

Abstract
Human angiotensin-converting enzyme is an important drug target for which little structural information has been available until recent years. The slow progress in obtaining a crystal structure was due to the problem of surface glycosylation, a difficulty that has thus far been overcome by the use of a glucosidase-1 inhibitor in the tissue culture medium. However, the prohibitive cost of these inhibitors and incomplete glucosidase inhibition makes alternative routes to minimizing the N-glycan heterogeneity desirable. Here, glycosylation in the testis isoform (tACE) has been reduced by Asn-Gln point mutations at N-glycosylation sites, and the crystal structures of mutants having two and four intact sites have been solved to 2.0Å and 2.8Å, respectively. Both mutants show close structural identity with the wild-type. A hinge mechanism is proposed for substrate entry into the active cleft, based on homology to human ACE2 at the levels of sequence and flexibility. This is supported by normal mode analysis that reveals intrinsic flexibility about the active site of tACE. Subdomain II, containing bound chloride and zinc ions, is found to have greater stability than subdomain I in the structures of three ACE homologues. Crystallisable glycosylation mutants open up new possibilities for co-crystallisation studies to aid the design of novel ACE inhibitors.
Keywords: angiotensin-converting enzyme, X-ray crystal structure, testis ACE, normal mode, hinge-bending
Since its isolation in 1956 as “hypertensin-converting enzyme”, human angiotensin-converting enzyme (ACE1) has been known to play a key role in the regulation of blood pressure (1). ACE is a membrane-bound zinc metalloprotease of the M2 family, acting as a dicarboxypeptidase on a range of oligopeptide substrates. Its hypertension-inducing activity is mediated through cleavage of two of these substrates: inactive decapaptide angiotensin I is converted into the active vasopressor angiotensin II, while cleavage of bradykinin inactivates this vasodilatory nonapeptide (reviewed in 2). Inhibitors of ACE have been shown to be effective in the treatment of hypertension and cardiac disease and a number of these drugs are commonly prescribed today (reviewed in 3).
Two isoforms of ACE occur in mammals: a somatic form (sACE) that is expressed in most mammalian tissues but primarily in the lungs, vascular endothelia and kidneys, and a germinal form (tACE) that arises only in the adult male testis (4,5). sACE has two homologous domains, each containing an active site, while tACE has a single active domain arising by tissue-specific transcription from a promoter in intron 12 of the sACE gene (6,7). tACE is thus identical to the C-terminal domain of sACE, except for an additional 67-residue N-terminal signal peptide and O-glycosylated region (8). Both forms are shed from the cell membrane by an ACE sheddase that cleaves at a site in the juxtamembrane stalk region (9).
Despite ACE's importance as a drug target, the first crystal structure of an ACE isoform, human tACE, was only solved recently (10). The major obstacle to the solution of this structure had been the presence of a number of N-linked glycan chains on the surface of the enzyme, which prevented crystallisation (11). Crystallisable material was eventually obtained by expressing truncated tACE (lacking the N-terminal O-glycosylated and transmembrane regions) in the presence of an α-glucosidase-1 inhibitor, N-butyl-deoxynojirimycin (NB-DNJ), which enforces uniform glycosylation to the simple high-mannose oligosaccharide level (12). This approach was later also used in the crystallisation and solution of the structure of the N domain of human sACE (Ndom) (13).
Another approach yielding promising initial results is the elimination of N-linked glycosylation sites by point mutation. It has been demonstrated that selected, but not all, N-linked glycosylation sites of human tACE can be removed, while maintaining functional integrity and crystal morphologies similar to the wild-type (14). This method bears further investigation since the extensive co-crystallisation studies desirable for a structure-based drug design process would be prohibitively expensive given the cost of glucosidase-1 inhibitors such as NB-DNJ.
The crystal structures of tACE alone and bound to inhibitors lisinopril, captopril and enalaprilat revealed a largely α-helical globular enzyme, divided down the middle by a deep cleft (10,15). This cleft is largely closed off from the external milieu, leaving only small pores less than 3Å in diameter through which substrates could gain access to the active site. While the substrates of ACE are limited to short oligopeptides, even these would require an enlarging of these openings in order to enter the cleft and access the active site, and a flexibility or breathing motion has been suggested to allow substrate binding (11). A similar closed conformation was also observed for Ndom, the Drosophila homologue AnCE, and human homologue ACE2, all at least 40% homologous to tACE (13,16,17). However, in the case of ACE2, the structure determined without bound inhibitor was in an open conformation in which the sides of the active cleft hinged apart by ∼16° (17). Given the structural similarity between Ndom, tACE and ACE2, and the need for movement to allow substrate entry, it seems likely that a similar hinge mechanism occurs in both domains of sACE during substrate binding. Such a hinge mechanism would also explain the large contribution of entropy that has been observed in the energetics of inhibitor-binding by sACE, since closing of the active site would result in the association of numerous residues that would otherwise be bound to ordered solvent molecules when in an open conformation (18).
Here, we present the crystal structures of two glycosylation mutants of human tACE and show that the removal of intact glycan chains does not affect the three-dimensional structure. In addition, we draw together lines of evidence from Normal Mode Analysis (NMA) and the crystal structures of ACE and ACE homologues that support the hypothesis of a conserved hinge mechanism for substrate entry.
MATERIALS AND METHODS
Protein purification
Mutants tACE-G13 and tACE-G1234 were previously constructed and expressed in Chinese hamster ovary (CHO) cells (14). Their sequences differ from that of wild-type human tACE, in that they lack the N-terminal O-glycosylated region (residues1-36), and have had some of their N-glycosylation sites knocked out by Asn-Gln mutation (Fig. 1). Specifically, tACE-G13 lacks all but the first and third sites, while tACE-G1234 lacks the fifth and sixth sites. The seventh potential glycosylation sequon was not mutated as it lies close to the cleavage site for shedding and has been shown to be unglycosylated (12). Soluble tACE-G13 and tACE-G1234 were expressed as previously described, and purified from harvested medium by lisinopril-Sepharose affinity chromatography (5,14,19).
Figure 1.
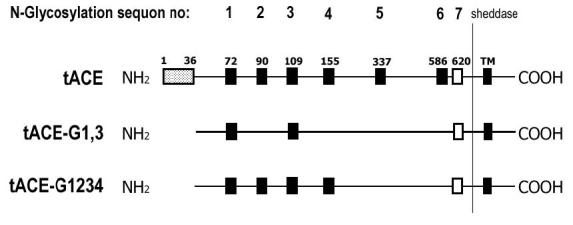
Schematic diagram of mutations introduced in tACE-G13 and tACE-G1234. Full-length wild-type tACE has an N-terminal O-glycosylated region (1-36), N-glycosylated sites 1-6 (filled squares), one unglycosylated site (open square), and a C-terminal transmembrane domain (TM). Both mutants lack the N-terminal 36 residues. Additionally, tACE-G13 lacks all but the first and third N-glycosylation sites, while tACE-G1234 lacks the fifth and sixth sites. Expressed protein is shed into the medium by cleavage between Arg 627 and Ser 628.
ACE activity was determined by measuring the hydrolysis of Hippuryl-Histidyl-Leucine (Sigma) in a fluorimetric assay (20). Protein concentration was determined by a Bradford protein assay (Bio-Rad protein micro-assay).
Crystallisation, data collection and processing
Crystals of tACE-G13 and tACE-G1234 were grown as for the wild-type structure, published previously (10). Diffraction data to 2.9Å for tACE-G13 were collected at 100K at the in-house X-ray source in the Department of Biotechnology, University of the Western Cape [comprising a Rigaku RUH3R copper rotating-anode X-ray source operated at 40 kV, 22mA; a Rigaku R-axis IV+ image plate camera; an X-stream 2000 low-temperature system; and an AXCO-PX50 glass capillary optic with a 0.1 mm focus]. Data for tACE-G1234 and an additional dataset for tACE-G13 were collected to 2.8Å and 2.0Å respectively, on station PX14.1 of the synchrotron radiation source (Daresbury, U.K.) using a Quantum 4 CCD (Area Detection Systems, Poway, CA).
DENZO/SCALEPACK were used to process the 2.9Å tACE-G13 data (21). Synchrotron data were processed and scaled using the HKL2000 software package, and data reduction was carried out using the CCP4 program TRUNCATE (HKL Research, Charlottesville, VA, 21, 22). The symmetry and systematic absences in all cases were consistent with the P212121 space group with one protein molecule per asymmetric unit.
Structure determination, model building and refinement
The tACE-G13 structure was solved twice, using independent methodology. The 2.9Å dataset was solved by molecular replacement with EPMR2.5 using protein atoms from the structure of minimally glycosylated wild-type tACE (PDB ID 1O8A) as a model (23). A correlation coefficient of 0.703 was obtained. CNS was used to perform energy minimisation and B factor refinement using bulk-solvent correction (24). 4.05% of reflections were reserved for Rfree calculation. Small adjustments to side chains and loops were made, and carbohydrates and acetates added using O 9.0.7, against composite omit maps calculated by CNS composite_omit_map using torsion angle simulated annealing (starting temperature 1000K; drop, 50K per set; 5.0% omission) (24,25).
The 2.0Å tACE-G13 dataset was solved by molecular replacement with Refmac through the CCP4i interface, again using the wild-type tACE structure as a model (26,27). 3% of reflections were kept aside for Rfree calculation (28). Small alterations to side chains and a few loops were made, and carbohydrate chains and acetate molecules were added using Coot (29). Water picking was achieved with the implementation of the ARP/wARP module in Refmac and peak picking in Coot (27,30).
The structure of tACE-G1234 was solved by refinement of the 2.0Å tACE-G13 structure with CNS (24). 8.4% of reflections were kept aside for Rfree calculation (28). Small alterations to side chains and loops were made, and carbohydrates and acetate added using Coot (29). Water picking was done manually and using the peak picking option in Coot (29). Structure validation was carried out using PROCHECK and SFCHECK from the CCP4 suite (27,31).
R.m.s deviations between structures were determined using the r.m.s. deviation function of CNS (24).
Identification of hinge regions in ACE2
The open and closed structures of ACE2 (ACE2o and ACE2c, PDB IDs 1R4L and 1R42, respectively) were compared in order to identify residues involved in the hinge-bending mechanism. Structure alignments and root mean square deviations for Cα atoms were computed using ALIGN (32). Hinge regions in ACE2 were defined as those residues to one side of which the Cα atoms of ACE2o deviated from those of ACE2c by >3Å, and to the other side of which the structures were closely aligned. These residues were compared with the corresponding residues in tACE (PDB ID 1O8A), Ndom (PDB ID 2C6N) and AnCE (PDB ID 1J38), based on the sequence alignments of (33) and (13).
Analysis of temperature factors
As an indication of structural order or disorder, the temperature factors (B-factors) of the 2.0Å tACE-G13 structure were compared with those from ACE homologues. Structures of wild-type tACE, Ndom, human ACE2 (open and closed forms), and Drosophila AnCE were used for comparison (PDB entries 1O8A, 2C6N, 1R4L, 1R42 and 1J38, respectively). 〈B〉 and σ(B) were calculated for all Cα atoms, and the B-factor of each individual Cα atom was expressed as a function of σ(B).
Modelling of a putative open form of tACE
Open and closed homology models of tACE (tACEo and tACEc) for NMA were built based on ACE2o and ACE2c. Amino acid sequences were aligned according to (17) and 25 open and 25 closed models were generated using MODELLER6v2 (34). PROCHECK was used to analyse the models. Based on their Ramachandran plots, one model was selected from each group as having the fewest residues falling into unfavourable regions. SCWRL was used to optimise the side-chain packing of these models (35).
Normal mode analysis
NMA of ACE2 and tACE was carried out according to the elastic network model, as implemented in the elNémo webserver (36, 37). The block size was set to three residues, and the Cα-Cα linkage cutoff to 8Å, being the recommended default values for these parameters.
The crystallographic structures of ACE2o, ACE2c (minus the collectrin-like domain) and unliganded wild-type tACE were analysed, as well as models tACEo and tACEc. In each case, the ten lowest-frequency normal modes were calculated, and the degree of cumulative overlap between the normal modes and the structural change was determined. Overlap is a measure of the degree of similarity between the direction of the observed conformational change and the one described by the normal mode in question, with a value of 1 indicating complete agreement between the normal mode and the observed change (37).
For the modes showing the highest overlap with the observed structural change, the amplitude of perturbation required for maximal agreement between the resulting structure and the comparison structure was determined, and perturbed models were generated accordingly. Cα-Cα distance fluctuations were calculated by elNémo and used as an indication of the residues involved in the motion described by the mode in question. The normal modes were also assessed in terms of collectivity, a measure of the proportion of atoms displaying large-amplitude displacements in the motion described by the mode (36).
RESULTS AND DISCUSSION
Structure of tACE-G13
The crystal structure of tACE-G13 was determined to a final resolution of 2.0Å with an R factor of 19.0% and an R free of 22.01% (see Table 1). The quality of the map allowed rebuilding of residues 40 to 623, which includes an additional six C-terminal residues that were disordered in the wild-type structure. Two conformations were modelled for a few surface residues where Fobs-Fcalc maps clearly indicated a second conformation. The catalytic zinc ion and two chlorides could be modelled, as well as one N-acetyl-glucosamine residue at N72 and ordered partial glycan chain at N109 (Fig. 2). The structures solved independently using the 2.9Å and 2.0Å datasets are in close agreement (Table 2).
Table 1.
Data collection and model refinement statistics
| tACE-G13 (2.9Å) | tACE-G13 (2.0Å) | tACE-G1234 | |
|---|---|---|---|
| Crystal data | |||
| Space group | P212121 | P212121 | P212121 |
| Unit cell parameters (Å) | a = 59.81, b = 85.16, c = 135.58 | a = 56.63, b = 84.72, c = 134.47 | a = 56.44, b = 85.15, c = 133.42 |
| Molecules in asymmetric unit | 1 | 1 | 1 |
| Diffraction data used in refinement* | |||
| Resolution limits (Å) | 50.0 - 2.90 (3.0-2.9) | 50.0 - 2.0 (2.07-2.00) | 50.0 - 2.8 (2.9-2.8) |
| No. of observations | 60751 | 515005 | 541512 |
| Redundancy | 4.0 | 12.4 | 39.0 |
| Completeness (%) | 89.9 (71.6) | 96.2 (86.4) | 83.6 (80.4) |
| Rsym (%)‡ | 20.0 (41.3) | 6.1 (21.4) | 11.4 (33.7) |
| Average I/σ(I) | 5.88 (1.86) | 26.8 (6.7) | 7.7 (2.8) |
| Refinement and model statistics | |||
| No. reflections used in refinement | 14338 | 40225 | 13572 |
| Rcryst§ [% reflections used] | 21.62 [95.95] | 18.04 [97.0] | 20.06 [91.6] |
| Rfree∥ [% reflections used] | 24.29 [4.05] | 22.01 [3.0] | 23.69 [8.4] |
| Mean B factor (Å2): | |||
| protein | 20.0 | 19.0 | 20.0 |
| carbohydrate | 46.0 | 50.4 | 58.6 |
| water (no. molecules) | 14.8 (74) | 25.0 (362) | 15.3 (34) |
| Root mean square deviations: | |||
| bond lengths (Å) | 0.009 | 0.009 | 0.010 |
| bond angles (°) | 1.57 | 1.12 | 1.46 |
| Ramachandran plot % residues in: | |||
| most favoured regions | 88.5 | 94.8 | 91.6 |
| additional allowed regions | 10.5 | 5.0 | 8.2 |
| generously allowed regions | 1.0 | 0.2 | 0.2 |
| disallowed regions | 0.0 | 0.0 | 0.0 |
Values in parentheses are for the last resolution shell.
Rsym = ΣhΣi|Ii(h) -〈I(h)〉|/ ΣhΣiIi(h), where Ii(h) and 〈I(h)〉 are the ith and the mean measurements of the intensity of reflection h, respectively.
Rcryst = Σh|Fo - Fc|/ Σh Fo, where Fo and Fc are the observed and calculated structure-factor amplitudes of reflection h, respectively.
Rfree is equal to Rcryst for a randomly selected 3% (g13) or 8.4% (g1234) of reflections not used in the refinement
Figure 2.
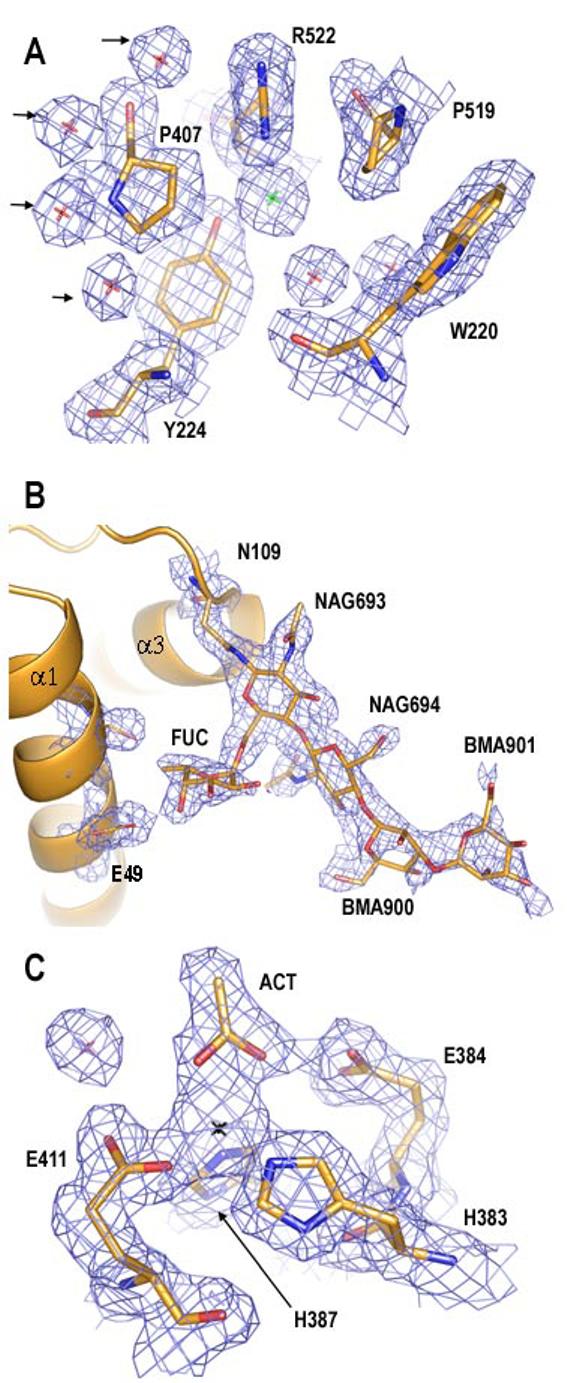
tACE-G13 in 2Fobs-Fcalc map density to 2.0Å, contoured at 1.0σ (blue mesh). A) Residues at the CL2 binding site. Green star = CL2; red stars = water; arrows indicate putative CL2 pore waters. B) Glycan residues at N109, with density for selected protein residues only. NAG = N-acetyl glucosamine; FUC = fucose; BMA = β-D-mannose; α1 and α3 = helices α1 and α3 in cartoon representation. C) Zinc-binding residues at the active site. Black star = ZN; ACT = acetate. This figure was generated using PYMOL 0.98 (DeLano Scientific).
Table 2.
Structural identity between tACE glycosylation mutants, unliganded wild-type tACE DB ID 1O8A) and lisinopril-bound wild-type tACE (PDB ID 1O8F).
| Structure | Comparison structure |
R.M.S Deviations (Å)* |
|||||
|---|---|---|---|---|---|---|---|
| All protein atoms |
Main chain |
Side chain |
Zn- binding residues |
CL1- associate d |
CL2- associate d |
||
| 1O8F | 1O8A | 0.36 | 0.15 | 0.49 | 0.10 | 0.15 | 0.05 |
| tACE-G13 (2.0Å) |
tACE-G13 (2.9Å) |
0.79 | 0.39 | 1.04 | 0.27 | 0.77 | 0.33 |
| tACE-G1234 | tACE-G13 (2.0Å) |
0.43 | 0.28 | 0.54 | 0.16 | 0.48 | 0.19 |
| 1O8F | tACE-G13 (2.0Å) |
0.53 | 0.69 | 0.27 | 0.14 | 0.40 | 0.14 |
| 1O8F | tACE-G1234 | 0.56 | 0.30 | 0.73 | 0.14 | 0.50 | 0.20 |
Zn-binding residues: 383, 384, 387 & 411; CL1-associated residues 186, 278, 485, 486, 489, 507; CL2-associated residues: 220, 224, 407, 519, 521, 522.
The structure of tACE-G1234 was determined to a final resolution of 2.8Å, including residues 40 to 592. The catalytic zinc ion and both chloride residues were modelled, but ordered glycan density was only seen at two of the four glycosylation sites (N72 and N109), and a single N-acetyl-glucosamine residue was modelled at each of these residues.
Density for the 310 helix 104-108 near the N109 glycosylation site, and for loop 434-440 was weak in both mutants and E436 could not be modelled. This pattern of loop disorder was also observed in the wild-type (10).
Like the wild-type structure, these mutants are largely α-helical, with a few short regions of β-strand. The molecules are ellipsoid in shape and divided down the middle by a deep cleft, in which the active site zinc ion is located. This cleft is closed off from the external milieu by the N-terminal two α-helices or lid-helices. R.m.s. deviations computed between tACE-G13, tACE-G1234 and the crystal structures of unliganded and lisinopril-bound wild-type tACE revealed very close structural identity between these structures, especially around the active site zinc ion (Table 2). Each mutant structure contains two chloride ions occurring at the same sites as in wild-type tACE, and four of the five conserved water molecules proposed to represent an entrance pore for CL2 are also present in tACE-G13 (38, Fig. 2 A). The lower resolution of the tACE-G1234 data set prevented the detection of any water molecules at this site. The close structural agreement further indicates that the difference in glycosylation between tACE-G13, tACE-G1234 and wild-type tACE does not affect their structures in this crystal form.
N-linked Glycans
Ordered N-linked glycan density was present only for sites 1 and 3 (N72- and N109-linked) in both mutants, with very little density being observed at sites 2 and 4 (N90- and N155-linked) in the tACE-G1234 structure, although this may be attributable to its lower resolution. The glycan chain at site 3 showed a particularly high degree of order in tACE-G13, allowing the positioning of two N-acetylglucosamine residues (NAG), two β-D-mannose residues and one NAG-linked fucose residue (Fig. 2 B). O4 of the fucose residue is in hydrogen bonding contact (2.6Å) with OE2 of E49 at the N-terminal end of α1, thus forming a glycan-mediated link between the N-terminus and the N-terminal end of α3 (Fig. 2 B). Glycan density for site 1, which lies in the loop between α1 and α2, was weaker, allowing the placement of only one NAG in both mutants. This NAG is in close proximity (3.2-4.5Å) to the side chains of T74 and E76 at the C-terminal end of α1. It thus appears that these two more ordered glycan chains, lying as they do at either end of the lid helices, may play a role in stabilising the interactions between the lid helices and the rest of the enzyme. This is consistent with the minimum requirement for the presence of an intact glycosylation site at either site 1 or site 3 in order for tACE to be properly expressed in an enzymatically active form (14).
Density in the active site
In both mutants, the active-site zinc ion is tetra-coordinated by H383, H387, E411 and the carboxyl oxygen of an acetate molecule (Fig. 2 C). This acetate, also modelled in unliganded wild-type tACE, makes hydrogen-bonding contact with E384 of the zinc-binding motif, and superimposes onto the zinc-binding carboxyl group of the inhibitor lisinopril when aligned with the lisinopril-bound structure (PDB ID 1O8F). The active site of tACE-G13 contains an additional acetate molecule coordinated by K511, Q281 and Y520, that superimposes onto the carboxyl terminus of lisinopril. An actetate was also observed in this position in the unliganded N domain structure. In tACE-G1234, additional density at this site was modelled as an N-carboxyalanine moiety that was also present in the unliganded wild-type tACE structure.
The presence of additional density or acetate molecules in the active sites of both tACE mutants and the wild-type inhibitor-free structures of tACE and Ndom suggests that these may not be true unliganded conformations of ACE domains, but rather that the “unliganded” structures thus far observed are equivalent to another liganded state.
ACE2 hinge regions
In order to investigate potential domain hinging in ACE, the residues involved in the motion observed in ACE2 were identified and compared with the equivalent residues in tACE. The structural change between the closed and open forms of ACE2 (ACE2c and ACE2o) can be described as a hinge movement that opens up the active site. The lid helices α1 and α2 swivel to one side, and the cleft opens up more on one end than the other (Fig. 3 A). Looking down onto the active site cleft with the lid helices (α1 and α2) on top, the hinge axis stretches from under the N-terminus to the middle of the underside of the active site (Fig. 3 A).
Figure 3.

Hinging in the open model of tACE. A) The open model of tACE (tACEo) used for normal mode analysis, showing ACE2 hinge residue equivalents. Hinge regions with low temperature factors and high sequence-conservation (400-409, 535-537, 569-578) are coloured blue; those with high temperature factors and low sequence-conservation (98-125, 296-297, 434-439) are coloured red. Note that S435-G438 were omitted from the model, as they were absent in the wild-type tACE structure. Deletion of the equivalent residues in ACE2 had no effect on the modes calculated (data not shown). B) Alignment of the tACEo model perturbed in the direction of the lowest-frequency normal mode calculated (grey) with wild-type tACE (green). The amplitude of perturbation chosen is that which generated the best fit to the closed structure. Hinge residues in tACEo are coloured as for A. This figure was generated using PYMOL 0.98 (DeLano Scientific).
Based on the structural alignment of ACE2o with ACE2c, six hinge regions were identified: 98-125, 296-297, 400-409, 434-439, 535-537 and 569-578 (tACE numbering; Fig. 3 A). These regions were compared with their equivalents in tACE, tACE-G13, tACE-G1234, Ndom and Drosophila homologue AnCE in terms of sequence conservation and flexibility as evidenced by their crystallographic temperature factors (B-factors).
Based on their structural location these hinge regions can be divided into two groups. The first group comprises two loop regions (296-297 and 434-439; tACE numbering) that stretch one above the other, across the opening end of the active site cleft (Fig. 3 A). These residues are not “hinge” residues as such, but rather allow the active cleft to open by becoming elongated in the unliganded open state. The sequence of these residues is not conserved, but their temperature factors are high in all of the structures considered, indicating the functional conservation of a degree of flexibility (Fig. 4 - data for wild-type tACE and tACE-G1234 not shown). In the case of wild-type tACE the S435-S439 loop was disordered in the wild-type crystal structure and could not be modelled.
Figure 4.
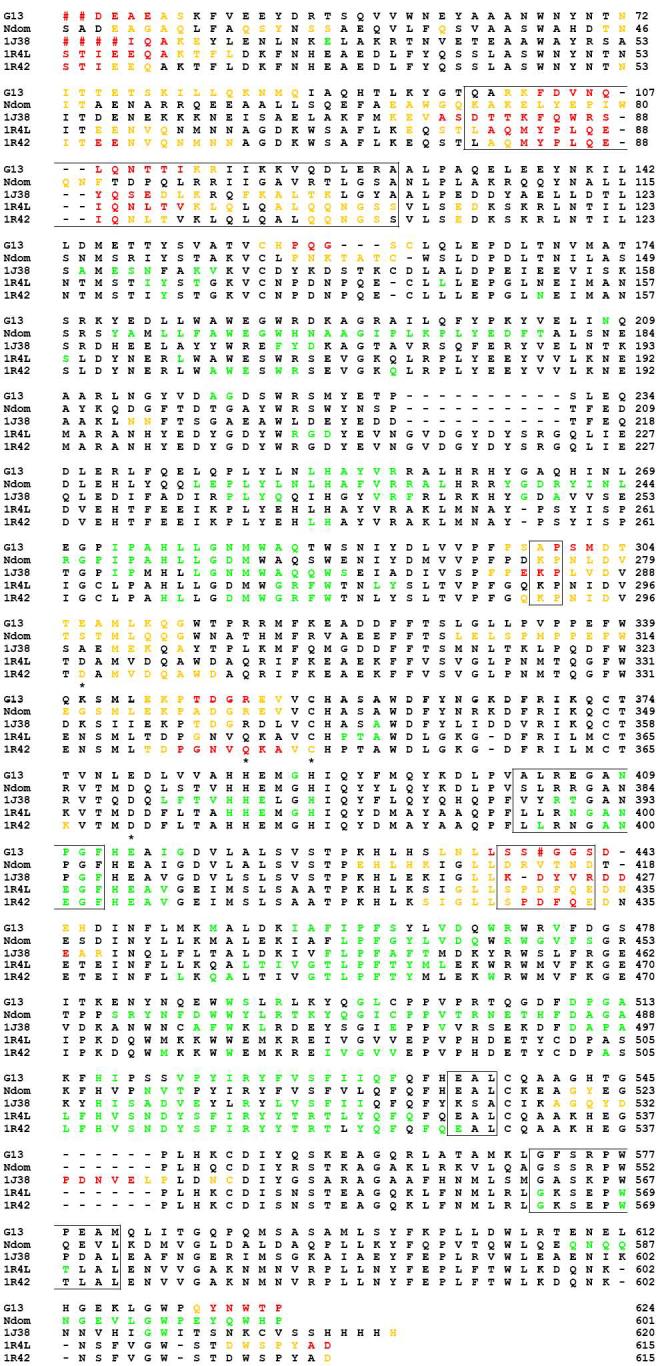
Temperature factor (B-factor) and sequence comparison between tACE-G13 (G13), the N domain of human sACE (Ndom, 2C6N), Drosophila AnCE (1J38) and the open (1R42) and closed (1R4L) structures of ACE2. Red hashes represent residues that were not modeled in the crystal structures due to disorder. Residues are coloured according to their temperature factors: ORANGE: B-factors greater than σ(B); RED: B-factors greater than 2σ(B); GREEN: B-factors less than -σ(B). Hinge regions in ACE2 are boxed; zinc ligands are marked with stars.
The second group comprises four regions (98-125, 400-409, 535-537 and 569-578; tACE numbering) all located along the hinge axis from the N-terminus side of the active site cleft to its underside (Fig. 3 A). This group could thus be described as the residues about which the hinge opens. Three of these regions (400-409, 535-537 and 569-578) show a high degree of sequence conservation; including several conserved glycines, alanines and serines; and low temperature factors, indicative of a high degree of order in the crystal structures (Fig. 4). The fourth (98-125) lies close to the surface between the α2 lid helix and α4, and has low sequence conservation and high temperature factors, indicating flexibility. 569-578 of this group lie between the conserved HEMGH of the zinc-binding motif (H383-H384 of tACE) and the additional conserved downstream zinc-binding Glu (E411 of tACE). Although hinging about this region does not alter the relative orientation of the zinc-coordinating residues, it does move the zinc ion away from the opposite wall of the active site cleft by ∼3.8Å, thus opening up the catalytic site.
Three of the six regions involved in hinging thus show low sequence-conservation and a high degree of disorder or flexibility in the crystal structures tACE, tACEG13, tACE-G1234, AnCE, ACE2 and Ndom as evidenced by their temperature factors. The other three regions display a high degree of order in the crystal structures and are highly conserved in the homologues considered. This indicates that the end points of the ACE2 hinge motion represent two stable conformations of these regions that have been captured under the crystallographic conditions. The presence of glycines, serines and alanines in these regions may allow for more than one stable conformation. Moreover, their sequence conservation, together with the conservation of functional flexibility in the loop regions, suggests a conservation of the hinge mechanism among these homologues.
Conservation of temperature factors
The conservation of thermal stability or flexibility in these structures extends beyond the hinge regions, with the pattern of B-factors being conserved in tACE-G13, tACE-G1234, wild-type tACE, Ndom, ACE2 and AnCE structures (Fig. 4 - wild-type tACE and tACE-G1234 data not shown). Furthermore, residues having lower-than-average temperature factors (less than -σ(B)) are all located in the interior of subdomain II, surrounding the CL1 binding-site and including chloride and substrate ligands, whereas the residues having high temperature factors are all on the surface, and fall largely into subdomain I. The only residues in subdomain II that have high temperature factors are at the end of α4 (which flanks the lid helices), and in the C-terminal loop which was disordered in the wild-type tACE structure. Thus subdomain II, containing the zinc and chloride binding sites, has a greater degree of structural rigidity than subdomain I in all of the known structures of ACE homologues.
Since most of the residues involved in intermolecular contacts in tACE crystals lie on the surface of subdomain II, it could be argued that the higher thermal stability of this subdomain is an artefact of lattice packing. However, the other structures studied were crystallised in different space groups and under different conditions to tACE, indicating that this is more likely to be a genuine conservation, which may have functional significance.
Normal mode analysis reveals intrinsic flexibility
In order to investigate the possibility of hinge-movement in tACE further, NMA of tACE and ACE2 was carried out. Since open structures of both were required for comparison, an open model of tACE (tACEo) was generated, based on the unliganded ACE2o, and a model of tACE (tACEc) based on ACE2c served as a control for errors introduced by the modelling process (Fig. 3). Low frequency normal modes have been shown to depend more on overall shape or mass distribution than on atomic structure, with coarse-grained approaches such as that employed by elNemo being as effective as more detailed approaches at identifying biologically relevant modes (39,40). Thus despite the relatively low homology between tACE and ACE2 (40%), these models can be regarded as suitable for NMA.
NMA of both open structures tACEo and ACE2o yielded a single normal mode (mode 1, having the lowest frequency, in both cases) having a high degree of overlap with the putative or observed structural change (Table 3; Fig. 5 A, E & F). In both cases, this mode has moderate collectivity and describes a closing of the active site cleft, with similar amplitudes of perturbation required for maximal overlap with the closed forms (Table 3). The residues on the underside of the active site cleft, including most of subdomain II, show little or no movement relative to neighbouring residues. The motion corresponds closely to the hinging observed in the ACE2 crystal structures, as well as to that predicted by the tACEo model, as is evidenced by the low r.m.s. deviations between the resulting perturbed end-state models and the closed form comparison structures (Table 3).
Table 3.
Normal mode analysis of tACE and ACE2 - statistics for the mode having the highest overlap with the proposed structural transition for each structure studied.
| Structure analysed |
Comparison structure |
Mode | Overlap | Collectivity | |Amplitude| (DQa) | RMSDb (Å) |
|---|---|---|---|---|---|---|
| ACE2o | ACE2c | 1 | 0.733 | 0.386 | 224 | 1.77 |
| tACEo | tACEc | 1 | 0.709 | 0.427 | 248 | 1.86 |
| tACEo | wt tACE | 1 | 0.643 | 0.427 | 245 | 2.19 |
| ACE2c | ACE2o | 2 | 0.117 | 0.132 | 85 | 3.43 |
| tACEc | tACEo | 6 | 0.133 | 0.500 | 96 | 3.62 |
| wt tACE | tACEo | 2 | 0.129 | 0.468 | 107 | 3.89 |
The relative amplitude of perturbation along the direction of the normal mode concerned, required to achieve the best alignment with the comparison structure; arbitrary units.
R.m.s. deviation for all Cα-atoms between the comparison structure and a model perturbed according to the mode in question and the stated amplitude.
Figure 5.
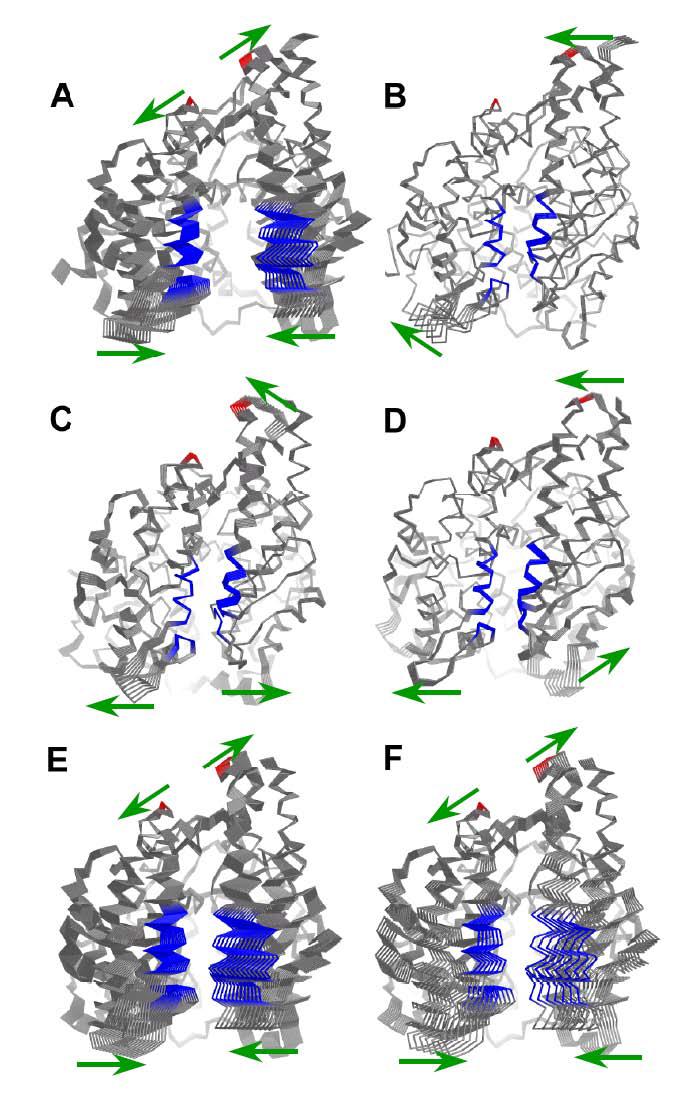
Normal mode analysis of tACE and ACE2: models perturbed according to the modes showing highest overlap with the putative structural change, the amplitude of perturbation being that which resulted in the closest alignment with the comparison structure. A) ACE2o (vs. ACE2c); B) ACE2c (vs. ACE2o); C) wild-type tACE (vs. tACEo); D) tACEc (vs. tACEo); E) tACEo (vs. wild-type tACE); F) tACEo (vs. tACEc). Colours are according to the residues showing the greatest relative displacements in ACE2o and tACEo: red residues move apart and blue residues move together as the cleft closes in the modes for these two structures. Green arrows indicate the major directions of movement in the mode in question. This figure was generated using PYMOL 0.98 (DeLano Scientific).
It could be argued that the similarity in behaviour of the normal modes of tACEo and ACE2o is an artefact due to the modelling of the open structure based on ACE2o. However, the close alignment of the perturbed model of tACEo with the wild-type tACE structure suggests that this model may represent a reasonable approximation of a real open form of tACE (Table 3; Fig. 3 B).
In contrast with these results, the closed structures (ACE2c, tACEc and wild-type tACE) did not yield normal modes having high overlap with the structural change (Table 3; Fig. 5 B, C & D). This is probably because the moving residues are close together in the starting structure, becoming linked in the force field so that large Cα-Cα fluctuations between them are not favoured. This tendency of closed structures to yield normal modes that do not describe the structural transition as well as those of the corresponding open form has been documented for a number of cases (40). While the motions described by the normal modes showing the highest overlap do involve partial opening of the active site cleft, the amplitudes of displacement required for maximum agreement with the comparison structure are low, and the resulting structures do not align closely with the open forms (Table 3). However, since these results were similar for both wild-type tACE and ACE2c, they do not exclude the possibility that tACE might undergo a hinge movement. Rather, the fact that NMA of the wild-type structure of tACE does yield some normal modes having high collectivity is a further indication that some kind of concerted movement about the active site is likely to occur.
This evidence; taken together with the necessity of opening of the active cleft to allow substrate access, the entropically-driven nature of substrate binding, the hinging of homologue ACE2, the presence of unexpected small molecules in the active sites of unliganded tACE-G13, tACE-G1234, wild-type tACE, and Ndom, and the conservation of the sequence or at least flexibility of the proposed hinge regions in these homologues; suggests that tACE and tACE homologues do have an open form and that this open form is similar to ACE2o. Moreover, since tACE is essentially identical to the C domain of sACE, this evidence points to a similar hinge motion in both domains of sACE.
CONCLUSIONS
Glycosylation mutants tACE-G13 and tACE-G1234 crystallise under the same conditions and in the same space group as wild-type tACE expressed in the presence of α-glucosidase-1 inhibitor NB-DNJ. The structures of the glycosylation mutants do not differ significantly from that of wild-type tACE, despite the presence of more complex glycan chains at the remaining glycosylation sites. It is thus possible to solve tACE structures without the use of expensive α-glucosidase-1 inhibitors, which in turn has important implications for future structural studies of ACE inhibitor binding. Elucidation of glycan chains at N72 and N109 revealed that interactions of glycan residues at these sites with adjacent protein residues may be important for the stabilisation of the lid helices α1 and α2.
Based on analysis of temperature factors, subdomain II of tACE-G13, containing bound chloride ions and the zinc-binding site, displays a high degree structural rigidity while subdomain I appears to be more flexible. This domain stability is conserved in tACE-G1234, wild-type tACE, Ndom, ACE2 and AnCE, and may have functional significance.
The residues involved in the hinge motion of ACE2 are conserved, at least at the level of functional flexibility, in tACE, Ndom and AnCE. Normal mode analysis of the closed tACE structure and a modelled open form demonstrated that the intrinsic flexibility of this structure about the active site cleft is similar to that of ACE2. This suggests that hinging is a common mechanism for substrate entry in ACE homologues. This hypothesis is further supported by the observation that some kind of motion must occur to allow substrate access, the calorimetric evidence for a large entropic contribution to substrate binding, and the presence of unexpected small molecules in the active sites of the unliganded wild-type tACE, Ndom, and tACE glycosylation mutant structures. (10,16,18).
Footnotes
This work was supported by the Carnegie Corporation of New York, the University of Cape Town, the South Afican National Research Foundation and the Wellcome Trust (UK) grants 070060 and 071047.
The atomic coordinates and structure factors for glycosylation mutants tACE-G13 (codes 2iul and r2iulsf) and tACE-G1234 (codes 2iux and r2iuxsf) have been deposited in the RCSB Protein Data Bank, www.pdb.org.
Abbreviations: ACE, angiotensin converting enzyme 1; sACE, somatic ACE; tACE, testis ACE; NBDNJ, N-butyl-deoxynojirimycin; Ndom, N-terminal domain of sACE; ACE2, human angiotensin-converting enzyme 2; NMA, normal mode analysis; tACE-G13, mutant of human tACE lacking the N-terminal 36 residues and all but the first and third N-glycosylation sites; tACE-G1234, mutant of human tACE lacking the N-terminal 36 residues and all but the first four N-glycosylation sites; ACE2o, open unliganded structure of ACE2; ACE2c, closed liganded structure of ACE2; tACEo, model of human tACE based on ACE2o; tACEc, model of human tACE based on ACE2c; NAG, N-acetyl glucosamine.
REFERENCES
- 1.Skeggs LT, Jr., Kahn JR, Shumway NP. The preparation and function of the hypertensin-converting enzyme. J. Exp. Med. 1956;103:295–299. doi: 10.1084/jem.103.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Acharya KR, Sturrock ED, Riordan JF, Ehlers MR. ACE revisited: a new target for structure-based drug design. Nat. Rev. Drug Discovery. 2003;2:891–902. doi: 10.1038/nrd1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zaman MA, Oparil S, Calhoun DA. Drugs targeting the renin-angiotensin-aldosterone system. Nat. Rev. Drug Discovery. 2002;1:621–636. doi: 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- 4.Ehlers MR, Riordan JF. Angiotensin-converting enzyme: new concepts concerning its biological role. Biochemistry. 1989;28:5311–5318. doi: 10.1021/bi00439a001. [DOI] [PubMed] [Google Scholar]
- 5.Ehlers MR, Fox EA, Strydom DJ, Riordan JF. Molecular cloning of human testicular angiotensin-converting enzyme: the testis isozyme is identical to the C-terminal half of endothelial angiotensin-converting enzyme. Proc. Natl. Acad. Sci. U. S. A. 1989;86:7741–7745. doi: 10.1073/pnas.86.20.7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc. Natl. Acad. Sci. U. S. A. 1988;85:9386–9390. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hubert C, Houot AM, Corvol P, Soubrier F. Structure of the angiotensin I-converting enzyme gene. Two alternate promoters correspond to evolutionary steps of a duplicated gene. J. Biol. Chem. 1991;266:15377–15383. [PubMed] [Google Scholar]
- 8.Ehlers MR, Chen YN, Riordan JF. The unique N-terminal sequence of testis angiotensin-converting enzyme is heavily O-glycosylated and unessential for activity or stability. Biochem. Biophys. Res. Commun. 1992;183:199–205. doi: 10.1016/0006-291x(92)91628-4. [DOI] [PubMed] [Google Scholar]
- 9.Ehlers MR, Schwager SL, Scholle RR, Manji GA, Brandt WF, Riordan JF. Proteolytic release of membrane-bound angiotensin-converting enzyme: role of the juxtamembrane stalk sequence. Biochemistry. 1996;35:9549–9559. doi: 10.1021/bi9602425. [DOI] [PubMed] [Google Scholar]
- 10.Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 11.Sturrock ED, Natesh R, van Rooyen JM, Acharya KR. Structure of angiotensin I-converting enzyme. Cell Mol. Life Sci. 2004;61:2677–2686. doi: 10.1007/s00018-004-4239-0. [DOI] [PubMed] [Google Scholar]
- 12.Yu XC, Sturrock ED, Wu Z, Biemann K, Ehlers MR, Riordan JF. Identification of N-linked glycosylation sites in human testis angiotensin-converting enzyme and expression of an active deglycosylated form. J. Biol. Chem. 1997;272:3511–3519. doi: 10.1074/jbc.272.6.3511. [DOI] [PubMed] [Google Scholar]
- 13.Corradi HR, Schwager SL, Nchinda AT, Sturrock ED, Acharya KR. Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J. Mol. Biol. 2006;357:964–974. doi: 10.1016/j.jmb.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 14.Gordon K, Redelinghuys P, Schwager SL, Ehlers MR, Papageorgiou AC, Natesh R, Acharya KR, Sturrock ED. Deglycosylation, processing and crystallization of human testis angiotensin-converting enzyme. Biochem. J. 2003;371:437–442. doi: 10.1042/BJ20021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natesh R, Schwager SL, Evans HR, Sturrock ED, Acharya KR. Structural details on the binding of antihypertensive drugs captopril and enalaprilat to human testicular angiotensin I-converting enzyme. Biochemistry. 2004;43:8718–8724. doi: 10.1021/bi049480n. [DOI] [PubMed] [Google Scholar]
- 16.Kim HM, Shin DR, Yoo OJ, Lee H, Lee JO. Crystal structure of Drosophila angiotensin I-converting enzyme bound to captopril and lisinopril. FEBS Lett. 2003;538:65–70. doi: 10.1016/s0014-5793(03)00128-5. [DOI] [PubMed] [Google Scholar]
- 17.Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andujar-Sanchez M, Camara-Artigas A, Jara-Perez V. A calorimetric study of the binding of lisinopril, enalaprilat and captopril to angiotensin-converting enzyme. Biophys. Chem. 2004;111:183–189. doi: 10.1016/j.bpc.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Ehlers MR, Chen YN, Riordan JF. Purification and characterization of recombinant human testis angiotensin-converting enzyme expressed in Chinese hamster ovary cells. Protein Expression Purif. 1991;2:1–9. doi: 10.1016/1046-5928(91)90001-y. [DOI] [PubMed] [Google Scholar]
- 20.Friedland J, Silverstein E. A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am. J. Clin. Pathol. 1976;66:416–424. doi: 10.1093/ajcp/66.2.416. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [20] [DOI] [PubMed] [Google Scholar]
- 22.French S, Wilson K. On the treatment of negative intensity observations. Acta Crystallogr., Sect. A: Found. Crystallogr. 1978;34:517–525. [Google Scholar]
- 23.Kissinger CR, Gehlhaar DK, Fogel DB. Rapid automated molecular replacement by evolutionary search. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999;55:484–491. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- 24.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Jones TA. A graphics model building and refinement system for macromolecules. J. Appl. Crystallogr. 1978;11:268–272. [Google Scholar]
- 26.Murshudov GN. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 27.Collaborative Computational Project, N. 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 28.Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: Model building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Perrakis A, Morris RM, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thronton JM. PROCHECK -A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.Cohen GE. ALIGN: A program to superimpose protein coordinates, accounting for insertions and deletions. J. Appl. Crystallogr. 1997;30:1160–1161. [Google Scholar]
- 33.Guy JL, Jackson RM, Acharya KR, Sturrock ED, Hooper NM, Turner AJ. Angiotensin-converting enzyme-2 (ACE2): comparative modeling of the active site, specificity requirements, and chloride dependence. Biochemistry. 2003;42:13185–13192. doi: 10.1021/bi035268s. [DOI] [PubMed] [Google Scholar]
- 34.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 35.Canutescu AA, Shelenkov AA, Dunbrack RL., Jr. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tama F, Gadea FX, Marques O, Sanejouand YH. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins: Struct. Funct. Bioinformat. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 38.Tzakos AG, Galanis AS, Spyroulias GA, Cordopatis P, Manessi-Zoupa E, Gerothanassis IP. Structure-function discrimination of the N- and C- catalytic domains of human angiotensin-converting enzyme: implications for Cl- activation and peptide hydrolysis mechanisms. Protein Eng. 2003;16:993–1003. doi: 10.1093/protein/gzg122. [DOI] [PubMed] [Google Scholar]
- 39.Lu M, Ma J. The role of shape in determining molecular motions. Biophys J. 2005;89:2385–2401. doi: 10.1529/biophysj.105.065904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure. 2005;13:373–380. doi: 10.1016/j.str.2005.02.002. [DOI] [PubMed] [Google Scholar]