

Abstract
Parathyroid hormone-related protein (PTHrP) has been localized in human colon cancer tissue and cell lines. We have previously shown that PTHrP increases colon cancer cell proliferation, extracellular matrix adhesion, and cell-surface integrin α6β4 expression. Since cancer cell migration, invasion, and survival are crucial components of metastasis, and colon cancer has a high metastatic potential, in this study we used the human colon cancer cell line LoVo as a model system to study the effects of PTHrP on these parameters. PTHrP expression was modulated by stable transfection with a construct expressing PTHrP (−36 to +139). We report that PTHrP increases cell migration, invasion, and survival. PTHrP altered cell morphology, with PTHrP-overexpressing cells exhibiting increased spreading and several long protrusions. PTHrP also increased the steady-state mRNA levels of the integrin α6 and β4 subunits, indicating a direct and/or indirect effect of PTHrP on the transcriptional and/or posttranscriptional regulation of integrin α6 and β4 expression. Integrin α6β4 activates the phosphoinositol 3-kinase (PI3-K)/Akt pathway, leading to glycogen synthase kinase-3 (GSK-3) deactivation. PTHrP overexpression also led to an increase in active Akt and inactive GSK-3 levels, indicating that the PTHrP-mediated upregulation of integrin α6β4 expression may activate the PI3-K pathway, resulting in increased cell survival, migration and invasion.
Keywords: Parathyroid hormone-related protein, integrin α6β4, apoptosis, glycogen synthase kinase-3, laminin
Introduction
Colorectal adenocarcinoma accounts for over 90% of the malignant tumors of the large bowel, and is the second most common cause of death from malignant disease in the Western world [1,2]. As with other cancer cells, colon cancer invasion and metastasis requires that a cell acquire a motile phenotype to penetrate tissue and reach the vasculature and lymphatics [3,4]. Cancer cells also adopt survival mechanisms, thereby avoiding apoptosis [5]. These properties are dependent on inherent tumor cell characteristics [3,6,7], and on the presence of several growth factors in the metastatic microenvironment. One of these factors is parathyroid hormone-related protein (PTHrP).
PTHrP was originally identified as the causative agent of humoral hypercalcemia of malignancy, one of the most frequent paraneoplastic syndromes [8]. PTHrP undergoes extensive post-translational processing to generate secretory forms representing N-terminal, mid-region, and C-terminal isoforms. Each of these secretory forms appears to act via one or more distinct cell-surface receptors [9,10]. To date, only the parathyroid hormone/PTHrP (PTH1) receptor that binds parathyroid hormone (PTH), PTHrP, and its N-terminal analogs has been cloned [11].
Subsequently, PTHrP was found to be widely distributed in most fetal and adult tissues, including the gut mucosal epithelium [12-14]. Widespread expression of PTHrP and the PTH/PTHrP receptor genes and proteins has been found in gut villus epithelium [15,16], suggesting that PTHrP exerts a local regulatory role via an autocrine/paracrine pathway. PTHrP also functions via an intracrine pathway after translocation to the nucleus or nucleolus, and the 88-106 region contains multibasic clusters that have homology to nuclear and nucleolar import signals found in multiple transcription factors [17,18]. The mechanism via which PTHrP affects the proliferation of the rat intestinal crypt cell line IEC-6 was studied by overexpressing wild-type PTHrP or PTHrP deleted over the NLS. Using these PTHrP-overexpressing clones, it was concluded that PTHrP increases the proliferation of this cell line via an intracrine pathway [19].
PTHrP is also expressed by cancer cells not typically associated with hypercalcemia [20-24]. It has been shown that PTHrP expression correlates with the severity of colon carcinoma — specifically, with cell differentiation, depth of invasion, lymphatic invasion, lymph node and hepatic metastasis, and Dukes' classification [21]. We have previously shown that PTHrP increases the proliferation of the human colon cancer cell line LoVo [20]. This cell line was originated from a left supraclavicular region metastasis of a Dukes' type C, grade IV colorectal carcinoma. LoVo cells are tumorigenic when injected into nude mice [2,25]. PTHrP also increases the adhesion of LoVo cells to the extracellular matrix (ECM) proteins collagen type I, fibronectin, and laminin [20]. The PTHrP-mediated increase in cell proliferation and adhesion favors tumor progression. Since cancer cell migration, invasion, and survival are also crucial components of metastasis, here we asked whether PTHrP plays a role in these processes in colon cancer cells.
PTHrP upregulates the expression of the pro-invasive integrin α6β4 in multiple cell lines [20,24,26]. Integrins are transmembrane receptors comprised of αβ heterodimers that play a major role in cancer cell behavior through their ability to cooperate with growth factor signaling [27,28]. Integrins have been shown to regulate cell growth, differentiation, apoptosis, cell motility, and gene expression [29,30]. The extracellular domain of the β4 subunit associates exclusively with the α6 subunit to form α6β4 heterodimers [31]. Multiple studies have shown that integrin α6β4 contributes to the functions of carcinoma cells, including colon cancer cells [32-36]. Since integrin α6β4 activates phosphoinositol 3-kinase (PI3-K) [35], here we also investigated the effect of PTHrP on active levels of Akt, a downstream effector of PI3-K [37]. Akt activation results in the phosphorylation, and consequent inactivation, of glycogen synthase kinase-3 (GSK-3) [38]. Therefore, we also asked whether PTHrP modulates active GSK-3 levels.
Materials and Methods
Materials
Fetal bovine serum (FBS) was obtained from Atlanta Biologicals (Norcross, GA). Tissue culture supplies were purchased from Life Technologies, Inc. (Gaithersburg, MD). The R-PE-conjugated anti-p-Akt antibody (p-Thr 308), used for FACS analysis, was obtained from BD Pharmingen (San Diego, CA). The anti p-Akt antibody (p-Ser 473), used for Western blot and FACS analyses, was obtained from Cell Signaling Technologies (Danvers, MA). ). The anti-total Akt antibody was obtained from BD Pharmingen. The anti-p-GSK-3α/β (Ser21/Ser9) antibody and the anti-total GSK-3α/β antibody, used for FACS analysis and Western blot analysis, were obtained from Cell Signaling Technologies (Danvers, MA). The isotype control antibodies for p-Akt, p-GSK-3, total Akt and total GSK-3 were obtained from BD Pharmingen. The FluoroBlok inserts for analysis of migration and invasion were purchased from BD Pharmingen. Matrigel, mouse laminin, and NuSerum were obtained from BD Biosciences (San Diego, CA), and Calcein-AM was obtained from Molecular Probes (Eugene, OR).
Plasmid constructs
A cDNA encoding human PTHrP (obtained from Genentech, Inc., South San Francisco, CA) was digested with EcoRI and HindIII and subcloned in the sense orientation into the expression vector pcDNA3.1(+) (Invitrogen, San Diego, CA). The (+) refers to the orientation of the multiple cloning site within the vector, relative to the direction of transcription from the T7 promoter. This construct, as well as the empty vector control pcDNA 3.1(+), was transfected into LoVo cells using FuGENE™ 6 Transfection Reagent (Roche Molecular Biochemicals, Indianapolis, IN). pcDNA3.1 (+) was used as the empty vector control.
Cell culture and transfection
LoVo cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and were grown at 37°C in a humidified 95% air-5% CO2 atmosphere in F12 medium supplemented with 10% FBS and L-glutamine.
The cells were stably transfected using FuGENE™ 6 Transfection Reagent. Two days after transfection, 600 μg/ml G418 (Geneticin; Life Technologies Inc.) was added, and resistant clones were selected. Single clones of stably transfected cells, isolated by limiting dilution in 96-well plates, were transferred to individual flasks and cultured in medium containing 150 μg/ml G418. Individual PTHrP-overexpressing clones were tested for PTHrP secretion using an immunoradiometric assay (IRMA), and expressed mRNAs were measured by real-time Polymerase Chain Reaction (PCR) analysis. The characterization of the PTHrP-overexpressing LoVo cell clones has been described [20].
Analysis of PTHrP and integrin expression
PTHrP, integrin α2, integrin α6, and integrin β4 mRNA levels in cells transfected with the PTHrP-pcDNA3.1(+) construct were analyzed by real-time PCR. PTHrP secretion from cells transfected with the PTHrP-pcDNA3.1(+) expression vector was measured by IRMA (Nichols Institute, San Juan Capistrano, CA) [20].
Real-time Polymerase Chain Reaction Analysis of PTHrP, Integrin α2, Integrin α6, and Integrin β4 Gene Expression
Total RNA from LoVo cells was extracted using the RNAqueous® isolation kit (Ambion Inc., Austin, TX), per the manufacturer's protocol. RNA concentrations were determined by spectrophotometry.
Relative quantification of gene expression
Separate tube (singleplex) real-time PCR was performed using 0.5 μg of RNA as template to detect PTHrP, integrin α2, integrin α6 and integrin β4 mRNA transcripts and 18S rRNA (endogenous control for normalization purposes). PTHrP primers (Assays-on-Demand™, P/N 4331182, 20X assay mix of primers), the PTHrP TaqMan MGB probe (FAM™ dye-labeled, PTHLH (parathyroid hormone-like hormone), NM 002820, Hs00174969 m1: CGCCGCCTCAAAAGAGCTGTGTCTG), integrin α2 primers (ITGA2, NM_002203, Hs00158127_m1, TCAGTCAAGGCATTTTAAATTGTTG), integrin α6 primers (ITGA6, NM 000210, X59512, X53586, BC007002 Hs00173952_m1 AAGCGGCTGTTGCTCGTGGGGGCCC), integrin β4 primers (ITGB4, NM_001005619, NM_001005731, NM_000213, X51841, X52186, AF011375, Hs00173995_m1 GGTTCCTCTGCAATGACCGAGGACG) and the pre-developed 18S rRNA primers (VIC™-dye labeled probe, TaqMan® assay reagent, P/N 4319413E) were obtained from Applied Biosystems, as was the universal PCR master mix reagent kit (P/N 4304437). The cycling parameters for real-time PCR were: UNG activation at 50 °C for 2 min, AmpliTaq activation at 95 °C for 10 min, then denaturation at 95 °C for 15 sec and annealing/extension at 60 °C for 1 min (repeated 40 times) on an ABI7000 real-time PCR machine. Duplicate CT values were analyzed in Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems). The amount of target (2−ΔΔCT) was obtained by normalization to the endogenous reference (18S). Real-time PCR was performed using the facilities of UTMB's Sealy Center for Cancer Cell Biology's Real-Time PCR core facility; http://www.utmb.edu/scccb/pcr/index.htm. As negative controls, we carried out PCR reactions without RNA.
Flow cytometry
p-Akt, p-GSK-3, and total Akt and GSK-3 levels were measured by FACS, following BD Pharmingen's protocol. This methodology offers a novel, sensitive method to compare active and total kinase levels in cells. Cells at 50% to 80% confluence were rinsed twice with PBS and incubated with Accutase solution (Chemicon International, Temecula, CA) for 10 min at 37 °C. After centrifugation, the cells were fixed with 2% paraformaldehyde overnight at 4 °C, centrifuged, and then permeabilized by adding 1 ml of BD™ Phosflow Perm Buffer III and incubating for 30 min on ice. The cells were then washed twice with Stain Buffer (Becton Dickinson and Co). After centrifugation, the cell pellet was resuspended in 100 μl Stain Buffer containing the primary antibody. The following primary antibodies were used: R-PE-conjugated anti-p-Akt (Thr 308), anti-p-Akt (Ser 473), anti-total Akt, or the corresponding isotype control antibody (BD Pharmingen), anti-p-GSK-3α/β (Ser21/Ser9) (Cell Signaling Technologies), anti-total GSK-3α/β (Affinity Bioreagents), or the corresponding isotype control antibody (BD Pharmingen). After incubation for 30 min in the dark, the cells were washed twice with 1 ml Stain Buffer and centrifuged. Except in the case when the R-PE-labeled anti-p-Akt antibody was used as primary antibody, the cells were resuspended in Stain Buffer containing the secondary antibody, incubated for 30 min in the dark, and then washed twice with 1 ml Stain Buffer prior to FACS analysis, which was performed using a FACS Scan flow cytometer (Becton Dickinson and Co., Franklin Lakes, N.J.). Viable cells were electronically gated using forward and side scatter parameters. Flow cytometry data were analyzed using the CellQuest program (Becton Dickinson and Co., Franklin Lakes, N.J.).
Western blot analysis
Levels of p-Akt, p-GSK-3, total Akt, and total GSK-3 in total cellular extracts from LoVo cells transfected with the PTHrP-expressing construct or the empty vector (as control) were analyzed by Western blot analysis. Cells were grown to 70-80% confluence in 100 mm dishes. The cells were then washed with cold PBS on ice and lysed in RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 158 mM NaCl, 50 mM Tris-HCl pH 7.5; plus protease inhibitors (1 mM PMSF, 1 mM iodoacetamide, 25 μg/ml leupeptin, 10 μg/ml aprotinin and 1 mM EGTA). The lysate was centrifuged at 4 °C for 30 minutes at 12500 rpm. The clear supernatant was collected into fresh Eppendorf tubes. The total protein was estimated using the Bio-Rad protein assay.
For Western analysis, 60 μg of protein were resolved on 8 % SDS polyacrylamide gels and subsequently transferred to a nitrocellulose membrane (Schleicher & Schuell BioScience, Inc; Sanford, ME). The membranes were incubated with the anti-p-Akt antibody (Ser 473) (Cell Signaling Technologies), or anti-total Akt antibody (Santa Cruz Biotechnology Inc., Santa Cruz CA), anti-p-GSK-3 antibody or anti-total GSK-3 antibody (Cell Signaling Technologies), diluted in blocking buffer [5% non-fat dry milk in TBST (10 mM Tris-HCl, pH 7.6, 100 mM NaCl, 0.1% Tween-20)]. After three washes in TBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody (SouthernBiotech, Birmingham, AL) diluted to 1:7500 in blocking solution. The signals were detected using the SuperSignal West Pico Substrate kit (Pierce Biotechnology Inc., Rockford, IL).
Apoptosis
Early stage apoptosis was measured using annexin V-Fluos (Roche Molecular Biochemicals) in conjunction with 7-AAD (Sigma) to distinguish early apoptosis, late apoptosis, and necrosis [39,40]. Cells were plated in T-75 flasks in medium containing 10% FBS. After 48 h, the cells were transferred to medium containing 0.1% FBS. After 16 h, the cells were trypsinized and counted, a fraction of the cells (0.2 ×106) was collected by centrifugation, and the pellet was washed twice with PBS. After a further centrifugation, the cell pellet was resuspended in 100 μl labeling solution containing 2 μl annexin V-FITC labeling reagent in 100 μl HEPES buffer [10 mm HEPES/NaOH (pH 7.4), 140 mm NaCl, and 5 mm CaCl2]. For the negative control, the cell pellet was resuspended in HEPES buffer in the absence of annexin V. After a 15-min incubation on ice, the cell suspension was collected by centrifugation and washed twice with PBS containing 1mm CaCl2. The cell pellet was then resuspended in PBS and 1 mm CaCl2 containing 20 μg/ml 7-AAD. Apoptosis was measured by two-color flow cytometry on a FACScan flow cytometer (Becton Dickinson and Co.). Viable cells were electronically gated on forward and side scatter parameters. In addition, viable cells were gated on their ability to exclude the dye 7-AAD [39,40].
Cell migration and invasion
Cell migration was assessed using the FluoroBlok system (BD Pharmingen), per the manufacturer's specifications. Cells were trypsinized, washed once with medium containing 10% FBS, then once in serum-free medium, and resuspended in PBS. After counting, 0.5 × 106 cells were loaded with Calcein-AM (10 μM; Molecular Probes) and incubated for 45 min in a humidified incubator at 37°C in 95% air/5% CO2. After centrifugation, the cells were washed once in FBS-containing medium to quench the Calcein-AM, and once in serum-free medium to wash out the FBS. After resuspension in serum-free medium, the cells were plated onto FluoroBlok inserts (BD Pharmingen). In some experiments, the inserts were coated with laminin (derived from Engelbreth-Holm-Swarm mouse tumor; BD Pharmingen; 100 μg/ml) before plating the cells. As a control, cells were plated onto clear inserts. Medium containing 5% or 10% FBS was added to the lower chamber as a chemoattractant. As a negative control, medium containing 0.1% BSA was placed in the lower chamber. After incubation for 4 h in a humidified incubator at 37°C in 95% air/5% CO2, the plates were scanned with a bottom-reading Fluroskan Ascent CF Microplate Fluorometer (Thermo Labsystems, Franklin, MA), at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. Cell migration was calculated as follows: cell migration (%) = (total fluorescence intensity in the presence of FluoroBlok insert/total fluorescence intensity in the presence of clear insert) × 100. Pilot studies were performed to verify a linear and reproducible relationship between cell number and fluorescence intensity (data not shown).
Cell invasion was measured using FluoroBlok inserts coated with Matrigel, according to the manufacturer's specifications. Matrigel creates a matrix barrier which cells must degrade in order to traverse. The Matrigel was thawed overnight at 4 °C, and 100 μl of a 3 mg/ml stock was added using a pre-cooled pipette tip onto a pre-cooled FluoroBlok insert mounted onto a pre-cooled 24-well companion plate. The plates were kept in a humidified incubator at 37 °C in 95% air/5%CO2 for 2 h before adding cells. Cells were prepared and plated as described for the migration assays, and left in contact with the Matrigel for 4h. The number of invading cells was calculated as for the migration experiments.
Monolayer scratch assay
For this assay, cells were grown to confluence in 6-well dishes in medium containing 10% FBS. In some experiments, the cells were plated in 5% NuSerum (BD Biosciences) or 1% FBS in order the decrease the exposure of cells to ECM proteins, such as fibronectin and vitronectin, which are present in FBS. The confluent monolayer was scraped with a P200 pipette tip, and then rinsed with PBS to dislodge cellular debris. The cells were then further incubated under standard conditions for 72 h. Pictures were taken at time zero, and at 24 h, 48 h, and 72 h after wounding, using a Nikon phase contrast microscope. The extent of migration was analyzed using the NIH image software (http://rsb.info.nih.gov/nih-image/Default.html).
Statistics
Numerical data are presented as the mean ± SEM. The data were analyzed by ANOVA, followed by a Bonferroni post-test to determine the statistical significance of differences. All statistical analyses were performed using Instat Software (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered significant.
Results
PTHrP overexpression alters LoVo cell morphology
We compared the morphology of PTHrP-overexpressing LoVo cells to that of cells transfected with the empty vector pcDNA3.1 (controls). PTHrP expression was measured by IRMA [20] and by real-time PCR. PTHrP expression by the control cells was not significantly different from that of parental cells [20]. As PTHrP-overexpressing cells, we used four independent clones that express between 55-fold and 75-fold higher PTHrP levels than parental LoVo cells. Parental cells show a rounded morphology and grow in clusters (Fig. 1). Colonies of these cells do not show significant spreading, and the cells tend to pile and form multilayers in culture (Fig. 1). The empty vector transfectants show a similar morphology to that of the parental cells (Fig. 1). Overexpressing PTHrP induces a change in cell morphology to one in which the cells have an extended, spindle-shaped and flattened appearance with several long protrusions, and do not grow in tight colonies (Fig. 1). Fig. 1 shows the morphology of two PTHrP-overexpressing clones. The other two PTHrP-overexpressing clones showed a similar morphology (data not shown).
Figure 1.
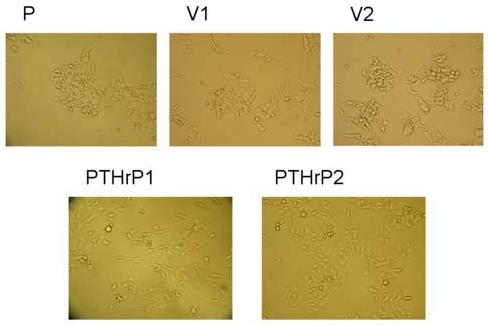
Effects of PTHrP on LoVo cell morphology. LoVo cells were stably transfected with a construct overexpressing PTHrP (−36 to +139) or with pcDNA3.1 (empty vector). PTHrP1 and PTHrP 2 = independent clones overexpressing PTHrP; V1 and V2 = empty vector transfectants; P = parental cells. Cells were plated in medium containing 10% FBS for 72 h, then analyzed using a Nikon phase-contrast microscope. Magnification, x10.
PTHrP increases LoVo cell migration and invasion
Overexpressing PTHrP in LoVo cells increased cell migration, as measured using the Fluoroblok system (Fig. 2A). For these experiments, we used the same four independent LoVo cell clones that express between 55-fold and 75-fold higher PTHrP levels than parental LoVo cells (described above). Cells transfected with the empty vector were used as control. The cells were plated onto the inserts in the absence of FBS, to avoid any contributions to migration by extracellular matrix proteins present in serum. The migration of PTHrP-overexpressing cells plated on Fluoroblok inserts was ∼ 1.5-fold to 2-fold of that of the control (empty vector-transfected) cells when 5% (data not shown) or 10% (Fig. 2A) FBS was used as the chemoattractant in the lower chamber. Transfection with the empty vector had no effect on cell migration, compared to that of parental cells (Fig. 2A). Migration in the absence of chemoattractant was significantly lower; ∼ 3% of the cells migrated (data not shown), vs. ∼ 20% of the parental cells in the presence of 10% FBS (Fig. 2A).
Figure 2.
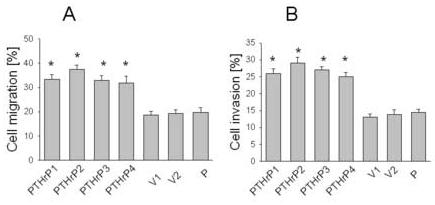
Cell migration (A) and invasion (B) of PTHrP-overexpressing and control LoVo cells. Cells were plated on FluoroBlok inserts in the absence of FBS. To measure invasion, the inserts were overlaid with Matrigel. Cells (0.5 × 106) were loaded with Calcein-AM and plated onto the inserts; FBS (10%) was used as the chemoattractant. Migration and invasion were measured as described in Materials and Methods. Data for each of four independent PTHrP-overexpressing clones (PTHrP1 to PTHrP4), two independent empty vector-transfected clones (V1 and V2; controls), and parental cells (P) are presented. The PTHrP-overexpressing clones secrete between 55-fold and 75-fold more PTHrP than the empty vector-transfected cells. Each bar is the mean ± SEM of three independent experiments. * = Significantly different from the control value (P < 0.001).
Integrin α6β4-mediated cell migration can occur via a ligand-dependent or ligand-independent pathway [41]. Laminin is a ligand for the α6β4 integrin [31]. To determine whether LoVo cell migration is dependent on ligand binding, we also measured cell migration using laminin-coated FluroBlok inserts. There was no significant difference in the migration of cells on laminin-coated vs. uncoated inserts (data not shown).
Cell migration was also assessed using the monolayer scratch assay, which measures the ability of cells to migrate and bridge a gap induced in a cell monolayer by scratching. Fig. 3A shows the extent to which two independent PTHrP-overexpressing and empty vector-transfected LoVo cell clones cultured in 10% FBS were able to migrate over a 72 h period to bridge the gap produced by wounding the cell monolayer. Repair of the cell monolayer was significantly faster for PTHrP-overexpressing cells than for empty vector-transfected (control) cells, and was almost compete 72 h after wounding (Fig. 3A). Repair of the control monolayer was significantly slower (Fig. 3A). There was no significant difference in cell monolayer repair between parental and empty vector-transfected cells (data not shown). A significant difference in cell migration between PTHrP-overexpressing and control cells was also observed 24 h and 48 h after wounding the cell monolayer. Quantitation of these data is presented in Fig. 3B, which shows the percent migration of PTHrP-overexpressing and empty vector-transfected cells at 24 h, 48 h, and 72 h after wounding the cell monolayer.
Figure 3.
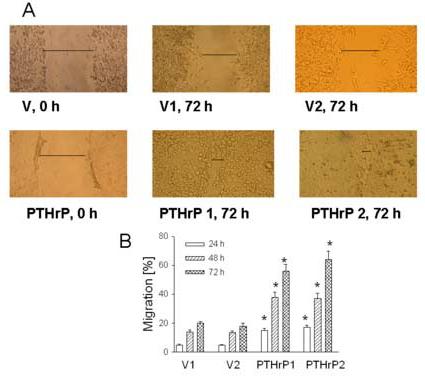
Cell migration of PTHrP-overexpressing vs. control LoVo cells Migration was analyzed using a monolayer scratch assay, performed as described in Materials and Methods. (A) Images at 0 and 72 h after scratch are shown. PTHrP 1 and 2 represent two independent PTHrP-overexpressing clones; V1 and 2 represent two independent empty vector transfectants (control). The horizontal line marks the edges of the monolayer. Magnification, x10. (B) Quantitation of cell migration 24 h, 48 h, and 72 h after wounding the cell monolayer. Each bar is the mean ± SEM of two independent experiments for each of four individual PTHrP-overexpressing and two independent empty vector-transfected clones. * = Significantly different from the control value (P < 0.001).
The monolayer scratch assay was also performed with cells cultured in 5% NuSerum (BD Biosciences), a growth medium supplement that provides a low-protein alternative to FBS, and so contains a decreased concentration of such ECM components as fibronectin and vitronectin. Under these conditions, repair of the cell monolayer was also significantly faster for PTHrP-overexpressing cells (∼ 65% complete 72 h after wounding) than for control cells (∼ 15% complete 72 h after wounding) (data not shown). Similar results were obtained when the cells were cultured in 1% FBS, with PTHrP-overexpressing cells exhibiting increased cell migration compared to control cells (data not shown). However, in this case, repair of the monolayer was significantly slower — ∼ 65% bridging of the gap in the cell monolayer took 6 days, vs. ∼ 3 days in the presence of 10% FBS. The control cell monolayer showed only ∼10% repair 6 days after wounding (data not shown). These data indicate that PTHrP increases cell migration under different growth factor and ECM concentrations.
We also compared the invasiveness of PTHrP-overexpressing vs. control LoVo cells. Cell invasion was measured using FluoroBlok inserts coated with Matrigel, thus creating a matrix barrier which cells must degrade in order to traverse. For these experiments, we used the same PTHrP-overexpressing and empty vector-transfected clones that were used in the migration experiments. PTHrP also increased the invasiveness of LoVo cells; Matrigel invasion by PTHrP-overexpressing cells was ∼ 1.5-fold to 2-fold that of the controls (empty vector transfectants) (Fig. 2B). There was no significant difference in invasiveness in the presence of 5% or 10% FBS as chemoattractant, and invasion was negligible in the absence of FBS (data not shown). As in the migration experiments, there was no difference in the invasiveness of cells transfected with the empty vector vs. parental cells (Fig. 2B).
PTHrP increases LoVo cell survival
To ask whether PTHrP modulates LoVo cell survival, we compared apoptosis in cells overexpressing PTHrP with that in parental cells, using the Annexin V labeling method. When coupled with 7-AAD, the Annexin V method differentiates between apoptotic and necrotic cells [39,40]. Apoptosis was assessed in cells overexpressing between 55-fold and 75-fold higher PTHrP levels than parental cells. Overexpressing PTHrP significantly decreased apoptosis by ∼ 6-fold, vs. parental cells (Fig. 4). Transfection with the empty vector had no significant effect on apoptosis, compared to parental cells (Fig. 4). Conversely, there was no significant difference in the number of necrotic cells in PTHrP-overexpressing vs. control cells (data not shown), indicating a selective effect of PTHrP on apoptosis.
Figure 4.
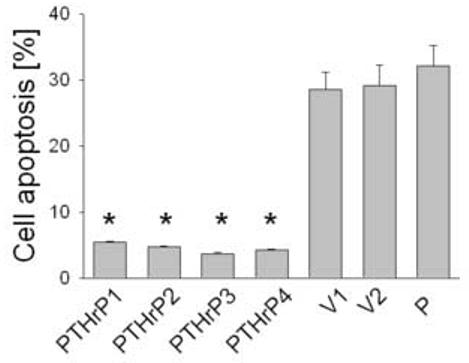
Apoptosis of LoVo cells after modulating PTHrP expression. Apoptosis was measured after staining with Annexin V. Cells were plated in medium containing 10% FBS, then transferred to medium containing 0.1% FBS after 48 h. Apoptosis was measured after 16 h as described in Materials and Methods. Data for each of four independent PTHrP-overexpressing clones (PTHrP1 to PTHrP4), two independent empty vector-transfected clones (V1 and V2; controls), and parental cells (P) are presented. The PTHrP-overexpressing clones secrete between 55-fold and 75-fold more PTHrP than the empty vector-transfected cells. Each bar is the mean ± SEM of three independent experiments. * = Significantly different from the control value (P < 0.001).
PTHrP upregulates integrin α6 and β4 mRNA levels
We have previously shown that overexpressing PTHrP (−36 to +139) increases the cell-surface protein expression of the α6 and β4 integrin subunits in LoVo cells [20]. To determine whether PTHrP regulates the expression of these integrin subunits at the mRNA level, integrin α6 and β4 expression was measured in four independent clones overexpressing between 55-fold and 75-fold higher PTHrP levels than parental cells. As shown in Fig. 5, integrin α6 and β4 mRNA levels were significantly higher in PTHrP-overexpressing cells than in control cells. Transfection with the empty vector had no effect on integrin mRNA levels, vs. parental cells (Fig. 5). In contrast, PTHrP had no effect on the mRNA levels of the integrin α2 subunit (data not shown).
Figure 5.
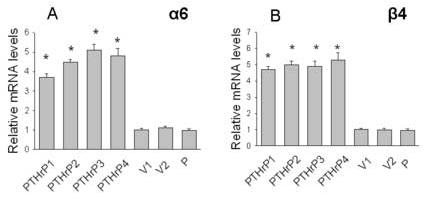
Expression of the integrin α6 (A) and β4 (B) subunits in PTHrP-overexpressing and control LoVo cells. mRNA levels were measured by real-time PCR as described in Materials and Methods. Data for each of four independent PTHrP-overexpressing clones (PTHrP1 to PTHrP4), two independent empty vector-transfected clones (V1 and V2; controls), and parental cells (P) are presented. The PTHrP-overexpressing clones secrete between 55-fold and 75-fold more PTHrP than the empty vector-transfected cells. Each bar is the mean ± SEM of three independent experiments. * = Significantly different from the control value (P < 0.001).
PTHrP modulates p-Akt and p-GSK-3 levels in LoVo cells
Since integrin α6β4 expression is linked to activation of the PI3-K pathway [35], we compared the levels of p-Akt, a downstream effector of the PI3-K pathway in PTHrP-overexpressing and control (empty vector-transfected) LoVo cells, utilizing conventional Western blot analysis and FACS analysis. Since activation of Akt is linked to phosphorylation (inactivation) of GSK-3, we also measured p-GSK-3 levels in PTHrP-overexpressing vs. control LoVo cells. The levels of p-Akt and p-GSK-3 were significantly higher in PTHrP-overexpressing than in control cells (Fig. 6, A-D). Since Akt is known to be phosphorylated at two independent sites (Thr 308 and Ser 473), we measured p-Akt levels in PTHrP-overexpressing and control cells using antibodies directed at these two sites. PTHrP produced a comparable increase in p-Akt levels when either of these antibodies was used (data not shown), indicating that both regions of the Akt molecule are phosphorylated in response to PTHrP. PTHrP had no effect on total Akt and GSK-3 levels (Fig. 6, B and D, and data not shown). Transfection with the empty vector caused no change in p-Akt and p-GSK-3 levels, or in total Akt and GSK-3 levels, when compared to parental cells (Fig. 6, A and C).
Figure 6.
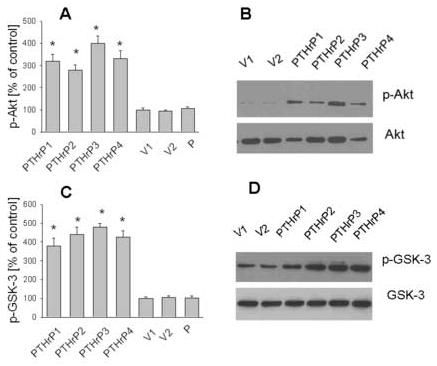
p-Akt and p-GSK-3 levels in PTHrP-overexpressing vs. control LoVo cells. (A) p-Akt and total Akt levels were measured by FACS analysis, as described in Materials and Methods. Positive cells are those whose log fluorescence intensity (MFI) is greater than that of isotype control antibody-stained cells. (B) p-Akt and total Akt levels were measured by Western blot analysis. (C) p-GSK-3 and total GSK-3 levels were measured by FACS analysis, as described in Materials and Methods. Positive cells are defined as in (A). (D) p-GSK-3 and total GSK-3 levels were measured by Western blot analysis. In (A) to (D), data for each of four independent PTHrP-overexpressing clones (PTHrP1 to PTHrP4) and two independent empty vector-transfected clones (V1 and V2; controls) are presented. In (A) and (C), p-Akt and p-GSK-3 levels for parental cells (P) are presented. The PTHrP-overexpressing clones secrete between 55-fold and 75-fold more PTHrP than the empty vector-transfected cells. In (A) and (C), each bar is the mean ± SEM of three independent experiments. * = Significantly different from the control value (P < 0.001).
Discussion
In this study, we examined the role of PTHrP in regulating colon cancer cell survival, migration, and invasion, using the human colon cancer cell line LoVo as a model. This cell line expresses PTHrP, as well as functional PTH/PTHrP receptors [20]. We previously showed that PTHrP modulates LoVo cell proliferation and ECM adhesion [20]. PTHrP also enhances the adhesion of the human colon cancer cell line HT-29 to collagen type I and increases the proliferation of the IEC-6 cell line [19,42], demonstrating an effect of PTHrP in different gastrointestinal tract-derived cell lines. Our main findings in this study, discussed in detail below, are that PTHrP increases LoVo cell survival, migration, and invasion. In addition, the protein also alters LoVo cell morphology, with the cells exhibiting increased spreading and several long protrusions. Accompanying these PTHrP-associated morphological changes, we found that PTHrP upregulates expression of the integrin α6 and β4 subunits at the mRNA level, and increases the levels of phosphorylated (and therefore active) Akt and phosphorylated (and thus inactive) GSK-3.
The role of PTHrP in mediating the metastasis of breast and prostate cancer cells to the bone has been firmly established [43-46]. Expression of PTHrP by breast cancer bone metastases is higher than at the primary site or by non-bone metastases [47-49]. In addition, nude mice injected with the breast cancer cell lines MCF-7 and MDA-MB-231 engineered to overexpress PTHrP (1-139) developed large bone metastases with distinct osteolysis; parental cells produced significantly fewer bone metastases [44,49]. Colon cancer does not typically metastasize to the bone, but shows a preference for the liver [2]. Few studies have addressed the role of PTHrP in cancers that do not metastasize to bone. This study demonstrates that PTHrP modulates the behavior of tumor cells that are not typically associated with bone metastasis, increasing their survival, migration, and invasion, indicating that PTHrP may play a universal role in the invasive behavior of tumor cells. PTHrP acts via both autocrine/paracrine and intracrine pathways [9,10,18,26], and the PTHrP-mediated effects reported here may be mediated via either or both of these pathways. We have previously shown that PTHrP modulates cell proliferation via both the autocrine/paracrine and intracrine pathways in MCF-7 human breast cancer cells and PC-3 human prostate cancer cells [50,51]. Similar effects on proliferation have been reported for vascular smooth muscle cells [52]. The same may apply to LoVo cells. However, the effects of PTHrP on the migration, invasion, and apoptosis of MCF-7 cells appear to be mediated via an intracrine pathway [26,51]. Thus, PTHrP may regulate the various parameters which define tumor aggression via different pathways, and the pathway(s) utilized may be cell-specific.
Integrins play a major role in cancer cell behavior, and alterations in their expression and function during transformation facilitate cancer progression. Integrin α6β4 is a receptor for the laminins, and is essential for the organization and maintenance of normal epithelial structures [53]. However, integrin α6β4 also plays a pivotal role in the biology of invasive carcinoma [53]; its expression persists in many tumor cells that do not form adhesive contacts but rather exhibit a motile phenotype characteristic of invasion and metastasis [53,54]. Thus, expression of the α6β4 integrin has been linked to tumor aggressiveness of colorectal, breast, thyroid, bladder, and gastric tumors, among others (reviewed in [55]). Integrin α6β4 expression correlates with colon cancer cell invasiveness, and stable expression of integrin α6β4 in β4-deficient colon cancer cells results in a significant increase in cell invasiveness [32,35]. Since PTHrP upregulates integrin α6β4 expression, the observed increase in migration and invasion of PTHrP-overexpressing LoVo cells vs. parental cells may be mediated via upregulation of the expression of these integrin subunits by PTHrP. In support of this possibility, we observe an increase in α6β4 expression, and cell migration and invasion in cells overexpressing PTHrP.
Integrin α6β4 mediates the migration of invasive colon carcinoma cells on laminin-1 through its ability to associate with the actin cytoskeleton and promote the formation and stabilization of filipodia and lamellae [55]. Inhibition of integrin α6β4 function results in the collapse of lamellae and filipodia, and an impediment of cell movement [55, 56]. Thus, the observed effects of PTHrP on cell morphology may be a result of the increased integrin α6β4 expression. The stimulation of cell migration and invasion by integrin α6β4 can also be independent of ligand binding, as engagement of this integrin by laminin substrates is not essential for its pro-migratory and pro-invasive effects [41]. We observed that the migration of both parental and PTHrP-overexpressing LoVo cells occurs to the same extent in the presence or absence of laminin. This “ligand-independent” property of integrin α6β4 implies that it can enhance cell migration on multiple matrix environments in vivo [57]). Integrin α6β4 may be functioning via this “ligand-independent” pathway in LoVo cells.
PTHrP upregulates integrin α6 and β4 subunits mRNA levels. Therefore, one mechanism via which PTHrP increases cell-surface protein expression of these integrin subunits may be via regulation at the mRNA level. PTHrP may be exerting its effects directly or indirectly via a transcription and/or posttranscriptional mechanism of action. The protein may either be functioning via an intracrine pathway to influence integrin α6 and β4 gene expression, or may function via an autocrine/paracrine pathway to ultimately regulate the activity of nuclear factors involved in the expression of these integrin subunits. PTHrP also increases expression of the α2 integrin at the cell-surface protein level [20]. However, this increase is not accompanied by an increase in the mRNA level. Therefore, PTHrP may regulate integrin expression via multiple mechanisms; its effects on integrin α6 and β4 expression may involve a direct and/or indirect transcriptional/posttranscriptional effect or a combination of a transcriptional/posttranscriptional effect plus a direct effect on protein synthesis/degradation or protein mobilization. In contrast, PTHrP may affect integrin α2 expression only at the protein level. These data further strengthen the conclusion that PTHrP exerts pleiotropic effects in the cell.
Integrin α6β4 also exerts a pro-survival effect in many cells lines (reviewed in [28]). This effect of integrin α6β4 on apoptosis is linked to the p53 status of the cell. In cells expressing wild-type p53, such as LoVo cells [58], integrin α6β4 stimulates p53-dependent caspase-3 activity, resulting in the cleavage and inactivation of Akt [59,60] and a loss of the pro-survival effects. However, we report that there is no significant difference in total Akt levels between cells with elevated integrin α6β4 levels (PTHrP-overexpressing cells) vs. parental LoVo cells, with lower integrin α6β4 levels. Thus, PTHrP may be acting to inhibit the degradation of Akt which occurs via the integrin α6β4-mediated stimulation of p53-dependent caspase-3 activity. Alternatively, the PTHrP-mediated increase in integrin α6β4 expression may not be large enough to stimulate the p53-dependent caspase-3 activity, even though it does activate the PI3-K/Akt pathway, as manifest by the increased levels of p-Akt in PTHrP-overexpressing vs. parental LoVo cells. We have previously shown that the Bcl-2 pathway is involved in the pro-survival effects of PTHrP in MCF-7 cells. In this cell line, PTHrP modulates the ratios of apoptosis-regulating proteins (Bcl-2 to Bax and Bcl-XL) [50]. This mechanism likely also plays a role in the pro-survival effects of PTHrP in LoVo cells.
PTHrP increases the levels of phosphorylated (active) Akt in LoVo cells. Akt is a downstream effector of PI3-K, a ubiquitous lipid kinase involved in receptor signal transduction by tyrosine kinase receptors [61, 62]. There is increasing evidence that the activation of PI3-K/Akt is associated with colorectal carcinoma, and can convert differentiated human gastric or colonic mucosa to a less differentiated and more malignant phenotype [63]. The effects of PI-3K on tumor growth and progression are thought to be mediated by Akt [64]. Akt is overexpressed in several cancers, including those of the colon, pancreas, ovary, and breast [65]. Moreover, Akt phosphorylation in human colon carcinomas correlates with cell proliferation and inhibition of apoptosis, as well as different clinicopathological parameters such as invasive grade, vessel infiltration, lymph node metastasis, and tumor stage [66,67]. There are three members of the Akt gene family in humans: Akt1, Akt2, and Akt3. Both Akt1 and Akt2 have been implicated in colon cancer progression [68]. Integrin α6β4 is known to activate the PI-3K pathway [35]. Thus, the effects of PTHrP on colon cancer cell proliferation, migration and invasion may be mediated via upregulation of integrin α6β4 expression, with consequent activation of the PI-3K pathway. Alternatively, distinct signaling pathways may be mediating the observed effects. For example, integrin α6β4 may promote cell cycle progression by recruitment of the adapter Shc/Grb2 to the β4 cytoplasmic domain, with consequence activation of the Ras-MAPK cascade [69,70]. PI-3K activation by PTHrP may also occur independently of the α6β4 integrin. Thus, PTHrP may activate PI-3K via an autocrine/paracrine mechanism of action involving the PTH/PTHrP receptor, as has been reported in a mesenchymal cell line [71], and in renal tubulointerstitial and fibroblastic cells [72]. In this study, p-Akt levels were measured in cells plated on poly-D-lysine. It is also possible that activation of Akt may be dependent on the extracellular matrix component on which the cells are cultured, as has been observed for collagen type I and fibronectin [73,74].
The PTHrP-mediated increase in phosphorylated Akt levels is accompanied by phosphorylation (inactivation) of GSK-3, which is an Akt substrate. GSK-3, a component of the Wnt signaling pathway, has been implicated in several biological processes via phosphorylation of multiple substrates, including transcription factors such as c-Myc, c-Jun, and c-Myb, and the translation factor eIf2B [75,76]. Since GSK-3 activity suppresses cell proliferation and survival [77,78], this represents another pathway via which PTHrP may be exerting its effects in LoVo cells.
In conclusion, the results presented here demonstrate that PTHrP enhances LoVo cell migration, invasion, and survival, as well as integrin α6β4 expression. Integrin α6β4 expression is modulated at the mRNA level, indicating that PTHrP is likely to be exerting at least some of its effect, either directly or indirectly, via a transcriptional and/or posttranscriptional mechanism of action. PTHrP also increases phosphorylated (active) Akt levels, leading to a decrease in active (dephosphorylated) GSK-3 levels. The role of PTHrP in cancers that are not typically associated with hypercalcemia and/or bone metastasis has not been extensively addressed. This study, in combination with our previous study [20], shows that PTHrP produced by colon cancer cells may increase cell proliferation, survival, adhesion, migration and invasion, so that attenuating PTHrP expression may provide a new therapeutic approach for colon cancer patients.
Acknowledgements
We thank Drs. D. Konkel and M.L. Thomas for critical reading of the manuscript. This work was supported by NIH grants DK035608 and CA104748.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics. CA Cancer J. Clin. 1996;46:5–27. doi: 10.3322/canjclin.46.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Martin R, Paty P, Fong Y, Grace A, Cohen A, Dematteo R, Jarnagin W, Blumgart L. Simultaneous liver and colorectal resections are safe for synchronous colorectal liver cancer. Am. J. Coll. Surg. 2003;197:233–241. doi: 10.1016/S1072-7515(03)00390-9. [DOI] [PubMed] [Google Scholar]
- 3.Kopfstein L, Christofori G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cellular and Molecular Life Sciences. 2006;63:449–468. doi: 10.1007/s00018-005-5296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derycke L, van Marck V, Depypere H, Bracke M. Molecular targets of growth, differentiation, tissue integrity, and ectopic cell death in cancer cells. Cancer Biotherapy and Radiopharmaceuticals. 2005;20:579–588. doi: 10.1089/cbr.2005.20.579. [DOI] [PubMed] [Google Scholar]
- 5.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nature Reviews Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 6.Kohn EC, Liotta LA. Molecular insights into cancer invasion: strategies for prevention and intervention. Cancer Res. 1995;55:1856–1862. [PubMed] [Google Scholar]
- 7.Ozanne BW, Spence HJ, McGarry LC, Hennigan RF. Invasion is a genetic program regulated by transcription factors. Current Opinion in Genetics and Development. 2006;16:65–70. doi: 10.1016/j.gde.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 8.Strewler GI. Mechanisms of disease: the physiology of parathyroid hormone-related protein. New. Engl. J. Med. 2000;342:177–185. doi: 10.1056/NEJM200001203420306. [DOI] [PubMed] [Google Scholar]
- 9.de Papp AE, Yang KH, Soifer NE, Stewart AF. Parathyroid hormone-related protein as a prohormone: Posttranslational processing and receptor interaction. In: Negro-Villar A, editor. Endocrine Review Monograph Series A4. 1995. pp. 207–210. [DOI] [PubMed] [Google Scholar]
- 10.Mannstadt M, Jüppner H, Gardella TJ. Receptors for PTH and PTHrP. Am. J. Physiol. 1999;277:F665–F675. doi: 10.1152/ajprenal.1999.277.5.F665. [DOI] [PubMed] [Google Scholar]
- 11.Jüppner H, Abou-Samra A-B, Freeman M, Kong XF, Schipani E, Richards J, Kolakowski LF, Jr, Hock J, Potts JT, Jr, Kronenberg HM, Segre GV. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science. 1991;254:1024–1026. doi: 10.1126/science.1658941. [DOI] [PubMed] [Google Scholar]
- 12.Wysolmerski JJ, Stewart AF. The physiology of parathyroid hormone-related protein: an emerging role as a developmental factor. Annu. Rev. Physiol. 1998;60:431–460. doi: 10.1146/annurev.physiol.60.1.431. [DOI] [PubMed] [Google Scholar]
- 13.Malakouti S, Asadi FK, Kukreja SC, Abcarian HA, Cintron JR. Parathyroid hormone-related protein expression in the human colon: immunohistochemical evaluation. American Surgeon. 1996;62:540–544. [PubMed] [Google Scholar]
- 14.Sidler B, Alpert L, Henderson JE, Deckelbaum R, Amizuka N, Silva JE, Goltzman D, Karaplis AC. Amplification of the parathyroid hormone-related peptide gene in a colonic carcinoma. J. Clin. Endo. Metabolism. 1996;81:2841–2847. doi: 10.1210/jcem.81.8.8768840. [DOI] [PubMed] [Google Scholar]
- 15.Li H, Seitz PK, Thomas ML, Selvanayagam P, Rajaraman S, Cooper CW. Widespread expression of the parathyroid hormone-related peptide and PTH/PTHrP receptor genes in intestinal epithelial cells. Lab Invest. 1995;73:864–870. [PubMed] [Google Scholar]
- 16.Watson PH, Fraher LJ, Hendy GN, Chung UI, Kisiel M, Natale BV, Hodsman AB. Nuclear localization of the type 1 PTH/PTHrP receptor in rat tissues. J. Bone Mineral Res. 2000;15:1033–1044. doi: 10.1359/jbmr.2000.15.6.1033. [DOI] [PubMed] [Google Scholar]
- 17.Kaiser SM, Sebag M, Rhim JS, Kremer R, Goltzman D. Antisense-mediated inhibition of parathyroid hormone-related peptide production in a keratinocyte cell line impedes differentiation. Mol. Endo. 1994;8:139–147. doi: 10.1210/mend.8.2.8170470. [DOI] [PubMed] [Google Scholar]
- 18.Henderson JE, Amikuza N, Warshawsky H, Biasotto D, Lanske BMK, Goltzman D, Karaplis AC. Nucleolar localization of parathyroid hormone-related peptide enhances survival of chondrocytes under conditions that promote apoptotic cell death. Mol. Cell. Biol. 1995;15:4064–4075. doi: 10.1128/mcb.15.8.4064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye Y, Falzon M, Seitz PK, Cooper CW. Overexpression of parathyroid hormone-related protein promotes cell growth in the rat intestinal cell line IEC-6. Reg. Peptides. 2001;99:169–174. doi: 10.1016/s0167-0115(01)00248-8. [DOI] [PubMed] [Google Scholar]
- 20.Shen X, Falzon M. PTH-related protein enhances LoVo colon cancer cell proliferation, adhesion, and integrin expression. Reg. Peptides. 2005;125:17–27. doi: 10.1016/j.regpep.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 21.Nishihara M, Ito M, Tomioka T, Ohtsuru A, Tagashi T, Kanematsu T. Clinicopathological implications of parathyroid hormone-related protein in human colorectal tumours. J. Path. 1999;187:217–222. doi: 10.1002/(SICI)1096-9896(199901)187:2<217::AID-PATH210>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Iwamura M, Di Sant'Agnese A, Deftos LJ, Cockett ATK, Gershagen S. Characterization of the cell-specific expression of parathyroid hormone-related protein in normal and neoplastic prostate tissue. Urology. 1998;51:110–120. doi: 10.1016/s0090-4295(98)00077-6. [DOI] [PubMed] [Google Scholar]
- 23.Iwamura M, di Sant'Agnese PA, Wu G, Benning CM, Cockett ATK, Deftos LJ, Abrahamsson P-A. Immunohistochemical localization of parathyroid hormone-related protein in human prostate cancer. Cancer Res. 1993;53:1724–1726. [PubMed] [Google Scholar]
- 24.Shen X, Falzon M. PTH-related protein modulates PC-3 prostate cancer cell adhesion and integrin subunit profile. Mol. Cell. Endo. 2003;199:165–177. doi: 10.1016/s0303-7207(02)00287-3. [DOI] [PubMed] [Google Scholar]
- 25.Schelele J, Stangl R, Altendorf-Hofmann A, Gall FP. Indicators of prognosis after hepatic resection of colorectal secondaries. Surgery. 1991;110:13–29. [PubMed] [Google Scholar]
- 26.Shen X, Qian L, Falzon M. PTH-related protein enhances MCF-7 breast cancer cell adhesion, migration, and invasion via an intracrine pathway. Exp. Cell Res. 2004;294:420–433. doi: 10.1016/j.yexcr.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 27.Ruoslahti E. Integrins. J. Clin. Invest. 1991;87:1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruoslahti E, Noble NA, Kagami S, Border WA. Integrins. Kidney Int. 1994;45(Suppl 44):S17–S22. [PubMed] [Google Scholar]
- 29.Kumar CC. Signaling by integrin receptors. Oncogene. 1998;17:1365–1373. doi: 10.1038/sj.onc.1202172. [DOI] [PubMed] [Google Scholar]
- 30.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 31.Mercurio AM. Receptors for the laminins. Achieving specificity through cooperation. Trends Cell Biology. 1995;5:419–423. doi: 10.1016/s0962-8924(00)89100-x. [DOI] [PubMed] [Google Scholar]
- 32.Chao C, Lotz MM, Clarke A, Mercurio AM. A function for the integrin alpha6beta4 in the invasive properties of colorectal carcinoma cells. Cancer Research. 1996;56:4811–4819. [PubMed] [Google Scholar]
- 33.Tagliabue E, Ghirelli C, Squicciarini P, Aiello P, Colnaghi MI, Menard S. Prognostic value of α6β4 integrin expression in breast carcinomas is affected by laminin production from tumor cells. Clin. Cancer Res. 1998;4:407–410. [PubMed] [Google Scholar]
- 34.Pouliot N, Connolly LM, Moritz RL, Simpson RJ, Burgess AW. Colon cancer cells adhesion and spreading on autocrine laminin-10 is mediated by multiple integrin receptors and modulated by EGF receptor stimulation. Exp. Cell Res. 2000;261:360–371. doi: 10.1006/excr.2000.5065. [DOI] [PubMed] [Google Scholar]
- 35.Shaw LM, Rabinovitz I, Wang HH-F, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- 36.Rigot V, Andre F, Lehmann M, Lissitzky JC, Marvaldi J, Luis J. Biogenesis of α6β4 integrin in a human colonic adenocarcinoma cell line: involvement of calnexin. Eur. J. Biochem. 1999;261:659–666. doi: 10.1046/j.1432-1327.1999.00300.x. [DOI] [PubMed] [Google Scholar]
- 37.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 38.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 39.Schmid I, Krall WJ, Uittenbogaart CH, Braun J, Giorgi JV. Dead cell discrimination with 7-amino-actinomycin D in combination with dual color immunoflurescence in single laser flow cytometry. Cytometry. 1992;13:204–208. doi: 10.1002/cyto.990130216. [DOI] [PubMed] [Google Scholar]
- 40.Lecoeur H, Ledru E, Prevost MC, Gougeon ML. Strategies for phenotyping apoptotic periperal human lymphocytes comparing ISNT, annexin-V and 7-AAD cytofluorimetric staining methods. J. Immunol. Methods. 1997;209:111–123. doi: 10.1016/s0022-1759(97)00138-5. [DOI] [PubMed] [Google Scholar]
- 41.Lipscomb EA, Dugan AS, Rabinovitz I, Mercurio AM. Use of RNA interference to inhibit integrin (α6β4)-mediated invasion and migration of breast carcinoma cells. Clin. Exp. Metastasis. 2003;20:569–576. doi: 10.1023/a:1025819521707. [DOI] [PubMed] [Google Scholar]
- 42.Ye Y, Seitz PK, Cooper CW. Parathyroid hormone-related protein overexpression in the human colon cancer cell line HT-29 enhances adhesion of the cells to collagen type I. Reg. Peptides. 2001;101:19–23. doi: 10.1016/s0167-0115(01)00259-2. [DOI] [PubMed] [Google Scholar]
- 43.Guise TA. Molecular mechanisms of osteolytic bone metastases. Cancer. 2000;88(12 Suppl):2892–2898. doi: 10.1002/1097-0142(20000615)88:12+<2892::aid-cncr2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 44.Thomas RJ, Guise TA, Yin JJ, Elliott J, Horwood NJ, Martin TJ, Gillespie MT. Breast cancer cells interact with osteoblasts to support osteoclast formation. Endocrinology. 1999;140:4451–4458. doi: 10.1210/endo.140.10.7037. [DOI] [PubMed] [Google Scholar]
- 45.Dougherty KM, Blomme EAG, Koh AJ, Henderson JE, Pienta KJ, Rosol TJ, McCauley LK. Parathyroid hormone-related protein as a growth regulator of prostate carcinoma. Cancer Res. 1999;59:6015–6022. [PubMed] [Google Scholar]
- 46.Rabbani SA, Gladu J, Harakidas P, Jamison B, Goltzman D. Over-production of parathyroid hormone-related peptide results in increased osteolytic metastasis by prostate cancer cells in vivo. Int. J. Cancer. 1999;80:257–264. doi: 10.1002/(sici)1097-0215(19990118)80:2<257::aid-ijc15>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 47.Bundred NJ, Walker RA, Ratcliffe WA, Warwick J, Morrison JM, Ratcliffe JG. Parathyroid hormone related protein and skeletal morbidity in breast cancer. Eur. J. Cancer. 1992;28:690–692. doi: 10.1016/s0959-8049(05)80127-3. [DOI] [PubMed] [Google Scholar]
- 48.Mundy G. Mechanisms of bone metastasis. Cancer (Suppl.) 1997;80:1546–1556. doi: 10.1002/(sici)1097-0142(19971015)80:8+<1546::aid-cncr4>3.3.co;2-r. [DOI] [PubMed] [Google Scholar]
- 49.Guise TA, Yin JJ, Thomas RJ, Dallas M, Cui Y, Gillespie MT. Parathyroid hormone-related protein (PTHrP)-(1-139) isoform is efficiently secreted in vitro and enhances breast cancer metastasis to bone in vivo. Bone. 2002;5:670–676. doi: 10.1016/s8756-3282(02)00685-3. [DOI] [PubMed] [Google Scholar]
- 50.Tovar Sepulveda VA, Shen X, Falzon M. Intracrine parathyroid hormone-related protein protects against serum starvation-induced apoptosis and regulates the cell cycle in MCF-7 breast cancer cells. Endocrinology. 2002;143:596–606. doi: 10.1210/endo.143.2.8645. [DOI] [PubMed] [Google Scholar]
- 51.Tovar Sepulveda VA, Falzon M. Parathyroid hormone-related protein enhances PC-3 prostate cancer cell growth via both autocrine/paracrine and intracrine pathways. Regulatory Peptides. 2002;105:109–120. doi: 10.1016/s0167-0115(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 52.Massfelder T, Dann P, Wu TL, Vasavada R, Helwig J-J, Stewart AF. Opposing mitogenic and anti-mitogenic actions of parathyroid hormone-related protein in vascular smooth muscle cells: a critical role for nuclear targeting. Proc. Natl. Acad. Sci. USA. 1997;94:13630–13635. doi: 10.1073/pnas.94.25.13630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rabinovitz I, Mercurio AM. The integrin alpha6beta4 and the biology of carcinoma. Biochem. Cell Biol. 1996;74:811–821. doi: 10.1139/o96-087. [DOI] [PubMed] [Google Scholar]
- 54.Mercurio AM, Rabinovitz I. Towards a mechanistic understanding of tumor-invasion – lessons from the alpha6beta4 integrin. Seminars Cancer Biol. 2001;11:129–141. doi: 10.1006/scbi.2000.0364. [DOI] [PubMed] [Google Scholar]
- 55.Rabinovitz I, Mercurio AM. The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J. Cell Biol. 1997;139:1873–1884. doi: 10.1083/jcb.139.7.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rabinovitz I, Toker A, Mercurio AM. Protein kinase C-dependent mobilization of the alpah6beta4 integrin from hemidesmosomes and its association with actin-rich cell protrusions drive the chemotactic migration of carcinoma cells. J. Cell Biol. 1999;146:1147–1160. doi: 10.1083/jcb.146.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lipscomb EA, Mercurio AM. Mobilization and activation of a signaling component α6β4 integrin underlies its contribution to carcinoma progression. Cancer Metastasis Rev. 2005;24:413–423. doi: 10.1007/s10555-005-5133-4. [DOI] [PubMed] [Google Scholar]
- 58.Szapeshazi K, Schally AV, Halmos G, Armatis P, Hebert F, Sun B, Feil A, Kiaris H, Nagy A. Targeted cytotoxic somatostatin analogue AN-238 inhibits somatostatin receptor-positive experimental colon cancers independently of their p53 status. Cancer Research. 2002;62:781–788. [PubMed] [Google Scholar]
- 59.Bachelder RE, Ribick MJ, Marchetti A, Falcioni R, Soddu S, Davis KR, Mercurio AM. p53 inhibits alpha6beta4 integrin survival signaling by promoting the caspase 3-dependent cleavage of AKT/PKB. J. Cell. Biol. 1999;147:1063–1072. doi: 10.1083/jcb.147.5.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bachelder RE, Wendt MA, Fujita N, Tsuruo T, Mercurio AM. The cleavage of Akt/protein kinase B by death receptor signaling is an important event in detachment-induced apoptosis. J. Biol. Chem. 2001;276:34702–34707. doi: 10.1074/jbc.M102806200. [DOI] [PubMed] [Google Scholar]
- 61.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annual Rev. Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- 62.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 63.Semba S, Itoh N, Ito M, Youssef EM, Harada M, Moriya T, Kimura W, Yamakawa M. Down-regulation of PI3KCG, a catalytic subunit of phosphoinositol 3-OH kinase, by CpG hypermethylation in human colorectal carcinoma. Clin. Cancer Res. 2002:3824–3831. [PubMed] [Google Scholar]
- 64.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Barón M. PI3K/Akt signalling pathway and cancer Treat. Res. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 65.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, Smyrk TC. AKT proto-oncogene overexpression is an early event during sporadic colon carcinoma. Carcinogenesis. 2002;23:201–205. doi: 10.1093/carcin/23.1.201. [DOI] [PubMed] [Google Scholar]
- 66.Khaleghpour K, Li Y, Banville D, Yu Z, Shen S-H. Involvement of the PI 3-kinase signaling pathway in progression of colon adenocarcinoma. Carcinogenesis. 2004;25:241–248. doi: 10.1093/carcin/bgg195. [DOI] [PubMed] [Google Scholar]
- 67.Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002;94:3127–3134. doi: 10.1002/cncr.10591. [DOI] [PubMed] [Google Scholar]
- 68.Rychahou PG, Jackson LN, Silva SR, Rajaraman S, Evers BM. Targeted molecular therapy of the PI3k pathway. Annals of Surgery. 2006;243:833–844. doi: 10.1097/01.sla.0000220040.66012.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maniero F, Pep A, Wary KK, Spinardi L, Mohammadi M, Schlessinger J, Giancotti FG. Signal transduction of the α6β4 integrin: distinct β4 subunit sites mediate recruitment of the Shc/Grb2 and association wit the cytoskeleton of hemidesmosomes. EMBO J. 1995;14:4470–4481. doi: 10.1002/j.1460-2075.1995.tb00126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maniero F, Murgia C, Wary KK, Curatola AM, Pep A, Blumemberg M, Westwick JK, Der CJ, Giancotti FG. The coupling of α6β4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 1997;16:2365–2375. doi: 10.1093/emboj/16.9.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen HL, Demiralp B, Schneider A, Koh AJ, Silve C, Wang CY, McCauley LK. Parathyroid hormone and parathyroid hormone-related protein exert both proand anti-apoptotic effects in mesenchymal cells. J. Biol. Chem. 2002;277:19374–19381. doi: 10.1074/jbc.M108913200. [DOI] [PubMed] [Google Scholar]
- 72.Ortega A, Rámilla D, Ardura JA, Esteban V, Ruiz-Ortega M, Barat A, Gazapo R, Bosch RJ, Esbrit P. Role of parathyroid hormone-related protein in tubulointerstitial apoptosis and fibrosis after folic acid-induced nephrotoxicity. J. Amer. Soc. Nephrol. 2006:1594–1603. doi: 10.1681/ASN.2005070690. [DOI] [PubMed] [Google Scholar]
- 73.Fujisaki H, Hattori S. Keratinocyte apoptosis on type I collagen gel caused by lack of laminin 5/10/11 deposition and Akt signaling. Exp. Cell Research. 2002;280:255–269. doi: 10.1006/excr.2002.5639. [DOI] [PubMed] [Google Scholar]
- 74.Matsuo M, Sakurai H, Ueno Y, Ohtani O, Saiki I. Activation of MEK/ERK and PI3K/Akt pathways by fibronectin requires integrin αv-mediated ADAM activity in hepatocellular carcinoma: a novel functional target for gefitinib. Cancer Sci. 2006;97:155–162. doi: 10.1111/j.1349-7006.2006.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- 76.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Gycogen synthase kinase-3: functions in oncogenesis and development. Biochim. Biophys. Acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- 77.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-κB by the Akt/PKB kinase. Curr. Biol. 1999;9:601–604. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 78.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]