

G-protein-coupled receptors (GPCRs) are a conserved family of seven transmembrane receptors that is one of the largest classes of receptors to be targeted for drug therapy [1, 2]. Among an estimated 200 cardiac GPCRs, drugs targeting adrenergic and angiotensin GPCR signaling pathways alone account for the majority of prescriptions for cardiovascular diseases [3, 4]. However, heart failure remains a leading cause of global morbidity and mortality [4], and a better understanding of the components that make up GPCR signaling pathways in healthy and failing hearts may provide a mechanistic basis for improving heart failure treatment. The focus of this review is to highlight the most commonly studied and clinically targeted cardiac GPCRs, placing emphasis on their common signaling components implicated in cardiac disease.
I. GPCR Signaling Overview
GPCR signaling is activated by ligand binding to an extracellular active site of the receptor. Depending on the ligand and type of GPCR, the active site is located on either the N-terminal tail, extracellular loops, or exofacial transmembrane helices [5, 6]. Ligand binding induces a conformational change in the GPCR, which disrupts the ionic interactions between the third cytoplasmic loop and the sixth transmembrane segment and allows for coupling with heterotrimeric guanine-nucleotide regulatory proteins (G-proteins)[6–11].
G-proteins consist ofα, β, and γ subunits and, upon GPCR coupling, the G-protein converts GTP to GDP on its α subunit resulting in dissociation of the Gα from the Gβγ subunits to mediate downstream signaling [12, 13]. The four primary families of Gα-proteins (Gαs, Gαi, Gαq, and Gα11/12) [12] diverge at this point with respect to downstream signaling molecules and consequential physiological responses. Dissociated Gα subunits couple with an effector, an enzyme such as adenylyl cyclase (AC) and phospholipase C β (PLCβ), or an ion channel.[12]. Dissociated Gβγ subunits target a range of signaling pathways involved in desensitization, downregulation, apoptosis, and ion channel activation (IKAch)[12, 14, 15] (see Figure 1).
Figure 1. Overview of GPCR Signaling.
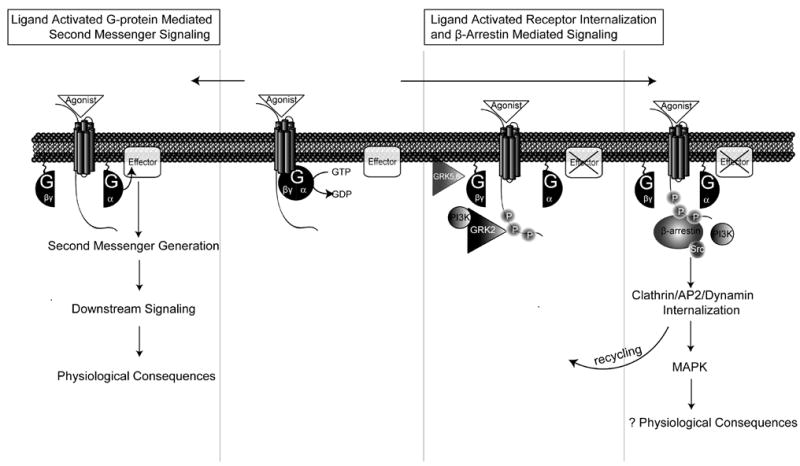
Agonist binding to receptor results in the coupling to a G-protein and the conversion of GTP to GDP to cause the dissociation of Gα and Gβγ subunits. Gα signaling is initiated with the activation of a membrane-bound effector molecule, such as adenylyl cyclase (AC) or phospholipase C β (PLCβ) to produce second messengers such as cyclic adenosine 3′,5′ monophosphate, (cAMP), diacylglycerol (DAG), or inositol 1,4, 5-triphosphate (IP3), which activate a variety of downstream signals with a range of physiological consequences in the heart such as the regulation of contractility, hypertrophy, and apoptosis. A simultaneous process involves the ligand-activated translocation of G protein-coupled receptor kinases (GRKs) and bound phosphoinositide-3-kinase (PI3K), to the membrane by dissociated Gβγ subunits leading to receptor phosphorylation on the C-terminal tail. Following GRK mediated receptor phosphorylation, β-arrestin is bound and acts to 1) desensitize G protein signaling by uncoupling the receptor from G protein, 2) scaffold for the recruitment of proteins involving in receptor trafficking, such as AP2, clathrin, and dynamin, and 3) form a signaling complex that initiates G protein-independent downstream signaling such as the activation of mitogen-activated protein (MAP) kinases.
This review will first discuss each G-protein family in terms of its signaling pathway in the healthy heart and follow with a description of individual signaling components and receptors in heart disease. Among the most commonly studied receptors are Gαs and Gαi-coupled β-adrenergic receptors; Gαq-coupled angiotensin, α-1-adrenergic, and endothelin receptors; Gαi-coupled adenosine-1, α2-adrenergic, and muscarinic-2 receptors; and common to all of these receptors is the corresponding Gβγ signaling, G-protein independent signaling, and the consequences of receptor downregulation and desensitization in the heart.
II. GS signaling and GS-coupled receptors
IIa. Overview of Gs signaling
Following ligand stimulation, the dissociated Gαs subunit activates signaling with the stimulation of AC (giving Gs or ‘Gstimulatory’ its name). AC exists in the human myocardium primarily as AC Types V and VI, though types IV and VII are also present at lower levels. AC V is primarily localized to the atria of the heart while AC VI is found in both atria and ventricles and is colocalized with β1 adrenergic receptors (β1ARs)[16]. AC is primarily responsible for increasing intracellular production of the second messenger cyclic adenosine 3′,5′ monophosphate, (cAMP) [12, 17]. cAMP modulates cardiac contractility via its binding to cAMP-dependent protein kinase (PKA). The catalytic subunit of PKA phosphorylates a range of substrates within the myocyte. In the heart, PKA functions to modulate contractility via its phosphorylation of myocyte proteins including the voltage-gated L-type Ca2+ channel, the cardiac ryanodine receptor (RyR2), phospholamban, and troponin I.
L-type Ca2+ channels activate RyR to mediate Ca2+-induced Ca2+ release (CICR); the primary source of intracellular Ca2+ to activate myofilament contraction [18]. Activated L-type Ca2+ voltage-dependent channels initiate the inward calcium entry required for CICR [19]. L-type Ca2+ channels are located primarily in transverse (T)-tubules, and in response to an action potential generate a small release of calcium into a cytosolic space known as the diadic cleft that occurs between the opposition of T-tubules with the junctional sarcoplasmic reticulum. This trigger Ca2+ activates the large release of Ca2+ by the RyR2 to generate what is known as the Ca2+ spark [20].
RyR2 is a macromolecular signaling complex located on the junctional sarcoplasmic reticulum (SR). It responds to trigger Ca2+ from the L-type Ca2+ channel by releasing larger Ca2+ stores into the cytoplasm from within the SR, which then activates myofilament contraction. RyR2 can be modulated through phosphorylation by PKA, cGMP-dependent protein kinase (PKG), and Ca2+/calmodulin kinase II (CaMKII) in addition to poly-S-nitrosylation [20–22].
Myocyte relaxation occurs through Ca2+ reuptake in the SR is achieved via the SR Ca2+-ATPase (SERCA) pump, which is modulated by phospholamban [23]. Phospholamban modulates Ca2+ affinity to SERCA2 which regulates the sequestration of cytosolic Ca2+ back into the SR [23]. Phospholamban phosphorylation is directly associated with increases in Ca2+ affinity for SERCA2 and enhanced rates of cardiac relaxation in intact beating hearts [24]. Phospholamban is also phosphorylated by CaMKII and can also regulate excitation-contraction coupling in the heart [23]. Troponin I phosphorylation by PKA reduces myofilament sensitivity to Ca2+, hence inhibiting contractile signaling and accelerating cardiac relaxation [25] (See Figure 2).
Figure 2. Gs/Gi-coupled receptor signaling.
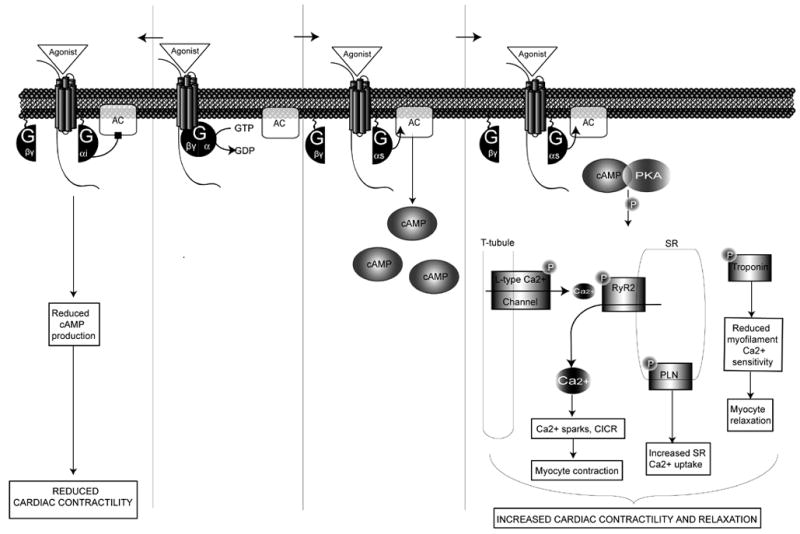
Gs- and Gi-coupled receptors target the second messenger adenylyl cyclase (AC). While Gs stimulates AC, activation of Gi inhibits AC, reducing the accumulation of cAMP and consequently leading to reduced PKA and agonist-stimulated cardiac contractility. Downstream effectors of Gαs activation result from the increase in PKA which phosphorylation substrates include: phospholamban (PLN), troponin, L-type Ca2+ channels, and the cardiac ryanodine receptor (RyR2).
IIb. Gs-coupled Receptors in the Heart
Adrenergic Receptors
Adrenergic receptors are an important class of GPCRs responsible for translating chemical messages from the sympathetic nervous system into cardiovascular responses. Three β-adrenergic receptor (βAR) subtypes are found in mammalian hearts. β1ARs account for approximately 80% of the βARs in the healthy heart and couple primarily to Gs-proteins to regulate heart rate and contractility [12, 26]. β2ARs couple to Gs-proteins, but can also switch to Gi-coupling after receptor phosphorylation by PKA [27]. β2ARs are also important for mediating smooth muscle relaxation and glycogenesis [17]. β3ARs couple to both Gs-and Gi-proteins and are the least abundant subtype in the heart. While β3ARs play a role in lipolysis and thermogenesis in adipose tissue, they have been shown to have both a positive inotropic effect in transgenic mice overexpressing human β3ARs [28] and a negative inotropic effect in the human heart [29].
β1 Adrenergic Receptors
β1ARs regulate the inotropic and chronotropic functions of the healthy heart primarily mediated by the effects of PKA. β1AR knockout mice have high embryonic lethality, and those that do survive exhibit no inotropic or chronotropic response to exercise or agonist stimulation despite having a normal basal heart rate and blood pressure [30]. In contrast, overexpression of human β1ARs in transgenic mice initially augments cardiac function but eventually leads to deleterious effects on the heart presumably due to chronic β1AR signaling [31, 32].
β2 Adrenergic Receptors
β2ARs were originally thought to signal exclusively through Gs. Like the β1AR, Gs-signaling leads to regulation of contractility via Ca2+ cycling as described above. The β2ARs differ importantly from the β1ARs in that they can also couple to the Ginhibitory, or Gi-protein [27]. While both βAR subtypes stimulate AC through Gs, stimulation via the β1AR activates L-type Ca2+ channels throughout the myocyte while β2AR only activates L-type Ca2+ channels in close proximity to the receptor [33]. However, when myocytes are treated with pertussis toxin to selectively inhibit Gi signaling, activation of L-type Ca2+ channels by β2ARs becomes more widespread and induces relaxation effects similar to β1AR stimulation [33]. This contrast between local and diffuse mediation of contractility at the cellular level suggests that there is a competition between β2AR mediated Gs and Gi signaling. This relationship is illustrated by the timecourse of β2AR –induced contractility in neonatal cardiomyocytes: agonist binding first induces a small increase followed by a long decrease in myocyte contraction rate, representing a short Gs-coupling followed by a prolonged Gi-coupling [17, 34]. Furthermore, while β2AR-overexpressing mice do not show an enhanced contractile response to βAR-agonists, treatment with pertussis toxin restores βAR contractile responsiveness [35]. These results suggest that β2AR stimulation initially activates Gs signaling pathways in a pattern analogous to β1AR activation. This stimulatory pathway is quickly overshadowed by the delayed activation of the Gi pathway, which negatively regulates contractility in response to prolonged βAR activation.
Transgenic mouse studies illustrate the physiological impact of these downstream β2AR signaling components in the heart. In vivo, modest cardiac-specific β2AR overexpression of 60-fold over basal results in enhanced contractile function without development of heart failure [36, 37]. However, mice with β2AR overexpression at 350 times greater than basal rapidly develop hypertrophy and heart failure [36]. Physiologically, these mice show a loss in their response to agonist, a reduction in L-type Ca2+ density, and a decreased systolic function [36]. In contrast, β2AR-deficient mice display normal development and resting cardiovascular function, and show increased exercise capacity compared to wild type mice [30]. However, they develop hypertension in response to either adrenergic system stimulation or exercise [30]. These data support the concept that β2ARs are important regulators of prolonged activation of the adrenergic system via stress or exercise, possibly due, in part, to their coupling to Gi.
IIc. Gs-coupled Receptors in Heart Disease
Gs Signaling in Heart Disease
Gs-coupled receptors signal via AC primarily to regulate contractility. Chronic activation of Gs signaling pathways leads to cardiac hypertrophy, fibrosis, and heart failure. In fact, transgenic mice with cardiac-specific overexpression of Gαs display increased heart rate and contractility in response to catecholamine stimulation, but eventually develop histological evidence of myocardial damage such as cellular hypertrophy, fibrosis, and necrosis [38, 39].
Recent data suggests that human heart failure may be mediated, in part, by defective regulation of RyR2, the SR channel responsible for gating Ca2+ efflux into the cytosol. Several human RyR2 mutations, which result in altered myocardial Ca2+ handling, have been linked to ventricular arrhythmias and sudden cardiac death [40–44]. Moreover, heart failure is associated with RyR2 hyperphosphorylation by PKA, leading to poor Ca2+ gating and uncoupling from neighboring RyR2 channels [45]. Such hyperphosphorylation causes a reduced ability to trigger Ca2+ release from the SR and a consequent loss of excitation-contraction coupling gain [45], which is a characteristic of hypertrophic and failing myocytes [46]. β-blockers such as metoprolol [47, 48], carvedilol [47], atenolol [47], and propranolol [49] suppress the hyperphosphorylation of RyR2 and restore channel function [50]. Recent data suggests that selective inhibition of RyR2 hyperphosphorylation stabilizes the RyR2, prevents SR Ca2+ leak and prevents development of heart failure in experimental models [51].
Phospholamban appears to decline during heart failure, possibly contributing to decreased myocyte contractility in the failing human heart [23]. Interestingly, transgenic mice lacking in phospholamban show an increase in the total level of Ca2+ in the SR, an increase in the number of spontaneous Ca2+ sparks, and a more rapid inactivation of L-type Ca2+ currents [52]. Mutations of phospholamban in humans results in a disturbance in Ca2+ reuptake to the SR, which is implicated in the development of dilated cardiomyopathy and heart failure [53]. Humans that lack phospholamban in their hearts also develop heart failure and early mortality [54, 55]. Transgenic overexpression of phospholamban in vivo is associated with altered systolic Ca2+ parameters and depressed ventricular systolic function [56, 57]. However, gene-targeted ablation of phospholamban enhances cardiac contractility [58], and can rescue experimental models of heart failure [59]. Thus altering phospholamban levels in heart failure may provide a way to relieve the impairment of cardiac relaxation by enhancing Ca2+ reuptake to the SR [23].
βARs in Heart Disease
During heart failure, heightened catecholamine stimulation of the adrenergic receptor system induces selective downregulation of β1ARs. Consequently, the β1AR to β2AR ratio changes from an 80:20 distribution in the healthy heart to a ratio of 60:40 in the failing heart [60, 61]. Chronic catecholamine stimulation also promotes cardiac hypertrophy and myocyte apoptosis while leading to a reduction in contractility [17].
Diminished contractile responsiveness to βAR stimulation and an increase in Gi signaling is a hallmark of heart failure [32]. In failing human ventricles, β2ARs are uncoupled from AC and show a marked loss of their ability to produce cAMP, which accounts for the reduced efficiency in producing a contractile response [61]. Thus, although β2ARs do not downregulate, they are considerably desensitized in the failing heart.
βAR signaling is a central component of heart failure, and its correlation with myocyte apoptosis has recently been clarified. Apoptotic myocytes are more prevalent in failing human hearts, showing 200-fold increases over basal compared to healthy human hearts [62]. Within individual failing hearts, there are more apoptotic myocytes in peri-infarcted regions than in cardiac regions distal to myocardial infarction [62]. Chronic βAR agonism via norepinepherine or isoproterenol induces apoptosis in cultured cardiomyocytes [63] and in vivo through chronic isoproterenol administration [64].
A recently recognized detrimental pathway is the β1AR mediated pro-apoptotic signaling of CaMKII. Both direct activation or cardiac overexpression of CaMKII induces myocyte apoptosis in vitro that can be blocked by CaMKII inhibition [33]. CaMKII levels are significantly elevated and show increased activation immediately following the induction of heart failure, and cardiac CaMKII overexpression induces dilated cardiomyopathy [65, 66]. Importantly, direct inhibition of CaMKII in vivo reduces adverse LV remodeling after β1AR agonism [67], and significantly diminishes the development of cardiac apoptosis in heart failure models [68]. Furthermore, β1AR-mediated development of apoptosis can be prevented with transgenic overexpression of a CaMKII inhibitor [68]. When these mice are crossed with phospholamban deficient mice to normalize Ca2+ stores in the heart and allow activation of CaMKII, the resistance to apoptosis is abolished [68]. These data support the current search for selective CaMKII inhibitors to directly test whether CaMKII inhibition can alter the development of heart failure.
IId. Other Gs-Protein-coupled Receptors in the Heart
Other Gs-coupled receptors in the heart include adenosine [69], glucagon [70], prostanoid [71], and histamine [72] receptors [3].
Adenonsine receptors are thought to be cardioprotective in myocardial ischemia by inducing coronary vasodilation, which increases oxygen supply by inhibiting oxygen radical-mediated cellular damage [73]. Overexpression of adenosine receptors 1 or 3 offers an overall increase in the response to adenosine agonism and consequently reduces the level of injury caused by myocardial ischemia [69, 74]. Moreover, direct stimulation of adenosine receptors via adenosine administration during reperfusion has been shown to improve left ventricular (LV) function and reduce infarct size [75, 76] implicating adenosine agonism as a potential therapy during myocardial ischemia.
Prostanoid receptors respond to prostaglandins in neonatal myocytes to play a role in cardiac cell growth [71]. Prostanoids regulate blood pressure by mediating vascular tone and renal sodium handling [77]. Reduction of prostacyclin in vivo results in an increase in blood pressure and consequent development of hypertension while reduction of the prostanoid receptor leads to the development of hypertension accompanied by robust cardiac hypertrophy and fibrosis [77].
Histamine is abundant in cardiac mast cells and, in the healthy heart, histamine infusion produces an increase in heart rate and atrial activity [78]. While histamine-2-receptors are coupled to Gs, histamine-1 receptors couple to Gq and histamine-3-receptors couple to Gi/o. Histamine-2-receptor knockout mice show a decrease in heart rate in response to histamine stimulation while histamine-3-receptor knockout mice show a decrease in blood pressure and reduced norepinephrine release during ischemia [79]. In addition, blockade of histamine receptors by the antihistamines used in allergy treatments have been shown to produce cardiotoxicity and atypical forms of arrhythmia such as torsade de pointes [80].
III. GQ signaling and GQ-coupled receptors
IIIa. Overview of Gq signaling
Receptors coupled to Gαq play important roles in mediating cardiac growth responses, myocyte apoptosis, and cardiac hypertrophy. Ligand-stimulated receptor signaling via Gq is initiated by the membrane-recruitment and activation of PLCβ. PLCβ is responsible for hydrolyzation of phosphatidylinositol 4, 5 biphosphate (PIP2) into two second messengers: diacylglycerol (DAG) and inositol 1, 4, 5-triphosphate (IP3)[81].
DAG activates several serine-threonine isoforms of protein kinase C (PKC)[82, 83]. PKC signaling as a whole is important for myocyte apoptosis, necrosis, and for triggering pro-hypertrophic gene transcription in the heart [84–86]. Recently, it has been shown that protein kinase D, a downstream effector of PKC, directly phosphorylates HDAC5 and stimulates its nuclear export, leading to activation of hypertrophic genes [87, 88].
There are three primary families of PKC: “conventional”, “novel”, and “atypical”[89]. “Conventional” PKCs are both Ca2+ and DAG dependent, “novel” PKCs are DAG activated but Ca2+ independent, and “atypical” PKCs are both DAG and Ca2+ independent. The four most functionally significant PKC isoforms in the heart are of the conventional (PKCα, β) and novel (PKCδ, ε) groups. PKCα is the most prevalent isoform in the human heart [90], and is primarily a regulator of contractility via modulation of phospholamban and SERCA phosphorylation [91]. PKCβ is important for the transduction of cardiac hypertrophy, contractility, and regulation of neonatal cardiac growth [92, 93]. PKCδ is a critical mediator of postischemic necrosis and contractile dysfunction [94, 95]. The role of PKCε in hypertrophy is disputed, but it is generally accepted to be a regulator of ischemia-reperfusion injury [94–96]. Upon activation, PKCε is translocated from the cytosol to the SR where it directly activates KATP channels to decrease ROS generation and calcium overload. These PKCε-mediate signaling events are critical for the cardioprotective effects of ischemic preconditioning [94–96].
The other second messenger of PLCβ, IP3, binds several receptors on the SR to induce the release of calcium stores into the cytoplasm [21, 40, 84–86, 97]. Unlike the short bursts of contraction and relaxation induced by CICR, IP3-mediated Ca2+ signaling results in a sustained Ca2+ release that allows for the activation of hypertrophic response genes [83]. This sustained release of Ca2+ results in calcineurin activation and consequent targeting of the nuclear factor of activated T-cells (NFAT) transcription factors [83]. CaMKII activation and subsequent NFAT3 signaling act in concert to promote pathologic hypertrophic signaling and cardiac growth [26, 83] (See Figure 3).
Figure 3. Gq-coupled receptor signaling.
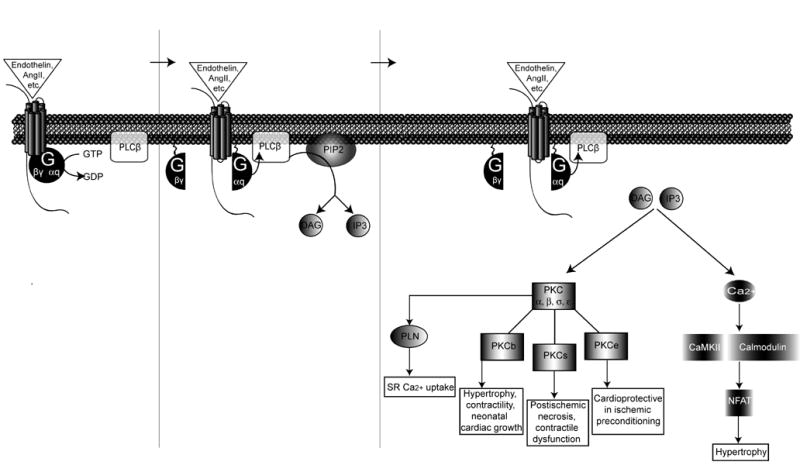
Gq-coupled receptors such as endothelin, angiotensin, or α-1-adrenergic receptors activate the second messenger phospholipase C β resulting in the hydrolysis of phosphatidylinositol 4, 5 biphosphate (PIP2) into two components: diacylglycerol (DAG) and inositol 1, 4, 5-triphosphate (IP3). DAG activates several isoforms of protein kinase C (PKC). PKCβ activation regulates hypertrophy, contractility, and neonatal cardiac growth. PKCσ activation regulates postischemic necrosis and contractile function. PKCε activation induces a cardioprotective effect in response to ischemic preconditioning. Activation of Gq-coupled receptors stimulates the release of calcium, which activates CaMKII and calcineurin to promote the nuclear translocation of nuclear factor of activated T-cells (NFAT) whereby the hypertrophic transcriptional machinery is activated.
IIIb. Gq-Coupled Receptors in the Heart
Angiotensin Receptors
Angiotensin receptor blockers and angiotensin-converting enzyme (ACE) inhibitors have been shown to reverse cardiac remodeling, reduce cardiac hypertrophy, slow the progression of heart failure, and improve survival in heart failure patients [98–102]. The main effector of this pathway is angiotensin II (Ang II), a peptide that stimulates vascular smooth muscle contraction, aldosterone release, and production of extracellular matrix proteins [103]. Ang II and angiotensin-converting enzyme (ACE) are both present at high levels after myocardial infarction and during cardiac hypertrophy [104–106].
Ang II signals primarily via the Gq-coupled AT1R [107]. Experimental studies have reported upregulation of AT1R in both hypertrophic and ischemic states [108–110] and downregulation with end-stage human heart failure [111, 112]. Part of the mechanism for AT1R downregulation may be attributed to the prolonged elevation of AngII levels with cardiac hypertrophy and during heart failure [113, 114]. Overexpression of ATIARs in the heart results in hypertrophy and fibrosis in otherwise healthy mice. These mice also show enhanced hypertrophic responses to pressure overload and die prematurely in response to heart failure [115]. In contrast, mice deficient in AT1ARs have an initially attenuated hypertrophic response to Ang II, and show diminished left ventricular dilatation and fibrosis after myocardial infarction with improved LV systolic function over time [116]. However, AT1AR-knockouts still develop hypertrophy in response to long-term pressure overload [117, 118]. This suggests the possibility that other angiotensin receptor subtypes such as the AT1BR or the angiotensin type II receptor may also play a role in the hypertrophic response to pathological stimuli.
α-1-Adrenergic Receptors
α1A, α1B, α1D-ARs activate Gαq signaling in response to sympathetic nervous system stimulation [119]. Signaling via the PLCβ/DAG/PKC pathway mobilizes Ca2+ entry into intracellular compartments, inducing cardiac growth [119]. α1A and α1BARs crosstalk with β1ARs to regulate cardiac inotropy and, although the mechanism is not clearly understood, will diminish the β1AR response to isoproterenol [120]. The abundance of αARs increases during heart failure [121], with varying consequences from each αAR subtype. Overexpression of α1AAR results in increased cardiac contractility without hypertrophy [94]. Cardiac-specific overexpression of α1BAR induces a blunted response to βAR stimulation mediated in part by enhanced Gi coupling [122], while α1BAR deficiency results in a worsened dilated cardiomyopathy only after pressure overload [123]. Deficiency in α1BAR additionally blocks increased blood pressure responses to the α1AR agonist phenylephrine [124]. Although animals deficient in individual α1AR subtypes do not show significant hypertrophy, knockout mice lacking both α1A/CAR and α1BAR genes show increased sympathetic innervation and the development of cardiac hypertrophy after weaning [125]. Together these studies demonstrate that α1AR subtypes interact with each other, and with other adrenergic receptors, to actively regulate blood pressure, contractile, and hypertrophic responses.
Endothelin Receptors
Another cardiac GPCR that promotes cardiac remodeling and hypertrophy via Gαq is the endothelin-1 receptor (ET1R). Endothelin is a powerful vasoconstrictor, and a chronotropic, inotropic, and hypertrophic promoting growth factor [126]. Of the three isoforms, endothelin-1 (ET-1) is expressed in the cardiovascular system and signals via the Gq-coupled ET1R [127].
Cardiac-specific murine overexpression of ET-1 results in myocardial inflammation, hypertrophy, and premature death from heart failure [128]. ET-1 overexpression also induces nuclear translocation of NF-kB, implicating a potential role of ET-1 in initiating an inflammatory cascade that ultimately leads to myocardial damage and heart failure [126, 128]. Various studies to date investigating changes in endothelin receptor levels in failing human hearts have shown contradicting results [126]: while first reports showed unchanged or diminished ETIR levels [129, 130], subsequent reports have shown elevated ETIR levels [131]. Furthermore, it is known that ET-1 signaling is activated in human heart failure [132, 133]. While ET1R antagonists are beneficial in experimental heart failure [132–136], and the elevated levels of ET1AR support it as a potential target for selective antagonism in human heart failure, recent clinical trials have failed to show significant benefits [137, 138].
IIIc. Gq–coupled Receptor Signaling in Heart Disease
Gq Signaling in Heart Disease
Modulation of Gαq itself can result in apoptotic and hypertrophic responses. While low overexpression of Gαq in mice does not significantly affect the heart; mild overexpression results in increased hypertrophic markers and minimal cardiac dysfunction [139, 140], and high overexpression of Gαq results in marked hypertrophy, heart failure, and increased mortality [139]. Gαq-overexpressing hearts that are exposed to pressure overload show increased apoptosis that can be rescued with a caspase inhibitor [141], implicating apoptotic pathways as mediators in the development of heart failure [142, 143]. Importantly, overexpression of an inhibitory peptide that interferes with Gq coupling prevents the development of cardiac hypertrophy and dysfunction in pressure-overloaded mice [144, 145]. These data support the concept that activation of Gq-signaling pathways in vivo are critical mediators of the hypertrophic response and the transition to heart failure.
PKC expression and activity are markedly increased in failing hearts compared to healthy human hearts [146]. Importantly, PKC activity levels are also markedly increased in failed hearts compared to nonfailed human hearts [146]. However, this overall trend varies by subtype: PKCα and β are both upregulated, PKCε is either upregulated or preferentially activated, and PKCδ levels are not changed [83]. Knocking out PKCα improves contractility while overexpression of PKCα diminishes contractile function in the heart without effecting cardiac growth [83, 91]. Animals with high overexpression of Gαq and resultant increased PKCε show overt heart failure and high mortality rates, implicating the critical role of PKCε activation in the development of hypertrophy and heart failure [139]. Transgenic animals lacking PKCε lose the ability to reduce infarct size in response to reactive oxygen species after ischemia reperfusion, suggesting that PKCε is also required for cardioprotective signaling pathways in ischemic preconditioning [128].
IIId. Other Gq-Protein-Coupled Receptors in the Heart
Other Gq-coupled receptors in the heart include: subtype 5-HT2b of the serotonin [147, 148], bradykinin [149], subtypes I, III, and V of the muscarinic [18], and subtype 1 of the histamine [150] receptors. Muscarinic receptor stimulation by acetylcholine mediates parasympathetic system activation of the heart to modulate heart rate, conduction velocity, and contractility [18]. More specifically, muscarinic receptors I, III, and V are involved in regulating anti-apoptotic pathways [151]. Serotonin is a known vasoconstrictor that has been shown to regulated embryonic cardiac development via its action on serotonin 5-HT2b receptors [148]. Bradykinin, part of the kallikreinkinin system, is found in vascular smooth muscle cells of the heart. Reduced bradykinin signaling may contribute to the onset of hypertension, cardiac failure, ischemia, myocardial infarction and left ventricular hypertrophy. In contrast to other Gq-receptors in the heart, the activation of the bradykinin system may be beneficial for heart function [152].
IV. GI signaling and GI-coupled receptors
IVa. Overview of Gi signaling
Ginhibitory, or Gi, signaling opposes Gs signaling by inhibiting AC and reducing AC mediated production of cAMP and its downstream targets. Specifically, a decrease in cAMP leads to a reduction in L-type Ca2+ currents which inhibits the force of contraction in myocytes [26]. Heart rate is slowed by the effects of activation of Gi-protein-coupled receptors on pacemaker cells [1]. Sustained Gi stimulation by genetic overexpression of a synthetic Gi-coupled receptor results in bradycardia and cardiomyopathy [153]. Elevated plevels of Gi are not known to cause cardiomyopathy, though levels are increased in patients with dilated cardiomyopathy and heart failure [154, 155]. Gi couples with A1 adenosine receptors (A1Rs), M2 muscarinic receptors (M2Rs), and α-2-adrenergic receptors (α2ARs) in the heart (See Figure 2).
IVb. Gi-Coupled Receptors in the Heart
Adenosine-1 Receptors
Adenosine receptors 1 and 3 (A1, A3) signal primarily through Gi to inhibit cAMP production while adenosine 2 receptors signal primarily via Gs. Adenosine receptor 1 agonists have been used as anti-arrhythmic agents or in unmasking atrial tachyarrhythmias [156]. The administration of adenosine prior to ischemia reduces infarction size and improves recovery of ventricular function during reperfusion[157]. It is postulated that the protective effect is secondary to A1 receptor-mediated phosphorylation and opening of mitochondrial potassium channels [157]. These data support the concept that the activation of adenosine-A1 receptors is cardioprotective and mediated by a reduction in heart rate, contractility, and an attenuation of catecholamine stimulation in the heart [158].
Muscarinic-2 Receptors
While most muscarinic receptors couple primarily to Gq, the predominant muscarinic isoform in the heart, M2, couples to Gi. M2 receptor stimulation mediates negative inotropic effects via Gαi, while the negative chronotropic effects are mediated by Gβγ subunit activation of the IKAch channel [159]. In vivo, injection of the muscarinic agonist carbachol reduces resting heart rate without affecting basal contractility [160]. In humans, acetylcholine infusion does not change the force of contraction basally, but attenuates increases in contractile force induced by βAR stimulation [161, 162]. M2 density in the heart has been found to decrease with age, accompanied with a reduced ability to respond to muscarinic agonists [129]. While M2 receptor density has been shown to increase in patients with congestive heart failure [163], a change in total muscarinic receptor density in patients with congestive heart failure has not been shown [26].
α2-Adrenergic Receptors
Presynaptic α2ARs are responsible for suppressing norepinephrine (NE) release from synaptic nerves of healthy hearts, and reducing the excessive amounts of NE in the synaptic terminals during increased adrenergic drive [129, 164, 165]. As increased sympathetic stimulation activates β1AR, β2AR, and α1AR signaling, the role of α2ARs in attenuating these pathways is of critical importance. In fact, mice deficient in αARs have elevated catecholamine levels and develop contractile dysfunction [17, 129]. Furthermore, individuals with a loss of function deletion in α2ARs show a severely attenuated ability to suppress norepinephrine release, leading to higher instances of heart failure [166]. Treatments that stimulate the α2AR to decrease adrenergic receptor stimulation at its source may provide a way of moderately attenuating βAR and α1AR signaling.
V. Turning off the Signal: Desensitization, Downregulation, & Gβγ
In synchrony with Gα-protein signaling is the simultaneous activation of Gβγ-dependent and β-arrestin-mediated pathways that are responsible for the desensitization, downregulation, and further signaling of GPCRs.
Desensitization describes the tendency of biological systems and receptors to lose their responsiveness to continuous or repeated stimulation [14]. Desensitization occurs via receptor downregulation, the net loss of receptors from the cell membrane, and via receptor uncoupling from its G-protein through a process of receptor phosphorylation [12].
Receptor desensitization induced by phosphorylation results in the rapid (minutes) waning of G-protein-dependent second messenger signaling. Second messengers such as cAMP or DAG activate protein kinases can phosphorylate GPCRs and induce desensitization in a ‘non-agonist-specific’ manner. This mechanism of desensitization is termed heterologous desensitization.
In contrast, ‘agonist specific’, or homologous desensitization, occurs when ligand-activated receptors recruit G-protein-coupled receptor kinases (GRKs). GRKs phosphorylate residues on the c-terminal tail of agonist-occupied GPCRs leading to the binding of the multifunctional scaffold protein, β-arrestin, which then sterically hinders further G-protein coupling. [12, 167]. In the case of βARs, β-arrestin blocks the activation of adenylyl cyclase, thus reducing βAR responsiveness to catecholamines [168]. An important new concept of GPCR desensitization and downregulation is that additional signaling pathways are activated at the same time G-protein-dependent signaling is turned off. These G-protein-independent pathways are mediated by β-arrestin (See Figure 1).
Gβγ subunits
Gβγ subunits signal via effectors such as PI3K, AC, PLC, potassium channels, and GRKs [22, 169–175]. In addition, Gβγ subunits promote GPCR desensitization via their action on GRK2. Upon agonist stimulation, the dissociation of Gβγ from the heterotrimeric G protein promotes the translocation and targeting of GRK2 to the membrane. GRK2 is a member of a protein kinase family of seven subtypes: GRKs1-7, that are primarily responsible for the phosphorylation and subsequent downregulation/desensitization of GPCRs.
G protein-coupled Receptor Kinases
Of the 7 GRK isoforms, GRK 2, 3, 4, and 6 are expressed in the heart [168]. GRK2, or βARK1 as it is commonly known, is best studied and seems to have the greatest impact on cardiac function. It has been shown that GRK2 mRNA levels are increased almost 3 fold in the failing heart, with enhanced activity and consequent decrease in β1AR levels [60]. Moreover, mice overexpressing GRK2 have a reduced response to βAR stimulation as a result of enhanced receptor desensitization [176]. In contrast, inhibition of GRK2 via the competitive peptide inhibitor βARKct enhances myocyte contractility and βAR responsiveness in vivo [144, 176]. GRK2-homozygous-deleted mice are embryonically lethal but exhibit pronounced hypoplasia of the ventricular myocardium and greater than 70% decrease in cardiac ejection fraction in utero, implicating a role of GRK2 in cardiac development [177]. GRK2-heterozygous deleted mice with a 50% reduction in GRK2, however, survive to adulthood with normal cardiac development but show enhanced contractile response to β adrenergic agonists, confirming the primary role of GRK2 in mediating adrenergic receptor desensitization [144].
Mice with cardiac specific overexpression of GRK5 show increased βAR desensitization and reduced βAR responsiveness with no effect on angiotensin receptor signaling [177]. GRK3 myocardial overexpression, in contrast, presents normal βAR and angiotensin receptor signaling but attenuated cardiac signaling via the thrombin receptor [144], demonstrating the substrate specificity of GRK3.
β-arrestins
β-arrestins 1 and 2 were originally characterized as scaffolding proteins responsible for receptor desensitization. Larger roles in GPCR signaling were soon found, as β-arrestins are now also known for their functions as endocytic adaptors and mediators of β-arrestin-dependent signaling pathways [168]. β-arrestin binding to GRK-phosphorylated receptors shuts off further G-protein signaling by interdicting G-protein coupling [168]. β-arrestin allows for the assembly of multi-protein signaling complexes such as ERK and tyrosine kinases [178, 179]. β-arrestin-mediated signaling downstream of the AT1AR requires GRK5 and 6 and is independent of G-protein signaling [10]. Transgenic mouse studies to date show that while β-arrestin-2 knockouts do not seem to show a cardiac phenotype [180], β-arrestin-1-heterozygous knockouts show increased contractile responsiveness to β1AR stimulation [181].
VI. Phosophoinositide-3-Kinase and Heart Failure
An important modulator of βAR trafficking and function is phosophoinositide-3-kinase (PI3K)[182]. PI3Ks are a family of enzymes containing both lipid and protein kinase activities that mediate cell proliferation, cytoskeletal trafficking, and endoycytosis via such downstream effectors as PKB/Akt, GSK3β, and p70S6K[182].
One isoform, PI3Kα, which is activated by receptor tyrosine kinases, is important in the regulation of cardiomyocyte size [183]. Cardiac-specific overexpression of a constitutively active form of PI3Kα results in increased heart size and PKB/Akt levels, while a dominant negative overexpression of PI3Kα reduces heart size [184]. The other isoform, PI3Kγ, is activated by Gβγ subunits and phosphorylates phosphoinositides on the cell membrane to ultimately activate PKB/Akt signaling. Furthermore, PI3Kγ regulates contractility by negatively regulating AC and PLB activity [185], as cardiac-deletion of PI3Kγ increases cAMP levels and enhances contractility [183].
PI3K is important for βAR recycling in the healthy heart, but its role in heart failure may be detrimental. Recent studies have shown that PI3K binds to GRK2 to regulate βAR endoycytosis [186, 187]. When the PI3Kγ/βARK interaction is disrupted by a PI3K blocking protein, heart failure is ameliorated in a number of experimental models [182, 187, 188]. Moreover, inhibition of the endogenous PI3Kγ interaction with GRK2, results in normalization of βAR levels and improved βAR responsiveness to agonist stimulation [182, 187, 188]. This is also accompanied by marked improvement of cardiac function and increased survival. Hence, disruption of PI3K-GRK2 interactions improves βAR function and may improve cardiac function and survival in failing hearts.
Conclusions
With over 200 GPCRs in the heart, we have discussed the key GPCRs that have been implicated in heart failure, such as the β and α adrenergic receptors, the angiotensin receptors, endothelin receptors, and the muscarinic receptors. We have discussed their downstream signaling pathways, including those mediated by PKA, PKC, and PI3K, in both healthy and failing hearts. GPCR signaling cascades are complex, involving receptor cross-talk and convergence of a variety of pathways. Interestingly, the G-protein signaling pathways of many of the so called “orphaned” GPCRs remain to be identified [3]. Elucidating the detailed components of these pathways and their effects on cardiac function will likely provide exciting therapeutic options for treating heart failure.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierce KL, Premont RT, Lefkowitz RJ. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Hopkins AL, Groom CR. Nat Rev Drug Discov. 2002;1:727–30. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 3.Tang CM, Insel PA. Trends Cardiovasc Med. 2004;14:94–9. doi: 10.1016/j.tcm.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, Jessup M, Konstam MA, Mancini DM, Michl K, Oates JA, Rahko PS, Silver MA, Stevenson LW, Yancy CW, Antman EM, Smith SC, Jr, Adams CD, Anderson JL, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B. Circulation. 2005;112:e154–235. doi: 10.1161/CIRCULATIONAHA.105.167586. [DOI] [PubMed] [Google Scholar]
- 5.Gether U, Kobilka BK. J Biol Chem. 1998;273:17979–82. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- 6.Wess J. Faseb J. 1997;11:346–54. [PubMed] [Google Scholar]
- 7.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Science. 1996;274:768–70. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 8.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. J Biol Chem. 2001;276:29171–7. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. J Biol Chem. 2002;277:11441–9. doi: 10.1074/jbc.M111675200. [DOI] [PubMed] [Google Scholar]
- 10.Han SJ, Hamdan FF, Kim SK, Jacobson KA, Bloodworth LM, Li B, Wess J. J Biol Chem. 2005;280:34849–58. doi: 10.1074/jbc.M506711200. [DOI] [PubMed] [Google Scholar]
- 11.Han M, Groesbeek M, Sakmar TP, Smith SO. Proc Natl Acad Sci U S A. 1997;94:13442–7. doi: 10.1073/pnas.94.25.13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rockman HA, Koch WJ, Lefkowitz RJ. Nature. 2002;415:206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 13.Neves SR, Ram PT, Iyengar R. Science. 2002;296:1636–9. doi: 10.1126/science.1071550. [DOI] [PubMed] [Google Scholar]
- 14.Lefkowitz RJ. J Biol Chem. 1998;273:18677–80. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- 15.Krapivinsky G, Krapivinsky L, Wickman K, Clapham DE. J Biol Chem. 1995;270:29059–62. doi: 10.1074/jbc.270.49.29059. [DOI] [PubMed] [Google Scholar]
- 16.Wang T, Brown MJ. Mol Cell Endocrinol. 2004;223:55–62. doi: 10.1016/j.mce.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 17.Xiang Y, Kobilka BK. Science. 2003;300:1530–2. doi: 10.1126/science.1079206. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Han H, Zhang L, Shi H, Schram G, Nattel S, Wang Z. Mol Pharmacol. 2001;59:1029–36. doi: 10.1124/mol.59.5.1029. [DOI] [PubMed] [Google Scholar]
- 19.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. J Clin Invest. 2005;115:3306–17. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannell MB, Cheng H, Lederer WJ. Science. 1995;268:1045–9. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 21.Fabiato A, Fabiato F. Annu Rev Physiol. 1979;41:473–84. doi: 10.1146/annurev.ph.41.030179.002353. [DOI] [PubMed] [Google Scholar]
- 22.Rybin VO, Xu X, Lisanti MP, Steinberg SF. J Biol Chem. 2000;275:41447–57. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- 23.Brittsan AG, Kranias EG. J Mol Cell Cardiol. 2000;32:2131–9. doi: 10.1006/jmcc.2000.1270. [DOI] [PubMed] [Google Scholar]
- 24.Lindemann JP, Jones LR, Hathaway DR, Henry BG, Watanabe AM. J Biol Chem. 1983;258:464–71. [PubMed] [Google Scholar]
- 25.Zhang R, Zhao J, Mandveno A, Potter JD. Circ Res. 1995;76:1028–35. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 26.Brodde OE, Michel MC. Pharmacol Rev. 1999;51:651–90. [PubMed] [Google Scholar]
- 27.Daaka Y, Luttrell LM, Lefkowitz RJ. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 28.Kohout TA, Takaoka H, McDonald PH, Perry SJ, Mao L, Lefkowitz RJ, Rockman HA. Circulation. 2001;104:2485–91. doi: 10.1161/hc4501.098933. [DOI] [PubMed] [Google Scholar]
- 29.Gauthier C, Tavernier G, Charpentier F, Langin D, Le Marec H. J Clin Invest. 1996;98:556–62. doi: 10.1172/JCI118823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. J Biol Chem. 1999;274:16694–700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 31.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Proc Natl Acad Sci U S A. 1999;96:7059–64. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tilley DG, Rockman HA. Expert Rev Cardiovasc Ther. 2006;4:417–32. doi: 10.1586/14779072.4.3.417. [DOI] [PubMed] [Google Scholar]
- 33.Zheng M, Zhang SJ, Zhu WZ, Ziman B, Kobilka BK, Xiao RP. J Biol Chem. 2000;275:40635–40. doi: 10.1074/jbc.M006325200. [DOI] [PubMed] [Google Scholar]
- 34.Devic E, Xiang Y, Gould D, Kobilka B. Mol Pharmacol. 2001;60:577–83. [PubMed] [Google Scholar]
- 35.Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, Lefkowitz RJ, Koch WJ, Lakatta EG. Circ Res. 1999;84:43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 36.Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW., 2nd Circulation. 2000;101:1707–14. doi: 10.1161/01.cir.101.14.1707. [DOI] [PubMed] [Google Scholar]
- 37.Milano CA, Allen LF, Rockman HA, Dolber PC, McMinn TR, Chien KR, Johnson TD, Bond RA, Lefkowitz RJ. Science. 1994;264:582–6. doi: 10.1126/science.8160017. [DOI] [PubMed] [Google Scholar]
- 38.Gaudin C, Ishikawa Y, Wight DC, Mahdavi V, Nadal-Ginard B, Wagner TE, Vatner DE, Homcy CJ. J Clin Invest. 1995;95:1676–83. doi: 10.1172/JCI117843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwase M, Bishop SP, Uechi M, Vatner DE, Shannon RP, Kudej RK, Wight DC, Wagner TE, Ishikawa Y, Homcy CJ, Vatner SF. Circ Res. 1996;78:517–24. doi: 10.1161/01.res.78.4.517. [DOI] [PubMed] [Google Scholar]
- 40.Marks AR. J Mol Cell Cardiol. 2001;33:615–24. doi: 10.1006/jmcc.2000.1343. [DOI] [PubMed] [Google Scholar]
- 41.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 42.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Circulation. 2001;103:485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 43.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Hum Mol Genet. 2001;10:189–94. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 44.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Circulation. 1995;91:1512–9. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 45.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 46.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Science. 1997;276:800–6. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 47.Reiken S, Wehrens XH, Vest JA, Barbone A, Klotz S, Mancini D, Burkhoff D, Marks AR. Circulation. 2003;107:2459–66. doi: 10.1161/01.CIR.0000068316.53218.49. [DOI] [PubMed] [Google Scholar]
- 48.Reiken S, Gaburjakova M, Gaburjakova J, He KL, Prieto A, Becker E, Yi GH, Wang J, Burkhoff D, Marks AR. Circulation. 2001;104:2843–8. doi: 10.1161/hc4701.099578. [DOI] [PubMed] [Google Scholar]
- 49.Doi M, Yano M, Kobayashi S, Kohno M, Tokuhisa T, Okuda S, Suetsugu M, Hisamatsu Y, Ohkusa T, Kohno M, Matsuzaki M. Circulation. 2002;105:1374–9. doi: 10.1161/hc1102.105270. [DOI] [PubMed] [Google Scholar]
- 50.Yano M, Yamamoto T, Ikeda Y, Matsuzaki M. Nat Clin Pract Cardiovasc Med. 2006;3:43–52. doi: 10.1038/ncpcardio0419. [DOI] [PubMed] [Google Scholar]
- 51.Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Kohno M, Matsuzaki M. Circulation. 2003;107:477–84. doi: 10.1161/01.cir.0000044917.74408.be. [DOI] [PubMed] [Google Scholar]
- 52.Santana LF, Kranias EG, Lederer WJ. J Physiol. 1997;503( Pt 1):21–9. doi: 10.1111/j.1469-7793.1997.021bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Science. 2003;299:1410–3. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 54.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. Proc Natl Acad Sci U S A. 2006;103:1388–93. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. J Clin Invest. 2003;111:869–76. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brittsan AG, Carr AN, Schmidt AG, Kranias EG. J Biol Chem. 2000;275:12129–35. doi: 10.1074/jbc.275.16.12129. [DOI] [PubMed] [Google Scholar]
- 57.Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn GW, 2nd, Walsh RA, Kranias EG. J Clin Invest. 1996;97:533–9. doi: 10.1172/JCI118446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG. Circ Res. 1994;75:401–9. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
- 59.Minamisawa S, Hoshijima M, Chu G, Ward CA, Frank K, Gu Y, Martone ME, Wang Y, Ross J, Jr, Kranias EG, Giles WR, Chien KR. Cell. 1999;99:313–22. doi: 10.1016/s0092-8674(00)81662-1. [DOI] [PubMed] [Google Scholar]
- 60.Ungerer M, Bohm M, Elce JS, Erdmann E, Lohse MJ. Circulation. 1993;87:454–63. doi: 10.1161/01.cir.87.2.454. [DOI] [PubMed] [Google Scholar]
- 61.Bristow MR, Hershberger RE, Port JD, Minobe W, Rasmussen R. Mol Pharmacol. 1989;35:295–303. [PubMed] [Google Scholar]
- 62.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. N Engl J Med. 1997;336:1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 63.Communal C, Singh K, Pimentel DR, Colucci WS. Circulation. 1998;98:1329–34. doi: 10.1161/01.cir.98.13.1329. [DOI] [PubMed] [Google Scholar]
- 64.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. Am J Physiol. 1998;275:H961–8. doi: 10.1152/ajpheart.1998.275.3.H961. [DOI] [PubMed] [Google Scholar]
- 65.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. Circ Res. 2003;92:912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 66.Pieske B, Beyermann B, Breu V, Loffler BM, Schlotthauer K, Maier LS, Schmidt–Schweda S, Just H, Hasenfuss G. Circulation. 1999;99:1802–9. doi: 10.1161/01.cir.99.14.1802. [DOI] [PubMed] [Google Scholar]
- 67.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Nat Med. 2005;11:409–17. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 68.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, Yang J, Price EE, Gleaves L, Eren M, Ni G, Vaughan DE, Xiao RP, Anderson ME. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 69.Dougherty C, Barucha J, Schofield PR, Jacobson KA, Liang BT. Faseb J. 1998;12:1785–92. doi: 10.1096/fasebj.12.15.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mery PF, Brechler V, Pavoine C, Pecker F, Fischmeister R. Nature. 1990;345:158–61. doi: 10.1038/345158a0. [DOI] [PubMed] [Google Scholar]
- 71.Mendez M, LaPointe MC. Hypertension. 2002;39:382–8. doi: 10.1161/hy02t2.102808. [DOI] [PubMed] [Google Scholar]
- 72.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Cardiovasc Res. 1999;42:254–61. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 73.Ely SW, Berne RM. Circulation. 1992;85:893–904. doi: 10.1161/01.cir.85.3.893. [DOI] [PubMed] [Google Scholar]
- 74.Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP. Proc Natl Acad Sci U S A. 1997;94:6541–6. doi: 10.1073/pnas.94.12.6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Babbitt DG, Virmani R, Forman MB. Circulation. 1989;80:1388–99. doi: 10.1161/01.cir.80.5.1388. [DOI] [PubMed] [Google Scholar]
- 76.Olafsson B, Forman MB, Puett DW, Pou A, Cates CU, Friesinger GC, Virmani R. Circulation. 1987;76:1135–45. doi: 10.1161/01.cir.76.5.1135. [DOI] [PubMed] [Google Scholar]
- 77.Francois H, Athirakul K, Howell D, Dash R, Mao L, Kim HS, Rockman HA, Fitzgerald GA, Koller BH, Coffman TM. Cell Metab. 2005;2:201–7. doi: 10.1016/j.cmet.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 78.Genovese A, Adinolfi L, Quattrin S, Accinni A, Spadaro G, Dote I, Marone G. Agents Actions. 1985;17:42–5. doi: 10.1007/BF01966679. [DOI] [PubMed] [Google Scholar]
- 79.Koyama M, Seyedi N, Fung-Leung WP, Lovenberg TW, Levi R. Mol Pharmacol. 2003;63:378–82. doi: 10.1124/mol.63.2.378. [DOI] [PubMed] [Google Scholar]
- 80.Takahara A, Sugiyama A, Ishida Y, Satoh Y, Wang K, Nakamura Y, Hashimoto K. Br J Pharmacol. 2006;147:634–41. doi: 10.1038/sj.bjp.0706493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hubbard KB, Hepler JR. Cell Signal. 2006;18:135–50. doi: 10.1016/j.cellsig.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 82.Rohde S, Sabri A, Kamasamudran R, Steinberg SF. J Mol Cell Cardiol. 2000;32:1193–209. doi: 10.1006/jmcc.2000.1153. [DOI] [PubMed] [Google Scholar]
- 83.Dorn GW, 2nd, Force T. J Clin Invest. 2005;115:527–37. doi: 10.1172/JCI24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Simpson P. Circ Res. 1985;56:884–94. doi: 10.1161/01.res.56.6.884. [DOI] [PubMed] [Google Scholar]
- 85.Simpson P, McGrath A, Savion S. Circ Res. 1982;51:787–801. doi: 10.1161/01.res.51.6.787. [DOI] [PubMed] [Google Scholar]
- 86.Simpson PC, Kariya K, Karns LR, Long CS, Karliner JS. Mol Cell Biochem. 1991;104:35–43. doi: 10.1007/BF00229801. [DOI] [PubMed] [Google Scholar]
- 87.Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA. Mol Cell Biol. 2004;24:8374–85. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Harrison BC, Kim MS, van Rooij E, Plato CF, Papst PJ, Vega RB, McAnally JA, Richardson JA, Bassel-Duby R, Olson EN, McKinsey TA. Mol Cell Biol. 2006;26:3875–88. doi: 10.1128/MCB.26.10.3875-3888.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Naruse K, King GL. Circ Res. 2000;86:1104–6. doi: 10.1161/01.res.86.11.1104. [DOI] [PubMed] [Google Scholar]
- 90.Hahn HS, Marreez Y, Odley A, Sterbling A, Yussman MG, Hilty KC, Bodi I, Liggett SB, Schwartz A, Dorn GW., 2nd Circ Res. 2003;93:1111–9. doi: 10.1161/01.RES.0000105087.79373.17. [DOI] [PubMed] [Google Scholar]
- 91.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Nat Med. 2004;10:248–54. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 92.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, King GL. Proc Natl Acad Sci U S A. 1997;94:9320–5. doi: 10.1073/pnas.94.17.9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bowman JC, Steinberg SF, Jiang T, Geenen DL, Fishman GI, Buttrick PM. J Clin Invest. 1997;100:2189–95. doi: 10.1172/JCI119755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin F, Owens WA, Chen S, Stevens ME, Kesteven S, Arthur JF, Woodcock EA, Feneley MP, Graham RM. Circ Res. 2001;89:343–50. doi: 10.1161/hh1601.095912. [DOI] [PubMed] [Google Scholar]
- 95.Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D. Circulation. 2003;108:2304–7. doi: 10.1161/01.CIR.0000101682.24138.36. [DOI] [PubMed] [Google Scholar]
- 96.Inagaki K, Churchill E, Mochly-Rosen D. Cardiovasc Res. 2006;70:222–30. doi: 10.1016/j.cardiores.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 97.Marks AR. Circulation. 2002;106:8–10. doi: 10.1161/01.cir.0000021746.82888.83. [DOI] [PubMed] [Google Scholar]
- 98.Cohn JN, Tognoni G. N Engl J Med. 2001;345:1667–75. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 99.Lindholm LH, Ibsen H, Dahlof B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, Snapinn S. Lancet. 2002;359:1004–10. doi: 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- 100.Pitt B, Poole-Wilson PA, Segal R, Martinez FA, Dickstein K, Camm AJ, Konstam MA, Riegger G, Klinger GH, Neaton J, Sharma D, Thiyagarajan B. Lancet. 2000;355:1582–7. doi: 10.1016/s0140-6736(00)02213-3. [DOI] [PubMed] [Google Scholar]
- 101.Pfeffer MA, Braunwald E, Moye LA, Basta L, Brown EJ, Jr, Cuddy TE, Davis BR, Geltman EM, Goldman S, Flaker GC, et al. N Engl J Med. 1992;327:669–77. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 102.Kober L, Torp-Pedersen C, Carlsen JE, Bagger H, Eliasen P, Lyngborg K, Videbaek J, Cole DS, Auclert L, Pauly NC. N Engl J Med. 1995;333:1670–6. doi: 10.1056/NEJM199512213332503. [DOI] [PubMed] [Google Scholar]
- 103.Wollert KC, Drexler H. Cardiovasc Res. 1999;43:838–49. doi: 10.1016/s0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 104.Schunkert H, Jackson B, Tang SS, Schoen FJ, Smits JF, Apstein CS, Lorell BH. Circulation. 1993;87:1328–39. doi: 10.1161/01.cir.87.4.1328. [DOI] [PubMed] [Google Scholar]
- 105.Studer R, Reinecke H, Muller B, Holtz J, Just H, Drexler H. J Clin Invest. 1994;94:301–10. doi: 10.1172/JCI117322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun Y, Cleutjens JP, Diaz-Arias AA, Weber KT. Cardiovasc Res. 1994;28:1423–32. doi: 10.1093/cvr/28.9.1423. [DOI] [PubMed] [Google Scholar]
- 107.Touyz RM, Schiffrin EL. Pharmacol Rev. 2000;52:639–72. [PubMed] [Google Scholar]
- 108.Lambert C, Massillon Y, Meloche S. Circ Res. 1995;77:1001–7. doi: 10.1161/01.res.77.5.1001. [DOI] [PubMed] [Google Scholar]
- 109.Suzuki J, Matsubara H, Urakami M, Inada M. Circ Res. 1993;73:439–47. doi: 10.1161/01.res.73.3.439. [DOI] [PubMed] [Google Scholar]
- 110.Meggs LG, Coupet J, Huang H, Cheng W, Li P, Capasso JM, Homcy CJ, Anversa P. Circ Res. 1993;72:1149–62. doi: 10.1161/01.res.72.6.1149. [DOI] [PubMed] [Google Scholar]
- 111.Regitz-Zagrosek V, Friedel N, Heymann A, Bauer P, Neuss M, Rolfs A, Steffen C, Hildebrandt A, Hetzer R, Fleck E. Circulation. 1995;91:1461–71. doi: 10.1161/01.cir.91.5.1461. [DOI] [PubMed] [Google Scholar]
- 112.Rogg H, de Gasparo M, Graedel E, Stulz P, Burkart F, Eberhard M, Erne P. Eur Heart J. 1996;17:1112–20. doi: 10.1093/oxfordjournals.eurheartj.a015008. [DOI] [PubMed] [Google Scholar]
- 113.Regitz-Zagrosek V, Fielitz J, Dreysse R, Hildebrandt AG, Fleck E. Cardiovasc Res. 1997;35:99–105. doi: 10.1016/s0008-6363(97)00089-8. [DOI] [PubMed] [Google Scholar]
- 114.Makita N, Iwai N, Inagami T, Badr KF. Biochem Biophys Res Commun. 1992;185:142–6. doi: 10.1016/s0006-291x(05)80967-2. [DOI] [PubMed] [Google Scholar]
- 115.Paradis P, Dali-Youcef N, Paradis FW, Thibault G, Nemer M. Proc Natl Acad Sci U S A. 2000;97:931–6. doi: 10.1073/pnas.97.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harada K, Sugaya T, Murakami K, Yazaki Y, Komuro I. Circulation. 1999;100:2093–9. doi: 10.1161/01.cir.100.20.2093. [DOI] [PubMed] [Google Scholar]
- 117.Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Circulation. 1998;97:1952–9. doi: 10.1161/01.cir.97.19.1952. [DOI] [PubMed] [Google Scholar]
- 118.Harada K, Komuro I, Zou Y, Kudoh S, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y. Circ Res. 1998;82:779–85. doi: 10.1161/01.res.82.7.779. [DOI] [PubMed] [Google Scholar]
- 119.Exton JH. Am J Physiol. 1985;248:E633–47. doi: 10.1152/ajpendo.1985.248.6.E633. [DOI] [PubMed] [Google Scholar]
- 120.Rorabaugh BR, Gaivin RJ, Papay RS, Shi T, Simpson PC, Perez DM. J Mol Cell Cardiol. 2005;39:777–84. doi: 10.1016/j.yjmcc.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 121.Bristow MR, Minobe W, Rasmussen R, Hershberger RE, Hoffman BB. J Pharmacol Exp Ther. 1988;247:1039–45. [PubMed] [Google Scholar]
- 122.Akhter SA, Milano CA, Shotwell KF, Cho MC, Rockman HA, Lefkowitz RJ, Koch WJ. J Biol Chem. 1997;272:21253–9. doi: 10.1074/jbc.272.34.21253. [DOI] [PubMed] [Google Scholar]
- 123.O’Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC. J Clin Invest. 2006;116:1005–15. doi: 10.1172/JCI22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cavalli A, Lattion AL, Hummler E, Nenniger M, Pedrazzini T, Aubert JF, Michel MC, Yang M, Lembo G, Vecchione C, Mostardini M, Schmidt A, Beermann F, Cotecchia S. Proc Natl Acad Sci U S A. 1997;94:11589–94. doi: 10.1073/pnas.94.21.11589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.O’Connell TD, Ishizaka S, Nakamura A, Swigart PM, Rodrigo MC, Simpson GL, Cotecchia S, Rokosh DG, Grossman W, Foster E, Simpson PC. J Clin Invest. 2003;111:1783–91. doi: 10.1172/JCI16100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Asano K, Bohlmeyer TJ, Westcott JY, Zisman L, Kinugawa K, Good M, Minobe WA, Roden R, Wolfel EE, Lindenfeld J, David Port J, Perryman MB, Clevel J, Lowes BD, Bristow MR. J Mol Cell Cardiol. 2002;34:833–46. doi: 10.1006/jmcc.2002.2022. [DOI] [PubMed] [Google Scholar]
- 127.Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K, Kurihara H, Aikawa R, Takano H, Yazaki Y. J Biol Chem. 1996;271:3221–8. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- 128.Yang LL, Gros R, Kabir MG, Sadi A, Gotlieb AI, Husain M, Stewart DJ. Circulation. 2004;109:255–61. doi: 10.1161/01.CIR.0000105701.98663.D4. [DOI] [PubMed] [Google Scholar]
- 129.Ponicke K, Vogelsang M, Heinroth M, Becker K, Zolk O, Bohm M, Zerkowski HR, Brodde OE. Circulation. 1998;97:744–51. doi: 10.1161/01.cir.97.8.744. [DOI] [PubMed] [Google Scholar]
- 130.Serneri GG, Cecioni I, Vanni S, Paniccia R, Bandinelli B, Vetere A, Janming X, Bertolozzi I, Boddi M, Lisi GF, Sani G, Modesti PA. Circ Res. 2000;86:377–85. doi: 10.1161/01.res.86.4.377. [DOI] [PubMed] [Google Scholar]
- 131.Walker CA, Ergul A, Grubbs A, Zile MR, Zellner JL, Crumbley AJ, Spinale FG. J Card Fail. 2001;7:129–37. doi: 10.1054/jcaf.2001.24125. [DOI] [PubMed] [Google Scholar]
- 132.Cody RJ, Haas GJ, Binkley PF, Capers Q, Kelley R. Circulation. 1992;85:504–9. doi: 10.1161/01.cir.85.2.504. [DOI] [PubMed] [Google Scholar]
- 133.Wei CM, Lerman A, Rodeheffer RJ, McGregor CG, Brandt RR, Wright S, Heublein DM, Kao PC, Edwards WD, Burnett JC., Jr Circulation. 1994;89:1580–6. doi: 10.1161/01.cir.89.4.1580. [DOI] [PubMed] [Google Scholar]
- 134.Sakai S, Miyauchi T, Kobayashi M, Yamaguchi I, Goto K, Sugishita Y. Nature. 1996;384:353–5. doi: 10.1038/384353a0. [DOI] [PubMed] [Google Scholar]
- 135.Shimoyama H, Sabbah HN, Borzak S, Tanimura M, Shevlyagin S, Scicli G, Goldstein S. Circulation. 1996;94:779–84. doi: 10.1161/01.cir.94.4.779. [DOI] [PubMed] [Google Scholar]
- 136.Spinale FG, Walker JD, Mukherjee R, Iannini JP, Keever AT, Gallagher KP. Circulation. 1997;95:1918–29. doi: 10.1161/01.cir.95.7.1918. [DOI] [PubMed] [Google Scholar]
- 137.Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, Ruschitzka F, Luscher TF. Lancet. 2004;364:347–54. doi: 10.1016/S0140-6736(04)16723-8. [DOI] [PubMed] [Google Scholar]
- 138.Rich S, McLaughlin VV. Circulation. 2003;108:2184–90. doi: 10.1161/01.CIR.0000094397.19932.78. [DOI] [PubMed] [Google Scholar]
- 139.D’Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, Dorn GW., 2nd Proc Natl Acad Sci U S A. 1997;94:8121–6. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mende U, Kagen A, Cohen A, Aramburu J, Schoen FJ, Neer EJ. Proc Natl Acad Sci U S A. 1998;95:13893–8. doi: 10.1073/pnas.95.23.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH, Dorn GW, 2nd, Armstrong RC, Kitsis RN. Circulation. 2003;108:3036–41. doi: 10.1161/01.CIR.0000101920.72665.58. [DOI] [PubMed] [Google Scholar]
- 142.Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW., 2nd Circulation. 1998;97:1488–95. doi: 10.1161/01.cir.97.15.1488. [DOI] [PubMed] [Google Scholar]
- 143.Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, Chien KR, Brown JH, Dorn GW., 2nd Proc Natl Acad Sci U S A. 1998;95:10140–5. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ. Science. 1998;280:574–7. doi: 10.1126/science.280.5363.574. [DOI] [PubMed] [Google Scholar]
- 145.Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 146.Bowling N, Walsh RA, Song G, Estridge T, Sandusky GE, Fouts RL, Mintze K, Pickard T, Roden R, Bristow MR, Sabbah HN, Mizrahi JL, Gromo G, King GL, Vlahos CJ. Circulation. 1999;99:384–91. doi: 10.1161/01.cir.99.3.384. [DOI] [PubMed] [Google Scholar]
- 147.Nebigil CG, Hickel P, Messaddeq N, Vonesch JL, Douchet MP, Monassier L, Gyorgy K, Matz R, Andriantsitohaina R, Manivet P, Launay JM, Maroteaux L. Circulation. 2001;103:2973–9. doi: 10.1161/01.cir.103.24.2973. [DOI] [PubMed] [Google Scholar]
- 148.Nebigil CG, Maroteaux L. Trends Cardiovasc Med. 2001;11:329–35. doi: 10.1016/s1050-1738(01)00135-9. [DOI] [PubMed] [Google Scholar]
- 149.Nakamura F, Minshall RD, Le Breton GC, Rabito SF. Hypertension. 1996;28:444–9. doi: 10.1161/01.hyp.28.3.444. [DOI] [PubMed] [Google Scholar]
- 150.Genovese A, Spadaro G. Allergy. 1997;52:67–78. doi: 10.1111/j.1398-9995.1997.tb04813.x. [DOI] [PubMed] [Google Scholar]
- 151.Tobin AB, Budd DC. Biochem Soc Trans. 2003;31:1182–5. doi: 10.1042/bst0311182. [DOI] [PubMed] [Google Scholar]
- 152.Sharma JN, Thani RB. IDrugs. 2004;7:926–34. [PubMed] [Google Scholar]
- 153.Redfern CH, Degtyarev MY, Kwa AT, Salomonis N, Cotte N, Nanevicz T, Fidelman N, Desai K, Vranizan K, Lee EK, Coward P, Shah N, Warrington JA, Fishman GI, Bernstein D, Baker AJ, Conklin BR. Proc Natl Acad Sci U S A. 2000;97:4826–31. doi: 10.1073/pnas.97.9.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bohm M, Gierschik P, Jakobs KH, Pieske B, Schnabel P, Ungerer M, Erdmann E. Circulation. 1990;82:1249–65. doi: 10.1161/01.cir.82.4.1249. [DOI] [PubMed] [Google Scholar]
- 155.Flesch M, Schwinger RH, Schnabel P, Schiffer F, van Gelder I, Bavendiek U, Sudkamp M, Kuhn-Regnier F, Bohm M. J Mol Med. 1996;74:321–32. doi: 10.1007/BF00207509. [DOI] [PubMed] [Google Scholar]
- 156.Wilbur SL, Marchlinski FE. Am J Cardiol. 1997;79:30–7. doi: 10.1016/s0002-9149(97)00261-0. [DOI] [PubMed] [Google Scholar]
- 157.Donato M, Gelpi RJ. Mol Cell Biochem. 2003;251:153–9. [PubMed] [Google Scholar]
- 158.Hutchinson SA, Scammells PJ. Curr Pharm Des. 2004;10:2021–39. doi: 10.2174/1381612043384204. [DOI] [PubMed] [Google Scholar]
- 159.Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. Nature. 1987;325:321–6. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- 160.von Scheidt W, Bohm M, Schneider B, Reichart B, Erdmann E, Autenrieth G. Circulation. 1992;85:1056–63. doi: 10.1161/01.cir.85.3.1056. [DOI] [PubMed] [Google Scholar]
- 161.Landzberg JS, Parker JD, Gauthier DF, Colucci WS. Circulation. 1994;89:164–8. doi: 10.1161/01.cir.89.1.164. [DOI] [PubMed] [Google Scholar]
- 162.Newton GE, Parker AB, Landzberg JS, Colucci WS, Parker JD. J Clin Invest. 1996;98:2756–63. doi: 10.1172/JCI119101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Le Guludec D, Cohen-Solal A, Delforge J, Delahaye N, Syrota A, Merlet P. Circulation. 1997;96:3416–22. doi: 10.1161/01.cir.96.10.3416. [DOI] [PubMed] [Google Scholar]
- 164.Aggarwal A, Esler MD, Socratous F, Kaye DM. J Am Coll Cardiol. 2001;37:1246–51. doi: 10.1016/s0735-1097(01)01121-4. [DOI] [PubMed] [Google Scholar]
- 165.Altman JD, Trendelenburg AU, MacMillan L, Bernstein D, Limbird L, Starke K, Kobilka BK, Hein L. Mol Pharmacol. 1999;56:154–61. doi: 10.1124/mol.56.1.154. [DOI] [PubMed] [Google Scholar]
- 166.Small KM, Wagoner LE, Levin AM, Kardia SL, Liggett SB. N Engl J Med. 2002;347:1135–42. doi: 10.1056/NEJMoa020803. [DOI] [PubMed] [Google Scholar]
- 167.Sibley DR, Lefkowitz RJ. Nature. 1985;317:124–9. doi: 10.1038/317124a0. [DOI] [PubMed] [Google Scholar]
- 168.Shenoy SK, Lefkowitz RJ. Sci STKE. 2005;2005:cm10. doi: 10.1126/stke.2005/308/cm10. [DOI] [PubMed] [Google Scholar]
- 169.Ford CE, Skiba NP, Bae H, Daaka Y, Reuveny E, Shekter LR, Rosal R, Weng G, Yang CS, Iyengar R, Miller RJ, Jan LY, Lefkowitz RJ, Hamm HE. Science. 1998;280:1271–4. doi: 10.1126/science.280.5367.1271. [DOI] [PubMed] [Google Scholar]
- 170.Naga Prasad SV, Esposito G, Mao L, Koch WJ, Rockman HA. J Biol Chem. 2000;275:4693–8. doi: 10.1074/jbc.275.7.4693. [DOI] [PubMed] [Google Scholar]
- 171.Morris AJ, Malbon CC. Physiol Rev. 1999;79:1373–430. doi: 10.1152/physrev.1999.79.4.1373. [DOI] [PubMed] [Google Scholar]
- 172.Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. J Biol Chem. 1994;269:6193–7. [PubMed] [Google Scholar]
- 173.Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Proc Natl Acad Sci U S A. 1994;91:12706–10. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Touhara K, Lefkowitz RJ. J Biol Chem. 1995;270:16495–8. doi: 10.1074/jbc.270.28.16495. [DOI] [PubMed] [Google Scholar]
- 175.Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Krueger KM, Touhara K, Lefkowitz RJ. Adv Second Messenger Phosphoprotein Res. 1997;31:263–77. [PubMed] [Google Scholar]
- 176.Koch WJ, Rockman HA, Samama P, Hamilton RA, Bond RA, Milano CA, Lefkowitz RJ. Science. 1995;268:1350–3. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 177.Jaber M, Koch WJ, Rockman H, Smith B, Bond RA, Sulik KK, Ross J, Jr, Lefkowitz RJ, Caron MG, Giros B. Proc Natl Acad Sci U S A. 1996;93:12974–9. doi: 10.1073/pnas.93.23.12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2001;98:2449–54. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, Lefkowitz RJ, Luttrell LM. J Biol Chem. 2000;275:9572–80. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- 180.Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. Proc Natl Acad Sci U S A. 2002;99:7478–83. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Conner DA, Mathier MA, Mortensen RM, Christe M, Vatner SF, Seidman CE, Seidman JG. Circ Res. 1997;81:1021–6. doi: 10.1161/01.res.81.6.1021. [DOI] [PubMed] [Google Scholar]
- 182.Nienaber JJ, Tachibana H, Naga Prasad SV, Esposito G, Wu D, Mao L, Rockman HA. J Clin Invest. 2003;112:1067–79. doi: 10.1172/JCI18213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, Hirsch E, Suzuki A, Shioi T, Irie-Sasaki J, Sah R, Cheng HY, Rybin VO, Lembo G, Fratta L, Oliveira-dos-Santos AJ, Benovic JL, Kahn CR, Izumo S, Steinberg SF, Wymann MP, Backx PH, Penninger JM. Cell. 2002;110:737–49. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 184.Shioi T, Kang PM, Douglas PS, Hampe J, Yballe CM, Lawitts J, Cantley LC, Izumo S. Embo J. 2000;19:2537–48. doi: 10.1093/emboj/19.11.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Jo SH, Leblais V, Wang PH, Crow MT, Xiao RP. Circ Res. 2002;91:46–53. doi: 10.1161/01.res.0000024115.67561.54. [DOI] [PubMed] [Google Scholar]
- 186.Naga Prasad SV, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. J Biol Chem. 2001;276:18953–9. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- 187.Perrino C, Naga Prasad SV, Patel M, Wolf MJ, Rockman HA. J Am Coll Cardiol. 2005;45:1862–70. doi: 10.1016/j.jacc.2005.02.062. [DOI] [PubMed] [Google Scholar]
- 188.Perrino C, Prasad SVN, Mao L, Noma T, Yan Z, Kim HS, Smithies O, Rockman HA. J Clin Invest. 2006;116:1547–1560. doi: 10.1172/JCI25397. [DOI] [PMC free article] [PubMed] [Google Scholar]