exogenous hFSH or hLH for a period of one week. Wild typehpg males were similarly treated to provide data on the expected response in testes. As shown in Fig. 3, hLH increased Hsd17b3 expression almost 40-fold in the testes of wild typehpg males, while the response to hFSH was measurable but conspicuously lower (< 5-fold). Surprisingly, αERKOhpg ovaries exhibited a similar response to hLH by increasing Hsd17b3 expression over 25-fold, while wild typehpg ovaries exhibited a measurable but notably less robust increase in Hsd17b3 expression (Fig. 3). This difference in the induction of Hsd17b3 expression in the gonads of wild type and αERKO females is not due to a disparity in the effectiveness of hLH treatment since Cyp17a1, a known LH-induced gene, was increased 23- and 80-fold in each genotype, respectively (data not shown). hFSH treatment had no effect on Hsd17b3 expression in the ovaries of either genotype and only a minimal effect in the testes. However, the effectiveness of hFSH treatment in the ovaries of wild type and αERKO females was illustrated by induction of Hsd17b1 (Fig. 3), a known FSH regulated gene in the ovary (29, 30). FSH induced Hsd17b1 expression 4-fold in both wild typehpg and αERKOhpg ovaries, while LH had minimal effect (Fig. 3). Another known FSH-induced gene in the ovary, Cyp19a1, was also increased over 200-fold in both wild type and αERKO ovaries (data not shown). Neither gonadotropin induced Hsd17b1 expression in the testes of wild typehpg males, demonstrating that this gene is distinctly expressed in ovaries.
Ectopic Hsd17b3 expression occurs in the interstitial cells of αERKO ovaries
To gain insight into the functional compartment responsible for ectopic Hsd17b3 expression in αERKO ovaries, total RNA was prepared from ovaries that were first partitioned into fractions of enriched granulosa and stromal-interstitial cells. Northern blot analysis clearly indicated that Hsd17b3 mRNAs were concentrated in the stromal-interstitial compartment of αERKO ovaries with only nominal detection in the granulosa cell fraction, most likely due to cross-contamination by small amounts of stromal material (Fig. 4). The relative purity of the fractions was illustrated by reprobing the blot for Cyp19a1 mRNA,s which are generally localized to granulosa cells of large follicles (Fig. 4).
FIG. 4.
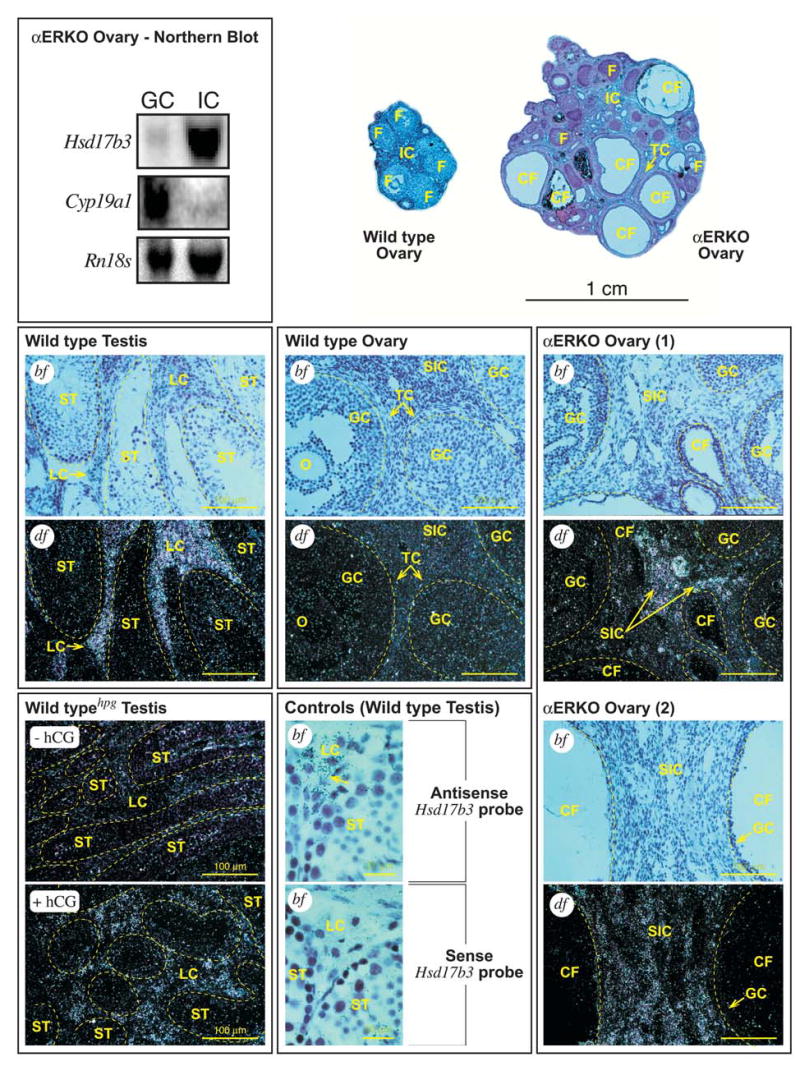
Northern blot and in situ hybridization (ISH) for Hsd17b3 mRNA in ovary and testes. (top, left) Northern blot analysis on total RNA from pooled αERKO ovaries that were fractionated into granulosa (GC) and stromal-interstitial (IC) cell compartments, demonstrating that Hsd17b3 expression in αERKO ovaries is localized to the stromal-interstitial compartment. The blot was stripped and reprobed for transcripts of a granulosa cell-specific gene (Cyp19a1) to indicate the cellular purity of each ovarian fraction. A photograph of ethidium-stained Rn18s (18S rRNA) bands demonstrates equal loading levels between lanes. (top, right) Cross-sections from representative adult wild type (WT) and αERKO ovaries photographed at low magnification. The WT ovary exhibits several healthy, large follicles (F) and a normal stromal-interstitial region (IC). In contrast, the αERKO ovary exhibits some smaller, relatively healthy follicles (F) but several enlarged, cystic follicles (CF). Also indicated in the αERKO ovary is a region of hypertrophied thecal cells (TC). (middle, bottom) Shown are representative photomicrographs (bf, bright field photomicrograph; df, dark-field photomicrograph) of tissue sections from wild type testes, wild type ovaries and αERKO ovaries (as labeled) that were subjected to ISH for Hsd17b3 mRNAs. [Wild type Testis] Photomicrographs of serial sections from a representative adult wild type testis show specific hybridization of the Hsd17b3 probe to the Leydig cells (LC) only, while hybridization levels among cells within the seminiferous tubules (ST), as indicated by dashed border, are at or below background. [Wild type Ovary] Photomicrographs of serial sections from a representative wild type ovary indicate no specific hybridization of the Hsd17b3 probe in the oocyte (O), granulosa (GC), secondary interstitial (SIC), thecal (TC) cells or any other cell type. Follicles are indicated by dashed border. [αERKO Ovary] Photomicrographs of a pair of serial sections from two representative αERKO ovaries indicates specific hybridization of the Hsd17b3 probe to the secondary interstitial cells (SIC) of the ovary, while hybridization levels among the granulosa cells of normal and cystic follicles (CF) are at or below background. Follicles are indicated by dashed border. [Wild typehpg Testis] Dark-field photomicrographs of testes sections from WThpg males following treatment with either vehicle (-hCG) or human choriogonadotropin (+hCG) to induce Hsd17b3 expression in the Leydig cells (see Materials and Methods). The untreated WThpg testis exhibits little detectable Hsd17b3 mRNA whereas the testis of the hCG-treated WThpg male exhibits significant induction of Hsd17b3 expression that is localized to the interstitial areas where Leydig cells (LC) are found. Seminiferous tubules are indicated by dashed border. [Controls - Wild type Testis] Photomicrographs of serial sections from a WT testis probed with either the Hsd17b3 sense (negative control) or anti-sense (positive control) probes to illustrate specific hybridization of the anti-sense probe only to the Leydig cells (LC), as indicated by the numerous silver grains (arrow).
The above findings were further supported by in situ hybridization analyses of adult αERKO ovaries. As shown in Fig. 4, Hsd17b3 mRNAs were distinctly localized to the stromal-interstitial regions of αERKO ovaries, while hybridization levels were below background in the granulosa cells of follicles of all sizes. More precisely, Hsd17b3 expression in αERKO ovaries was concentrated in the secondary interstitial (SI) cells, which populate the areas between the follicles and medullary portions of the ovary. SI cells are thought to have once been thecal interstitial (TI) cells, but following follicle atresia they become no longer associated with growing follicles and populate the ovarian stroma (11). Because growing follicles in αERKO ovaries characteristically exhibit poorly organized layers of TI cells, whether Hsd17b3 expression was distinct to the SI or shared between the two interstitial cell types was difficult to discern. No substantial hybridization of the Hsd17b3 probe was observed among any cell types in wild type ovaries (Fig. 4). As expected, parallel experiments in testes from adult wild type males indicated significant Hsd17b3 expression that was distinctly localized to Leydig cells (Fig. 4). Furthermore, hCG-mediated induction of Hsd17b3 expression in the testis of an adult wild typehpg male was again specifically localized to the interstitial (extratubuler) compartments of the testis where Leydig cells are located.
Ectopic HSD17B3 activity mediates testosterone synthesis in αERKO ovaries
Adult wild type and αERKO females were first treated with vehicle or hCG for 3 days to stimulate ovarian androgen production in vivo. Dispersed ovarian cell preparations were then generated and their acute steroidogenic capacity was assessed during a 4 h in vitro incubation in the presence or absence of hCG and/or a HSD17B3 inhibitor (DP3-3). The results of these experiments are summarized in Fig. 5. As expected, αERKO ovarian cells produced significantly more androstenedione and testosterone compared to wild type cells, regardless of in vivo or in vitro hCG exposure (Fig. 5a). In vivo hCG treatments enhanced in vitro androstenedione and testosterone synthesis in both genotypes; however, only αERKO ovarian cells exhibited an additive increase in overall androgen synthesis during in vitro hCG exposure. More importantly, these experiments provided two critical findings. First, consistent with the in vivo phenotype, testosterone was clearly the predominant androgen produced by αERKO ovarian cells in vitro, yielding an average T/A4 ratio of 3.32 ± 0.5 vs. 0.47 ± 0.1 in wild type cells (untreated both in vivo and in vitro). Secondly, inclusion of the HSD17B3 specific inhibitor (DP3-3) consistently reduced testosterone synthesis by 50-60% in αERKO ovarian cells and decreased the average T/A4 ratio to 0.73 ± 0.2, providing strong evidence that testosterone synthesis in αERKO ovaries is largely due to ectopic HSD17B3 activity. In contrast, the minimal but detectable testosterone synthesis by wild type cells was not affected by the HSD17B3 inhibitor (T/A4 ratio = 0.45 ± 0.02), indicating that it is likely mediated by the androgenic activity of rodent HSD17B1, which is innate to ovarian granulosa cells. A representative dose-response curve for HSD17B3 inhibition of testosterone synthesis by αERKO ovarian cells in vitro is shown in Fig. 5b.
Similar experiments were conducted to compare the steroidogenic capacity of dispersed ovarian cells from LH-CTP and αERKO females since these two models are more comparable in terms of chronic LH hyperstimulation of the ovary. Acute in vitro steroidogenic assays on dispersed cells from LH-CTP ovaries indicated substantial androstenedione production that was 2-fold that observed in αERKO ovarian cells (Fig. 6), consistent with their in vivo androgen profile (Fig. 1). Only when αERKO females were treated with exogenous hCG prior to tissue collection did the in vitro level of androstenedione synthesis become comparable to that of LH-CTP cells. This overabundance of androstenedione in LH-CTP cells is indicative of the increased Cyp17a1 expression shown earlier (Fig. 2). Still, even the highest level of in vitro androstenedione synthesis achieved in LH-CTP cells did not provide for αERKO-like levels of testosterone production, which were 3-fold higher in the latter genotype. Furthermore, the HSD17B3 inhibitor had no measurable effect on testosterone synthesis in LH-CTP cells, indicating it is likely mediated by the androgenic activity of HSD17B1.
αERKO ovaries harbor stromal Leydig-like cells
To determine if the interstitial portions of αERKO ovaries possessed Leydig-like ultrastructural features, we employed transmission electron microscopy (TEM) to compare normal Leydig cells of mouse testes to stromal-interstitial cells of adult αERKO ovaries. The typical whorl-arranged SER that is innate to Leydig cells of mouse testes (31) is illustrated in Fig. 7(a and b). The Leydig cell shown possesses multiple whorls of SER, one of which surrounds a lipid droplet. Also prominent are random tubular SER and numerous mitochondria, the latter possessing the tubular cristae that are characteristic of steroidogenic cells (Fig. 7b) (31). Remarkably, a parallel survey of stromal-interstitial regions of multiple αERKO ovaries identified cells that possessed similarly arranged SER, clearly organized as whorls and interspersed with large lipid droplets and tubular SER (Fig. 7c-e). The whorl-like SERs found in αERKO ovarian stroma-interstitial cells usually occupied a smaller intracellular area and were less structured relative to that in testicular Leydig cells (Fig. 7c and d) but were clearly Leydig-like in appearance. In addition, the whorled SERs observed among αERKO ovarian stromal-interstitial cells were often vesiculated (Fig. 7f), remarkably similar to that which occurs during the natural involution of Leydig cells in fetal or aged testes. These vesiculated structures in αERKO ovarian stromal-interstitial cells are likely sites of lipid degradation and may account for the substantial intracellular prevalence of lipofuscin observed in H & E stained tissue sections (Fig. 7h), which is also a common cytoplasmic component of Leydig cells in multiple species (31). Furthermore, the cells possessing Leydig-like SER and lipofuscin were limited to the interstitial regions in αERKO ovaries, the normal site of secondary interstitial cells and correlating with the areas of Hsd17b3 expression indicated by in situ hybridization. No similar structures were found in wild type ovaries (data not shown).
FIG. 7.
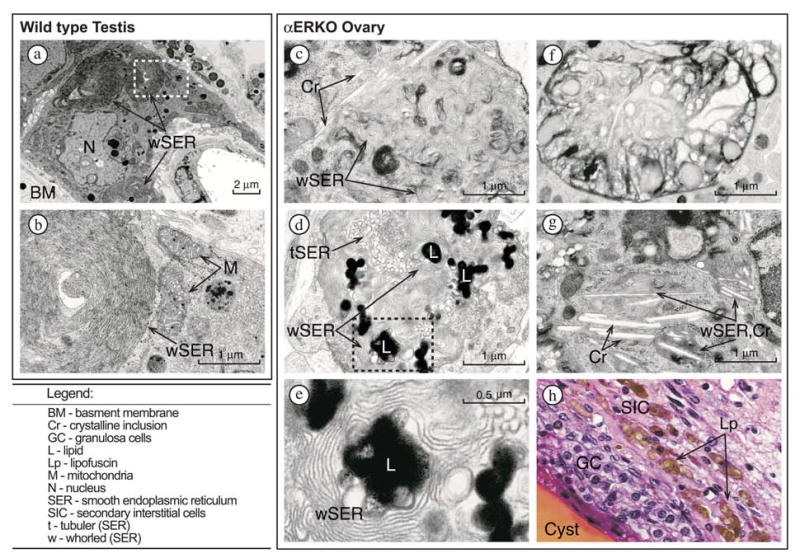
Transmission electron microscopy indicates secondary interstitial cells with Leydig-like ultrastructural features in αERKO ovaries. [Wild type Testes] (a) A representative Leydig cell in a wild type testis possesses 3 clusters of whorled smooth endoplasmic reticulum (wSER). (b) A higher magnification of panel a (indicated by outlined area) illustrates a wSER and two juxtaposed mitochondria (M) with tubular cristae that are characteristic of testicular Leydig cells. [αERKO Ovary] (c and d) Representative examples of Leydig-like wSER found in the secondary-thecal interstitial cells of αERKO ovaries. Numerous electron-dense lipid droplets (L) are surrounded by the wSER; tubuler SER (tSER) that is typical of ovarian steroidogenic cells is also present. (e) Shown is a higher magnification of panel d (indicated by outlined area). (f) Shown is an example of vesiculated wSER in a αERKO ovary. (g) A secondary-interstitial cell in a αERKO ovary containing wSER with multiple large crystalline (Cr) inclusions throughout the cytoplasm. (h) Photomicrograph of the follicle wall of a cystic follicle in a αERKO ovary section stained with H&E, illustrating the considerable accumulation of lipofuscin (brown-pigmented material) in the cytoplasmic vacuoles among the secondary-interstitial cells.
Along with the above findings, TEM revealed an additional noteworthy characteristic of αERKO ovarian secondary interstitial cells. As illustrated in Fig. 7(c and g), numerous crystalline-like cytoplasmic inclusions were found to repeatedly coexist in cells that harbored whorled, Leydig-like SER. These inclusions were long, cylindrical bodies of 0.1-2 μm in length that were often present in great numbers and almost always associated with whorled SER. Crystalline cytoplasmic inclusions are a common feature of normal Leydig cells in the testes of several species and are considered a definitive attribute of human Leydig cells and Leydig cell tumors, within which they are referred to as crystals of Reinke (31). However, there are no reports of similar cytoplasmic crystalline inclusions in Leydig cells or ovarian secondary interstitial cells of mice.
DISCUSSION
The collective findings from studies as early as the mid 1970’s impart a convincing argument for an inhibitory role of estrogens on Leydig cell development, differentiation and function (as reviewed in (14). For example, estrogens are documented to inhibit testosterone synthesis in adult rodent Leydig cells (14) as well as disrupt Leydig cell proliferation and differentiation in fetal and neonatal testes (32). Furthermore, estradiol inhibits the natural regeneration of Leydig cells that occurs in rodent testes following their destruction by acute exposure to ethane dimethylsulfonate (EDS), a Leydig cell-specific toxicant (33). In addition, the precisely timed periods of substantial Leydig cell proliferation and differentiation during testis development correlate inversely with periods of low endogenous estradiol levels (15). Remarkably, the concept that these findings might reveal a parallel, physiological role for estradiol-mediated suppression of Leydig cell differentiation and testosterone synthesis in ovaries has received little attention. Herein, we provide definitive evidence that estradiol is principally involved in preventing the development of functional Leydig-like cells in the stromal-interstitial portions of the mouse ovary and that these actions are exclusively mediated by ERα.
Our assertion that αERKO ovaries inappropriately possess functional Leydig-like cells is based on their exhibiting traits that are regarded as exclusive to testicular Leydig cells, including a) substantial Hsd17b3 expression and enzymatic activity, b) whorl-like SER and c) crystalline-like structures comparable to crystals of Reinke. Furthermore, the ectopic Hsd17b3 expression in αERKO ovaries is LH-dependent and thus exhibits a pattern of gonadotropin regulation that is comparable to that of mature testicular Leydig cells. Similar Hsd17b3 expression and cells possessing Leydig-like features are reportedly present in the ovaries of mice lacking endogenous estradiol due to targeted disruption of the Cyp19a1 gene; and the former phenotype is abolished by exogenous estradiol treatments (17). However, because estradiol treatment of CYP19A1-null females also results in normalization of the heightened LH levels that are present in these animals (17), these experiments do not discern whether the loss of ovarian Hsd17b3 expression is due to activation of intraovarian ERα or decreased gonadotropin stimulation. In the current study, we provide definitive evidence that ectopic Hsd17b3 expression in αERKO females is due solely to the loss of ERα-mediated actions within the ovary and simply requires LH stimulation to manifest. For example, the ovaries of transgenic LH-CTP female mice categorically lack Hsd17b3 expression and activity (18) and Fig. 2 and 6 herein) despite being chronically exposed to substantially high levels of circulating LH (34, 35). The ovaries of estrogen receptor (ERβ)-null female mice also lack ectopic Hsd17b3 expression (16) even when harboring the LH-CTP transgene to increase circulating LH levels (18). Likewise, reports of transgenic mice that constitutively express hCG, an LH analog, describe ovaries exhibiting interstitial-thecal cell hypertrophy but make no reference to Hsd17b3 expression or Leydig-like cells (36, 37). In contrast, Heikkilä et al. (38) recently reported that the ovaries of newborn Wnt4-null female mice exhibit ectopic induction of Hsd17b3 expression that is concurrent with a 8-fold reduction in ERα expression but no change in ERβ levels. All of these findings support our conclusion that ERα is critical to repressing the expression of this Leydig cell-specific steroidogenic gene in the ovary.
Evidence supporting the presences of substantial HSD17B3 enzymatic activities in αERKO ovaries was revealed by several findings. Foremost was the remarkably high T/A4 ratio in the plasma of adult αERKO females that is more comparable to that of normal male mice (39, 40) and adult men (41) as well as women clinically diagnosed with ovarian androgen excess (23). We have previously shown that increased androgen levels in αERKO females are at least partially due to hyperstimulation of the ovary by LH (16). Still, when wild type female mice were forced to possess a comparable hypergonadotropic condition via either daily injections with hCG, an LH analog, or transgenic methods, as achieved in LH-CTP mice, the resulting androgenic phenotype was markedly less severe and qualitatively different than that of αERKO females as androstenedione, and not testosterone, was the predominant steroid produced in the former models. Secondly, we conducted specific experiments to discern the individual contributions of type 1 and type 3 activities to the overall testosterone load in αERKO females. This was necessary because the rodent ortholog of HSD17B1, which is normally expressed in the ovary and mediates the reduction of estrone to estradiol (2, 42, 43), is equally efficient at reducing androstenedione to testosterone (44, 45). Furthermore, Hsd17b1 expression is increased almost 2-fold above normal in αERKO ovaries (16). By employing a method first described by Magoffin and Erickson that allows for the evaluation of the acute steroidogenic capacity of dispersed ovarian cells in vitro (21, 46) with the use of a specific inhibitor of HSD17B3 enzymatic activities (22), we were able to successfully discriminate type 1 and type 3 mediated HSD17B testosterone synthesis in dispersed wild type and αERKO ovarian cells. The resulting data strongly indicated that the male-like capacity for testosterone synthesis in αERKO ovaries is due predominantly to the presence of substantial HSD17B3 activities, whereas the minimal level of testosterone synthesis observed in hCG-treated wild type or LH-CTP ovaries is due to the androgenic activities of HSD17B1 that occur in the presence of increased androstenedione. Testosterone synthesis in αERKO females is undoubtedly further promoted by a parallel increase in CYP17A1 activities that provide for ample synthesis of androstenedione, the substrate for HSD17B3.
The mechanisms by which ERα actions suppress the testicular pathway of testosterone synthesis in the ovary are unclear. Ligand-activated ERα may act in a dynamic manner within thecal/secondary interstitial cells to persistently inhibit Hsd17b3 expression. ERα is indeed amply expressed in both testicular Leydig cells (47, 48) and ovarian thecal/secondary interstitial cells (49–51). Furthermore, estrogens are known to inhibit testosterone synthesis in both thecal and Leydig cells in vitro, although most evidence indicates that the upstream steroidogenic enzyme, CYP17A1, is the primary target (11, 14, 52). Indeed, CYP17A1 expression and activity are also increased in the ovaries (Fig. 2, (16), in vitro cultured follicles (F. Taniguchi, J.F. Couse and K.S. Korach, manuscript in preparation) and testes (53) of αERKO but not βERKO mice. However, ours is the first study to demonstrate that ERα represses ovarian testosterone synthesis via robust negative modulation of Hsd17b3 expression as well. Interestingly, a recent report that αERKO males exhibit a 2-fold increase in Hsd17b3 expression and type 3 activity (53) indicates that ERα may play an analogous role in testes.
In contrast to the above postulated dynamic mechanism for ERα-mediated inhibition of Hsd17b3 expression and testosterone synthesis in the ovary is a potential organizational role by which ERα might repress the development of a Leydig cell phenotype among the secondary/thecal interstitial cells. Support for such an organizational role for ER action during gonadal differentiation comes from multiple descriptions of permanently disrupted Leydig cell function in adult rodents following developmental exposure to estrogens, as well as estrogen-mediated inhibition of Leydig cell regeneration and proliferation in EDS-exposed testis (14). Herein, we employed TEM to produce evidence of Leydig-like cells in αERKO ovaries. The “single most identifying characteristic of the Leydig cell” is their unique, and often species-specific arrangements of smooth endoplasmic reticulum (SER) (31). The Leydig cells of adult mouse testes, as well as other species, characteristically exhibit a portion of SER that is arranged as “whorls” often surrounding a lysosome or lipid droplet (31 and references therein). This swirled form of SER is continuous with the tubular form and may comprise up to 7% of the total SER within a single Leydig cell (31). In the current study, we found that αER ovaries possess secondary-interstitial cells that exhibit similar “whorl”-arranged SER as well as other Leydig cell features, including crystalline-like cytoplasmic inclusions and an accumulation of lipofuscin. Furthermore, the αERKO ovarian cells exhibiting these Leydig-like features were located in the same ovarian regions that possessed significant levels of Hsd17b3 expression as indicated by in situ hybridization. These findings raise the prospect that a loss of ERα actions during secondary/thecal-interstitial cell differentiation in the ovary may allow for the inappropriate development of cells possessing a Leydig-like phenotype. The capacity of adult hypogonadal αERKO (αERKOhpg) ovaries to rapidly acquire Hsd17b3 expression following acute exogenous hLH treatment suggests that the Leydig-like cells may develop in an environment void of gonadotropins and are present as early as birth or prior to puberty.
Our finding that the Leydig-like cells in adult αERKO ovaries were located in the stroma and often bordered by secondary/thecal-interstitial cells distinguishes them from hilar interstitial (HI) cells, which are natural to the ovary and considered structurally and functionally similar to testicular Leydig cells (11). HI cells are normally limited to the connective tissues in the hilus of the ovary (11); whereas, the Leydig-like cells observed in αERKO ovaries are located in the stroma, surrounded by thecal/secondary interstitial cells and usually in close proximity to a follicle. Admittedly, poor organization of the medullary regions is characteristic of adult αERKO ovaries (54, 55) and may have altered the natural position of HI cells. Still, there is no evidence of Hsd17b3 expression in the HI cells of normal ovaries (8, 11), providing further evidence that the Leydig-like cells in αERKO ovaries are not of hilar origin.
The stromal location of the Leydig-like cells in αERKO ovaries in conjunction with their close proximity to hypertrophied secondary/thecal-interstitial cells suggests that they may have once been part of the latter cell population but have since acquired a male-like phenotype via transdifferentiation. This phenomenon is thought to underlie the formation of stromal-Leydig cell tumors and non-hilar, pure Leydig cell tumors in women (56, 57). Although extremely rare, these ovarian tumors are invariably androgenic and often bordered by hypertrophied ovarian stroma (56–59). Furthermore, there are clinical reports of HSD17B3-like activity in the ovaries of women suffering from ovarian androgen excess (60, 61), androgenic HSD17B immunoreactivity in a pure Leydig cell tumor (62), and HSD17B3 transcripts in a Sertoli-Leydig cell tumor and surrounding luteinized thecal tissue (63). In contrast, ovarian tissue and cell lines from women diagnosed with polycystic ovarian syndrome have been found to lack detectable HSD17B3 expression (64–66), indicating that this malady is not likely related to the ERα-null phenotype described here.
Thecal cell androgen synthesis is critical to ovarian function as androgens serve as both important hormonal signals during granulosa cell differentiation in the early stages of folliculogenesis and later, as substrates for estradiol synthesis in mature preovulatory follicles (67). However, when androgen levels and activity are allowed to surpass that required to support estradiol synthesis during the latter stages of folliculogenesis, the follicle invariably becomes atretic (68, 69). This condition is likely exacerbated if testosterone, rather than androstenedione, is the predominant androgen present, as the former steroid is a more potent androgen receptor agonist. Therefore, stringent regulation of the quantity and nature of thecal/secondary interstitial cell androgen synthesis is critical to the generation of healthy follicles capable of ovulation and fertilization. Our data provide strong evidence that ligand-dependent ERα actions are fundamental to this physiology of thecal/secondary interstitial cells by repressing their acquisition of HSD17B3 activities and perhaps their differentiation to a Leydig-like phenotype. Our description of ectopic Hsd17b3 expression and Leydig-like cells in the ovaries of ERα-null female mice represents the first data to support an integral intraovarian role for ERα. Further studies are required to discern whether ERα operates via activational or organizational mechanisms to repress the acquisition of a testis-like phenotype in the secondary/thecal interstitial cells of the ovary. A similar phenotype of granulosa to Sertoli cell transdifferentiation occurs in the ovaries of compound ER-null (ERα-null; ERβ-null) female mice (70, 71). From these collective data, we may infer that ER-mediated estradiol signaling is critical to the development and/or maintenance of the female phenotype in the endocrine somatic cell types of the ovary.
Acknowledgments
We are grateful to numerous colleagues who have supported our efforts over the course of these studies, including Linwood Koonce and Vickie Walker for animal handling and breeding, Dr. John H. Nilson for providing LH-CTP breeder mice, Ralph Wilson for generously allowing the use of his gamma counter, Dr. Abraham Nyska for consultation on pathology issues, and Drs. E. Mitch Eddy and William Schrader for their careful review of the manuscript. This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS).
Footnotes
Disclosure of Potential Conflicts of Interest: J.F.C., M.M.Y., K.F.R., J.A.J., D.P. and K.S.K. have nothing to declare.
References
- 1.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25:947–70. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- 2.Penning TM. Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr Rev. 1997;18:281–305. doi: 10.1210/edrv.18.3.0302. [DOI] [PubMed] [Google Scholar]
- 3.Mindnich R, Moller G, Adamski J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2004;218:7–20. doi: 10.1016/j.mce.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Sha JA, Dudley K, Rajapaksha WR, O'Shaughnessy PJ. Sequence of mouse 17beta-hydroxysteroid dehydrogenase type 3 cDNA and tissue distribution of the type 1 and type 3 isoform mRNAs. J Steroid Biochem Mol Biol. 1997;60:19–24. doi: 10.1016/s0960-0760(96)00165-3. [DOI] [PubMed] [Google Scholar]
- 5.Tsai-Morris CH, Khanum A, Tang PZ, Dufau ML. The rat 17β-hydroxysteroid dehydrogenase type III: molecular cloning and gonadotropin regulation. Endocrinology. 1999;140:3534–42. doi: 10.1210/endo.140.8.6944. [DOI] [PubMed] [Google Scholar]
- 6.Baker PJ, Sha JH, O'Shaughnessy PJ. Localisation and regulation of 17β-hydroxysteroid dehydrogenase type 3 mRNA during development in the mouse testis. Mol Cell Endocrinol. 1997;133:127–33. doi: 10.1016/s0303-7207(97)00159-7. [DOI] [PubMed] [Google Scholar]
- 7.Mustonen MV, Poutanen MH, Isomaa VV, Vihko PT, Vihko RK. Cloning of mouse 17β-hydroxysteroid dehydrogenase type 2, and analysing expression of the mRNAs for types 1, 2, 3, 4 and 5 in mouse embryos and adult tissues. Biochem J. 1997;325:199–205. doi: 10.1042/bj3250199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strauss JF, III, Hsueh AJW. Ovarian hormone synthesis. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Fourth Edition, 4th Edition ed. W.B. Saunders Company; Philadelphia: 2001. pp. 2043–2052. [Google Scholar]
- 9.Huhtaniemi I, Toppari J. Endocrine, paracrine and autocrine regulation of testicular steroidogenesis. Adv Exp Med Biol. 1995;377:33–54. doi: 10.1007/978-1-4899-0952-7_3. [DOI] [PubMed] [Google Scholar]
- 10.Sheffield JW, O'Shaughnessy PJ. Testicular steroid metabolism during development in the normal and hypogonadal mouse. J Endocrinol. 1988;119:257–64. doi: 10.1677/joe.0.1190257. [DOI] [PubMed] [Google Scholar]
- 11.Erickson GF, Magoffin DA, Dyer CA, Hofeditz C. The ovarian androgen producing cells: a review of structure/function relationships. Endocr Rev. 1985;6:371–99. doi: 10.1210/edrv-6-3-371. [DOI] [PubMed] [Google Scholar]
- 12.Magoffin DA. The ovarian androgen-producing cells: a 2001 perspective. Rev Endocr Metab Disord. 2002;3:47–53. doi: 10.1023/a:1012700802220. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Word RA, Fesmire S, Carr BR, Rainey WE. Human ovarian expression of 17 beta-hydroxysteroid dehydrogenase types 1, 2, and 3. J Clin Endocrinol Metab. 1996;81:3594–8. doi: 10.1210/jcem.81.10.8855807. [DOI] [PubMed] [Google Scholar]
- 14.Abney TO. The potential roles of estrogens in regulating Leydig cell development and function: a review. Steroids. 1999;64:610–7. doi: 10.1016/s0039-128x(99)00041-0. [DOI] [PubMed] [Google Scholar]
- 15.Ge R-S, Shan L-X, Hardy MP. Pubertal development of leydig cells. In: Payne AH, Hardy MP, Russell LD, editors. The Leydig Cell. Cache River Press; Vienna, IL: 1996. pp. 159–173. [Google Scholar]
- 16.Couse JF, Yates MM, Walker VR, Korach KS. Characterization of the hypothalamic-pituitary-gonadal axis in estrogen receptor (ER) Null mice reveals hypergonadism and endocrine sex reversal in females lacking ERα but not ERβ. Mol Endocrinol. 2003;17:1039–53. doi: 10.1210/me.2002-0398. [DOI] [PubMed] [Google Scholar]
- 17.Britt KL, Stanton PG, Misso M, Simpson ER, Findlay JK. The effects of estrogen on the expression of genes underlying the differentiation of somatic cells in the murine gonad. Endocrinology. 2004;145:3950–60. doi: 10.1210/en.2003-1628. [DOI] [PubMed] [Google Scholar]
- 18.Couse JF, Yates MM, Sanford R, Nyska A, Nilson JH, Korach KS. Formation of cystic ovarian follicles associated with elevated luteinizing hormone requires estrogen receptor-β. Endocrinology. 2004;145:4693–702. doi: 10.1210/en.2004-0548. [DOI] [PubMed] [Google Scholar]
- 19.Mason AJ, Hayflick JS, Zoeller RT, Young WS, 3rd, Phillips HS, Nikolics K, Seeburg PH. A deletion truncating the gonadotropin-releasing hormone gene is responsible for hypogonadism in the hpg mouse. Science. 1986;234:1366–71. doi: 10.1126/science.3024317. [DOI] [PubMed] [Google Scholar]
- 20.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Magoffin DA, Erickson GF. Primary culture of differentiating ovarian androgen-producing cells in defined medium. J Biol Chem. 1982;257:4507–13. [PubMed] [Google Scholar]
- 22.Poirier D. Inhibitors of 17 beta-hydroxysteroid dehydrogenases. Curr Med Chem. 2003;10:453–77. doi: 10.2174/0929867033368222. [DOI] [PubMed] [Google Scholar]
- 23.Wiebe RH, Morris CV. Testosterone/androstenedione ratio in the evaluation of women with ovarian androgen excess. Obstet Gynecol. 1983;61:279–284. [PubMed] [Google Scholar]
- 24.Nilson JH, Abbud RA, Keri RA, Quirk CC. Chronic hypersecretion of luteinizing hormone in transgenic mice disrupts both ovarian and pituitary function, with some effects modified by the genetic background. Recent Prog Horm Res. 2000;55:69–89. 89–91. [PubMed] [Google Scholar]
- 25.O'Shaughnessy PJ. Steroidogenic enzyme activity in the hypogonadal (hpg) mouse testis and effect of treatment with luteinizing hormone. J Steroid Biochem Mol Biol. 1991;39:921–8. doi: 10.1016/0960-0760(91)90350-e. [DOI] [PubMed] [Google Scholar]
- 26.O'Shaughnessy PJ, Bennett MK, Scott IS, Charlton HM. Effects of FSH on Leydig cell morphology and function in the hypogonadal mouse. J Endocrinol. 1992;135:517–25. doi: 10.1677/joe.0.1350517. [DOI] [PubMed] [Google Scholar]
- 27.Scott IS, Charlton HM, Cox BS, Grocock CA, Sheffield JW, O'Shaughnessy PJ. Effect of LH injections on testicular steroidogenesis, cholesterol side-chain cleavage P450 mRNA content and Leydig cell morphology in hypogonadal mice. J Endocrinol. 1990;125:131–8. doi: 10.1677/joe.0.1250131. [DOI] [PubMed] [Google Scholar]
- 28.Murono EP, Payne AH. Testicular maturation in the rat. In vivo effect of gonadotropins on steroidogenic enzymes in the hypophysectomized immature rat. Biol Reprod. 1979;20:911–7. doi: 10.1095/biolreprod20.4.911. [DOI] [PubMed] [Google Scholar]
- 29.Ghersevich S, Nokelainen P, Poutanen M, Orava M, Autio-Harmainen H, Rajaniemi H, Vihko R. Rat 17 beta-hydroxysteroid dehydrogenase type 1: primary structure and regulation of enzyme expression in rat ovary by diethylstilbestrol and gonadotropins in vivo. Endocrinology. 1994;135:1477–87. doi: 10.1210/endo.135.4.7925110. [DOI] [PubMed] [Google Scholar]
- 30.Ghersevich S, Poutanen M, Tapanainen J, Vihko R. Hormonal regulation of rat 17 beta-hydroxysteroid dehydrogenase type 1 in cultured rat granulosa cells: effects of recombinant follicle- stimulating hormone, estrogens, androgens, and epidermal growth factor. Endocrinology. 1994;135:1963–71. doi: 10.1210/endo.135.5.7956918. [DOI] [PubMed] [Google Scholar]
- 31.Russell LD. Mammalian Leydig cell structure. In: Payne AH, Hardy MP, Russell LD, editors. The Leydig Cell. Cache River Press; Vienna, IL: 1996. pp. 43–96. [Google Scholar]
- 32.Dhar JD, Setty BS. Epididymal response to exogenous testosterone in rats sterilized neonatally by estrogen. Endokrinologie. 1976;68:14–21. [PubMed] [Google Scholar]
- 33.Abney TO, Myers RB. 17 beta-estradiol inhibition of Leydig cell regeneration in the ethane dimethylsulfonate-treated mature rat. J Androl. 1991;12:295–304. [PubMed] [Google Scholar]
- 34.Risma KA, Clay CM, Nett TM, Wagner T, Yun J, Nilson JH. Targeted overexpression of luteinizing hormone in transgenic mice leads to infertility, polycystic ovaries, and ovarian tumors. Proc Natl Acad Sci USA. 1995;92:1322–1326. doi: 10.1073/pnas.92.5.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risma KA, Hirshfield AN, Nilson JH. Elevated luteinizing hormone in prepubertal transgenic mice causes hyperandrogenemia, precocious puberty, and substantial ovarian pathology. Endocrinology. 1997;138:3540–3547. doi: 10.1210/endo.138.8.5313. [DOI] [PubMed] [Google Scholar]
- 36.Rulli SB, Kuorelahti A, Karaer O, Pelliniemi LJ, Poutanen M, Huhtaniemi I. Reproductive disturbances, pituitary lactotrope adenomas, and mammary gland tumors in transgenic female mice producing high levels of human chorionic gonadotropin. Endocrinology. 2002;143:4084–95. doi: 10.1210/en.2002-220490. [DOI] [PubMed] [Google Scholar]
- 37.Matzuk MM, DeMayo FJ, Hadsell LA, Kumar TR. Overexpression of human chorionic gonadotropin causes multiple reproductive defects in transgenic mice. Biol Reprod. 2003;69:338–46. doi: 10.1095/biolreprod.102.013953. [DOI] [PubMed] [Google Scholar]
- 38.Heikkilä M, Prunskaite R, Naillat F, Itaranta P, Vuoristo J, Leppaluoto J, Peltoketo H, Vainio S. The partial female to male sex reversal in Wnt-4-deficient females involves induced expression of testosterone biosynthetic genes and testosterone production, and depends on androgen action. Endocrinology. 2005;146:4016–23. doi: 10.1210/en.2005-0463. [DOI] [PubMed] [Google Scholar]
- 39.Lau IF, Saksena SK, Chang MC. Effects of hCG on serum levels of testosterone, dihydrotestosterone and androstenedione in male mice. Horm Res. 1978;9:169–75. doi: 10.1159/000178910. [DOI] [PubMed] [Google Scholar]
- 40.Chandrashekar V, Bartke A, Awoniyi CA, Tsai-Morris CH, Dufau ML, Russell LD, Kopchick JJ. Testicular endocrine function in GH receptor gene disrupted mice. Endocrinology. 2001;142:3443–50. doi: 10.1210/endo.142.8.8298. [DOI] [PubMed] [Google Scholar]
- 41.Allan CA, McLachlan RI. Androgen deficiency syndrome. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 5th ed. 2001. pp. 3159–3191. [Google Scholar]
- 42.Andersson S, Moghrabi N. Physiology and molecular genetics of 17 beta-hydroxysteroid dehydrogenases. Steroids. 1997;62:143–7. doi: 10.1016/s0039-128x(96)00173-0. [DOI] [PubMed] [Google Scholar]
- 43.Peltoketo H, Vihko P, Vihko R. Regulation of estrogen action: role of 17 beta-hydroxysteroid dehydrogenases. Vitam Horm. 1999;55:353–98. [PubMed] [Google Scholar]
- 44.Nokelainen P, Puranen T, Peltoketo H, Orava M, Vihko P, Vihko R. Molecular cloning of mouse 17 beta-hydroxysteroid dehydrogenase type 1 and characterization of enzyme activity. Eur J Biochem. 1996;236:482–90. doi: 10.1111/j.1432-1033.1996.00482.x. [DOI] [PubMed] [Google Scholar]
- 45.Puranen T, Poutanen M, Ghosh D, Vihko R, Vihko P. Origin of substrate specificity of human and rat 17β-hydroxysteroid dehydrogenase type 1, using chimeric enzymes and site-directed substitutions. Endocrinology. 1997;138:3532–9. doi: 10.1210/endo.138.8.5303. [DOI] [PubMed] [Google Scholar]
- 46.Magoffin DA, Erickson GF. Direct inhibitory effect of estrogen on LH-stimulated androgen synthesis by ovarian cells cultured in defined medium. Mol Cell Endocrinol. 1982;28:81–9. doi: 10.1016/0303-7207(82)90042-9. [DOI] [PubMed] [Google Scholar]
- 47.Zhou Q, Nie R, Prins GS, Saunders PT, Katzenellenbogen BS, Hess RA. Localization of androgen and estrogen receptors in adult male mouse reproductive tract. J Androl. 2002;23:870–81. [PubMed] [Google Scholar]
- 48.Pelletier G, Labrie C, Labrie F. Localization of oestrogen receptor alpha, oestrogen receptor beta and androgen receptors in the rat reproductive organs. J Endocrinol. 2000;165:359–70. doi: 10.1677/joe.0.1650359. [DOI] [PubMed] [Google Scholar]
- 49.Sar M, Welsch F. Differential expression of estrogen receptor-β and estrogen receptor-α in the rat ovary. Endocrinology. 1999;140:963–971. doi: 10.1210/endo.140.2.6533. [DOI] [PubMed] [Google Scholar]
- 50.Saunders PTK, Maguire SM, Gaughan J, Millar MR. Expression of oestrogen receptor beta (ERβ) in multiple rat tissues visualized by immunohistochemistry. J Endocrinol. 1997;154:R13–R16. doi: 10.1677/joe.0.154r013. [DOI] [PubMed] [Google Scholar]
- 51.Saunders PT, Millar MR, Williams K, Macpherson S, Harkiss D, Anderson RA, Orr B, Groome NP, Scobie G, Fraser HM. Differential expression of estrogen receptor-alpha and -beta and androgen receptor in the ovaries of marmosets and humans. Biol Reprod. 2000;63:1098–105. doi: 10.1095/biolreprod63.4.1098. [DOI] [PubMed] [Google Scholar]
- 52.Papadopoulos V. Payne AH, Hardy MP, Russell LD. The Leydig Cell. Cache River Press; Vienna, IL: 1996. Pharmacologic influence on androgen biosynthesis; pp. 597–628. [Google Scholar]
- 53.Akingbemi BT, Ge R, Rosenfeld CS, Newton LG, Hardy DO, Catterall JF, Lubahn DB, Korach KS, Hardy MP. Estrogen receptor-α gene deficiency enhances androgen biosynthesis in the mouse Leydig cell. Endocrinology. 2003;144:84–93. doi: 10.1210/en.2002-220292. [DOI] [PubMed] [Google Scholar]
- 54.Schomberg DW, Couse JF, Mukherjee A, Lubahn DB, Sar M, Mayo KE, Korach KS. Targeted disruption of the estrogen receptor-α gene in female mice: characterization of ovarian responses and phenotype in the adult. Endocrinology. 1999;140:2733–44. doi: 10.1210/endo.140.6.6823. [DOI] [PubMed] [Google Scholar]
- 55.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 56.Sternberg WH, Roth LM. Ovarian stromal tumors containing Leydig cells. I. Stromal-Leydig cell tumor and non-neoplastic transformation of ovarian stroma to Leydig cells. Cancer. 1973;32:940–51. doi: 10.1002/1097-0142(197310)32:4<940::aid-cncr2820320428>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 57.Roth LM, Sternberg WH. Ovarian stromal tumors containing Leydig cells. II. Pure Leydig cell tumor, non-hilar type. Cancer. 1973;32:952–60. doi: 10.1002/1097-0142(197310)32:4<952::aid-cncr2820320429>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 58.Bohm J, Roder-Weber M, Hofler H, Kolben M. Bilateral stromal Leydig cell tumour of the ovary. Case report and literature review. Pathol Res Pract. 1991;187:348–52. 352–3. doi: 10.1016/S0344-0338(11)80801-0. [DOI] [PubMed] [Google Scholar]
- 59.Oler A, Singh M, Ural SH. Bilateral ovarian stromal hyperplasia concealing a nonhilar, pure stromal-Leydig cell tumor. A case report. J Reprod Med. 1999;44:563–6. [PubMed] [Google Scholar]
- 60.Barbieri RL. Human ovarian 17-ketosteroid oxidoreductase: unique characteristics of the granulosaluteal cell and stromal enzyme. Am J Obstet Gynecol. 1992;166:1117–23. 1123–6. doi: 10.1016/s0002-9378(11)90598-5. [DOI] [PubMed] [Google Scholar]
- 61.Haider SG, Pickartz H, Freundl G, Passia D. Demonstration of hydroxysteroid dehydrogenases and testosterone in the Sertoli-Leydig cell tumor (androblastoma) tissue of the human ovary: an enzyme histochemical and immunohistochemical study. Acta Anat (Basel) 1985;121:170–3. doi: 10.1159/000145960. [DOI] [PubMed] [Google Scholar]
- 62.Ichinohasama R, Teshima S, Kishi K, Mukai K, Tsunematsu R, Ishii-Ohba H, Shimosato Y. Leydig cell tumor of the ovary associated with endometrial carcinoma and containing 17 beta-hydroxysteroid dehydrogenase. Int J Gynecol Pathol. 1989;8:64–71. doi: 10.1097/00004347-198903000-00008. [DOI] [PubMed] [Google Scholar]
- 63.Barbieri RL, Gao X. Presence of 17 beta-hydroxysteroid dehydrogenase type 3 messenger ribonucleic acid transcript in an ovarian Sertoli-Leydig cell tumor. Fertil Steril. 1997;68:534–7. doi: 10.1016/s0015-0282(97)00233-1. [DOI] [PubMed] [Google Scholar]
- 64.Nelson VL, Legro RS, Strauss JF, 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946–57. doi: 10.1210/mend.13.6.0311. [DOI] [PubMed] [Google Scholar]
- 65.Nelson VL, Qin Kn KN, Rosenfield RL, Wood JR, Penning TM, Legro RS, Strauss JF, 3rd, McAllister JM. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925–33. doi: 10.1210/jcem.86.12.8088. [DOI] [PubMed] [Google Scholar]
- 66.Diao FY, Xu M, Hu Y, Li J, Xu Z, Lin M, Wang L, Zhou Y, Zhou Z, Liu J, Sha J. The molecular characteristics of polycystic ovary syndrome (PCOS) ovary defined by human ovary cDNA microarray. J Mol Endocrinol. 2004;33:59–72. doi: 10.1677/jme.0.0330059. [DOI] [PubMed] [Google Scholar]
- 67.Couse JF, Hewitt SC, Korach KS. Steroid receptors in the ovary and uterus. In: Neill JD, editor. Knobil and Neill's Physiology of Reproduction. 3rd / ed. Elsevier Science; San Diego, Calif. Oxford: 2006. pp. 593–678. [Google Scholar]
- 68.Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annu Rev Physiol. 1997;59:349–363. doi: 10.1146/annurev.physiol.59.1.349. [DOI] [PubMed] [Google Scholar]
- 69.Hsueh AJW, Billig H, Tsafriri A. Ovarian follicle atresia: a hormonally controlled apoptotic process. Endocr Rev. 1994;15:707–724. doi: 10.1210/edrv-15-6-707. [DOI] [PubMed] [Google Scholar]
- 70.Couse JF, Hewitt SC, Bunch DO, Sar M, Walker VR, Davis BJ, Korach KS. Postnatal sex reversal of the ovaries in mice lacking estrogen receptors α and β. Science. 1999;286:2328–31. doi: 10.1126/science.286.5448.2328. [DOI] [PubMed] [Google Scholar]
- 71.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERα) and beta (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277–91. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]