

Abstract
Axonal injury in white matter is an important consequence of many acute neurological diseases including ischemia. A role for glutamate-mediated excitotoxicity is suggested by observations from in vivo and in situ models that AMPA/kainate blockers can reduce axonal injury. We assessed axonal vulnerability in primary murine neuronal cultures, with axons isolated from their cell bodies using a compartmented chamber design. Transient removal of oxygen and glucose in the axon compartment resulted in irreversible loss of axon length and neurofilament labeling. This injury was not prevented by addition of ionotropic glutamate receptor blockers and could not be reproduced by glutamate receptor agonists. However, hypoxic injury was prevented by blockade of voltage-gated sodium channels, inhibition of calpain, and removal of extracellular calcium. These results suggest that isolated, unmyelinated axons are vulnerable to hypoxic injury which is mediated by influx of sodium and calcium, but is independent of glutamate receptor activation.
Keywords: glutamate, AMPA/kainate, NMDA, calpain, axon, excitotoxicity, ischemia, oxygen-glucose deprivation, white matter
INTRODUCTION
Ischemic damage to axons in cerebral white matter contributes to neurological dysfunction after stroke, cardiac arrest, and perinatal encephalopathy (Dewar et al., 1999; Volpe, 2001). Axonal injury is also important in brain and spinal cord trauma, multiple sclerosis, and neurodegenerative diseases (Bjartmar and Trapp, 2001; Stys, 2005). Many of the initial ionic events leading to hypoxia-induced loss of axon conduction have been well characterized, largely in isolated preparations of rodent optic nerve (Waxman, 1991). Energy failure causes depletion of ATP and axon depolarization. This is followed by accumulation of axoplasmic Na+ via non-inactivating voltage-gated Na channels (Stys and Lopachin, 1998), and of intra-axonal free Ca2+ by activation of voltage-sensitive Ca2+ channels (Fern et al, 1995), reversal of Na+/Ca2+ exchange (Li et al., 2000; Brown et al., 2001; Ouardouz et al., 2005), and release from intracellular stores (Ouardouz et al., 2003).
Recent evidence suggests that excessive glutamate receptor activation (or excitotoxicity) may also contribute to white matter injury in several conditions. The immediate target of this injury may be white matter glial cells. In particular, the central nervous system myelin-forming cells, oligodendrocytes, express functional glutamate receptors (Gallo et al., 1994) and can be injured in vitro by overactivation of ionotropic AMPA/kainate receptors (Yoshioka et al., 1995; McDonald et al., 1998; Fern and Moller, 2000). New studies suggest that distal oligodendrocyte processes express NMDA receptors which may also contribute to their injury (Salter and Fern, 2005; Karadottir et al., 2005; Micu et al., 2006). Supporting the role of glial excitotoxicity in vivo, AMPA/kainate antagonists, such as NBQX, have been shown to reduce oligodendrocyte or myelin loss in white matter in slice and animal models of brain and spinal cord injury (Rosenberg et al., 1999; Li et al., 2000; Follett et al., 2000; Wilke et al, 2004).
In these models AMPA/kainate blockade prevents injury of white matter axons as well as oligodendrocytes. For example, axonal protection has been observed in cortical brain slice (Tekkok and Goldberg, 2001), isolated spinal cord (Agrawal and Fehlings, 1997; Li et al., 2000) and in vivo rodent models including spinal cord trauma (Wrathall et al., 1994), spinal ischemia (Kanellopoulos et al., 2000), stroke (McCracken et al., 2002), and experimental allergic encephalomyelitis (Pitt et al., 2000). Glutamate receptor subunits have been observed in axons (Li et al., 2000), but it is not known whether functional receptors are expressed on axolemma. Therefore, these observations raise the following question: is AMPA/kainate receptor-dependent injury mediated by glutamate action directly on axons, or indirectly, through receptors located on neuronal cell bodies or white matter glial cells? The issue cannot be resolved using standard pharmacological approaches in intact models, nor in conventional dissociated cell culture systems. Therefore we examined the vulnerability of isolated axons using a compartmented chamber system (Ivins et al., 1998). Primary murine cortical cultures were plated on one side of a glass cover slip, and axons allowed to project under the cover slip to a different chamber. We examined the vulnerability of isolated cortical axons to oxygen-glucose deprivation (OGD), and assessed the roles of extracellular cations and glutamate. Axon injury was assessed by neurofilament immunocytochemistry and by fluorescence microscopy using neurons derived from mice expressing the green fluorescent protein derivative YFP.
METHODS
Cell culture
Cortical neurons were cultured using a two-compartment chamber design which was previously shown to promote neurite outgrowth from the side containing neuronal cell bodies, without chemical exchange between chambers (Ivins et al., 1998). Chambers were constructed of a 5 mm hemisected cylinder of Teflon tubing attached with sterile silicon grease to a glass coverslip barrier; the bottom of each chamber assembly was placed with silicon grease on a 35 mm polystyrene tissue culture dish previously treated with poly-D-lysine and laminin.
Cortices were isolated from E15 Swiss Webster (Charles Rivers, Wilmington, Maryland) or transgenic C57Bl/6 mice expressing YFP under the neuronal Thy1.1 promoter (Feng et al., 2000). The thy1-YFP-16 transgenic line provided high YFP expression in cortical neurons at early developmental ages. This tissue was briefly triturated yielding microexplants that were plated in the inner chamber of the culture array. Both the inner and the outer chamber were supplied with 10% serum containing minimal essential media (MEM)-based media (Gibco, Grand Island, New York) with 20 mM D-glucose, 2 mM glutamine for 14 days. Before experiments were performed, the external media was replaced with 0.04% mM trypan blue for two hours to establish the chambers’ integrity. Those with visible leaks were discarded.
Cell culture toxicity paradigms
Drugs and OGD treatments were applied to the outer, axonal compartment only. Neuronal cell bodies in the inner compartment were maintained in 10% serum-containing media (also containing oxygen and glucose) during axonal toxicity paradigms. Cultures were rinsed three times with serum-free MEM buffer containing 20 mM D-glucose warmed to 37 °C, and then exposed to drugs diluted in the same media. For experiments with recovery periods, cultures were washed three times with regular media and returned to 37 °C. OGD experiments were performed in an anaerobic chamber as described previously (Forma; Goldberg and Choi, 1993).
All drugs were purchased from Sigma (St. Louis, Missouri) except for (s)-AMPA (Tocris Cookson, Ballwin, Missouri) and ALLM (Calbiochem, La Jolla, California). Reagents were dissolved in water or DMSO as specified by the vendor and stored in stock solutions protected from light at −20 ºC for no more than three months until use.
Immunocytochemistry
Cultures were fixed with 4% paraformaldehyde and 0.025% glutaraldehyde in PBS for 30′. Cultures were permeablized with 0.125% Triton X-100 and blocked in 5% normal goat serum (NGS) in PBS for thirty minutes. Primary and secondary antibodies were diluted in 5% NGS and applied sequentially for 4 hours at room temperature or overnight at 4º.
Primary antibodies used in this study included mouse monoclonal neurofilament antibodies SMI31 and SMI32 (Sternberger Monoclonals, Baltimore, Maryland; 1:10,000 and 1:1,000, respectively), and rabbit polyclonal antibodies anti-MAP2 (Boehringer Mannhiem; 1:1,000), and anti-tau (ICN Biochemicals, Costa Mesa, California; 1:1,000). Secondary antibodies included Alexa-488 conjugated goat anti-mouse and goat anti-rabbit (Molecular Probes Eugene, Oregon; 1:1,000) and CY3 conjugated goat anti-mouse IgG (Jackson Immunoresearch, West Grove, Pennsylvania; 1:1,000). For double labeling, cultures were re-blocked and the second set of antibodies was applied similarly. Control experiments for single and double labeling demonstrated no cross-reactivity.
Microscopy
Cells were examined under epifluorescence illumination on a Nikon Eclipse TE300 (Nikon Inc., Melville, New York) inverted microscope using either a 10x, N.A. 0.30 or 20x, N.A. 0.45 objective. Digital images were acquired using an RT Color Spot Camera (Diagnostic Instruments, Hitschfel Instruments, Inc.).
Measurement of axon lengths
Axon integrity was assessed with immunofluorescence for axon cytoskeletal components including phosphorylated neurofilaments H and M (SMI31) and tau. We quantified the health of the axons by measuring their total length extended from the edge of the coverslip as the most consistent measure among control cultures. Fluorescent images at 100x were taken at 2–3 consecutive sites of axonal crossing in each dish and analyzed using image processing software (Metamorph, Universal Imaging, West Chester, Pennsylvania). 10 axons were randomly identified at the site where they protruded underneath the coverslip barrier and the lengths were traced from the edge of the coverslip to their ends. Calibration from pixels to microns was accomplished by reference to a hemocytometer image. At least three dishes from two different plating dates were used for all experiments. For YFP experiments, axon integrity was assessed over time. Images of the same sites were taken before (T0) and 24 hours after the paradigm (T24). Axon lengths were measured as described above. The average length was calculated as T24/T0. Four different cultures from three different plating dates were used for YFP experiments. Average values from each dish were identified as a single observation for statistical analysis.
RESULTS
Hypoxic injury in isolated cortical axons
To assess vulnerability of isolated axons, independent of effects upon neuronal cells bodies and dendrites, we made use of a chamber model (Ivins et al., 1998) modified for cortical neurons. Each chamber was constructed with a semicircular wall of Teflon tubing, and a glass coverslip divider fixed to the Teflon tubing and culture surface with sterile silicon grease (Figure 1). Cortical microexplants plated in the internal compartment projected neurites under the cover slip and into external compartment (Figure 1). The neurites that extended into the outer chamber were up to several hundred microns long, with continuous narrow calibers and no spines. All neurites in the external compartment were immunoreactive to SMI31 and never labeled with anti-MAP2 (data not shown) indicating that they were axons rather than dendrites. To observe axon morphology throughout the course of injury, some experiments were performed with tissue from transgenic animals expressing YFP in their neurons (Feng et al., 2000; Figure 1).
Figure 1. Compartmented chamber model.
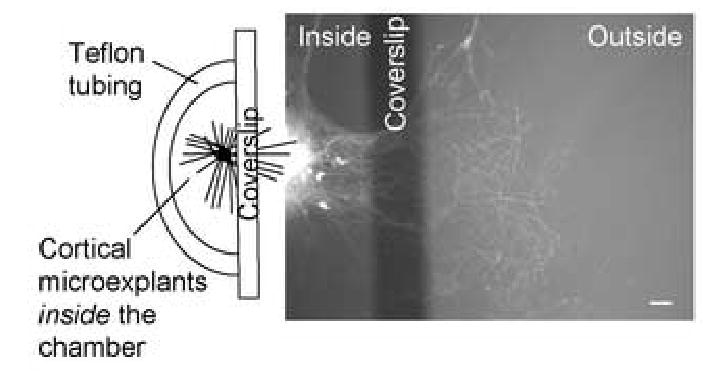
Modified Campenot chambers were built with teflon tubing, a glass coverslip barrier, and silicon grease to attach the assembly to the polystyrene culture surface (schematic on left). Cortical explant cultures were plated on the inside of this array. Axons grew out from the explants and some crossed the coverslip barrier entering the external chamber. Axon structural integrity was assessed by immunocytochemistry (not shown) or by use of explants derived from transgenic mice expressing neuronal YFP (right).
Axon injury was determined by measuring the total length axons extended from the outer edge of the coverslip. Under control conditions, the average axon lengths were reasonably consistent from culture to culture. There was considerable culture-to-culture variability in the number of axons crossing the coverslip barrier and in the degree of curvature of individual axons. Because of this variability it was not reliable to count the number of axons reaching a certain distance from the coverslip.
Isolated axons were exposed to OGD within an anaerobic chamber (Goldberg and Choi, 1993), by thorough solution exchange of the outer compartment with pre-heated oxygen- and glucose-free medium. The inner compartment did not undergo solution exchange. As also shown in previous experiments (Goldberg and Choi, 1993; Hasbani et al, 2001) neuronal cell bodies, dendrites, and axons are not injured by brief placement in the anaerobic chamber (because there is residual oxygen and glucose in the unexchanged medium) and there was no loss of cellular viability as assessed by dye exclusion.
OGD for 20′ to the axonal compartment led to no immediate change in YFP(+) axons. However, by 24 hours later the axons appeared 25–30% shorter (Figure 2). Increasing the duration of OGD to 60′ or 120′ did not potentiate this injury (Table 1). We also examined the cytoskeleton of YFP(+) axons with immunoreactivity for phosphorylated neurofilaments H and M with the monoclonal clone SMI31. Under control conditions, SMI31(+) axons were ~1 mm long. SMI31(+) axon length declined in length by ~40% in response to 20, 60, and 120′ of OGD (Figure 3 and Table 1). This response was observed as early as 2 hours after the time of injury (data not shown). Axons of neurons derived from Thy1-YFP and wild-type mice yielded similar results. There was no statistical difference between the two and the data have been pooled here (Table 1).
Figure 2. OGD reduces YFP(+) axon lengths (A–D).
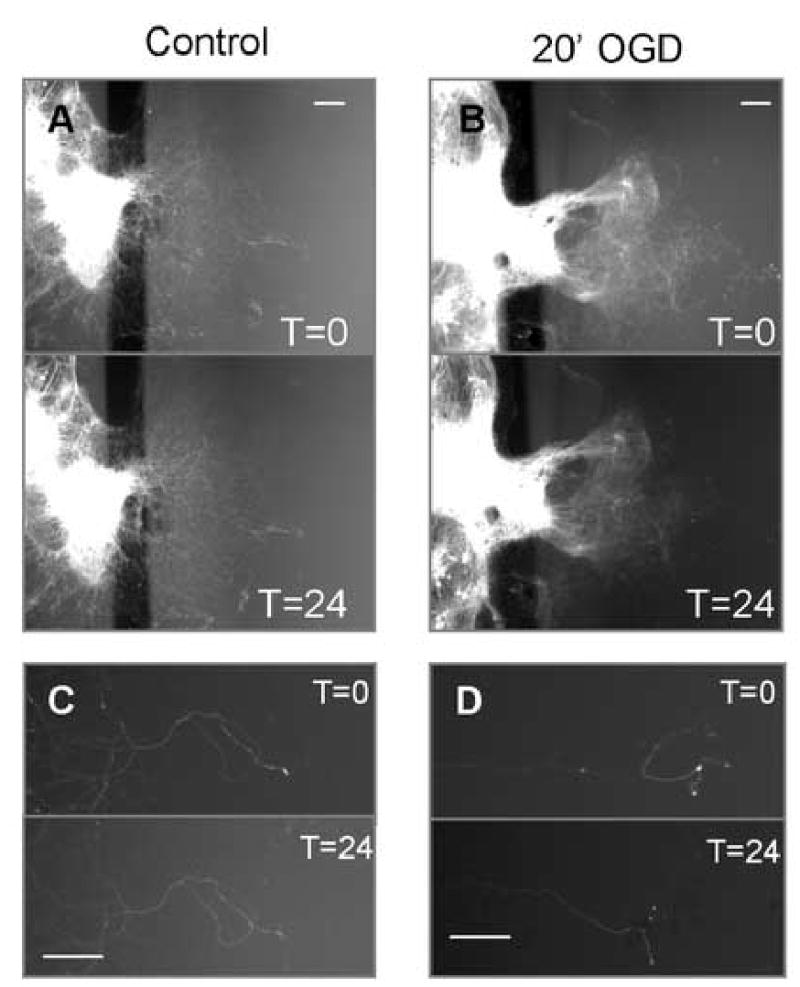
YFP(+) explant cultures in chamber culture extended axons into the outer compartment. Over 24 hours under wash conditions, no gross morphological changes were observed (figures A and C). Following 20′ of OGD, the distal ends of YFP axons disappeared by 24-hours (figures B and D). Higher magnification illustrates this loss (figure D). In both wash and control conditions, axons inside the chamber – those not exposed to any insult – remained intact. Scale bars indicate 100 μm.
Table 1. Pharmacology of axon vulnerability.
Axonal loss following 20′ of OGD was blocked by omission of extracellular calcium or by co-application of TTX (10 μM) or ALLM (1 μM) at the time of injury. Application of veratridine (10 μM, 20′) mimicked axonal injury seen during OGD. SMI31 and tau values are represented as percentages normalized to control values from sister cultures. YFP values are the T=24 values normalized to T=0 axon measurements from the same cultures. All experiments with OGD (20′) exhibited 50–60% loss for SMI31 and Tau labeling and 25–35% for YFP(+) axon lengths (data not shown). +/− values indicate SEM.
| SMI31 | Tau | YFP | |
|---|---|---|---|
| CONTROL | 100% ± 6% | 100% ± 7% | 104% ± 4% |
| 20′ OGD | 64% ± 5% * | 64% ± 6% * | 71% ± 10% * |
| 60′ OGD | 61% ± 9% * | 62% ± 6% * | 74% ± 7% * |
| 120′ OGD | 51% ± 6% * | 63% ± 3% * | 71% ± 7% * |
| CONTROL | 100% ± 3% | 100% ± 7% | 108% ± 4% |
| 20′ NaCN/2DG | 49% ± 5% * | 48% ± 2% * | 71% ± 11% * |
| 60′ NaCN/2DG | 39% ± 4% * | 37% ± 1% * | 55% ± 23% * |
| 120′ NaCN/2DG | 40% ± 4% * | 43% ± 6% * | 40% ± 4% * |
| Control | 100% ± 6% | 100% ± 8% | 101% ± 2% |
| 20′ OGD | 52% ± 4% * | 53% ± 3% * | 70% ± 14% * |
| + NBQX/MK-801 | 54% ± 7% * | 63% ± 6% * | 73% ± 11% * |
| +TTX | 102% ± 6% ** | 102% ± 5% ** | 97% ± 10% ** |
| -Calcium | 99% ± 7% ** | 90% ± 8% ** | 105% ± 10% ** |
| +ALLM | 103% ± 5% ** | 95% ± 7% ** | 102% ± 5% ** |
| CTL | 100% ± 6% | 100% ± 15% | 108% ± 12% |
| Veratridine | 33% ± 5% * | 56% ± 14% * | 39% ± 11% * |
| Control | 100% ± 7% | 100% ± 6% | 97% ± 3% |
| Kainate | 104% ± 6% | 108% ± 4% | 105% ± 7% |
| NMDA | 106% ± 5% | 99% ± 7% | 102% ± 8% |
Figure 3. OGD causes a loss of neurofilaments. (A–D).
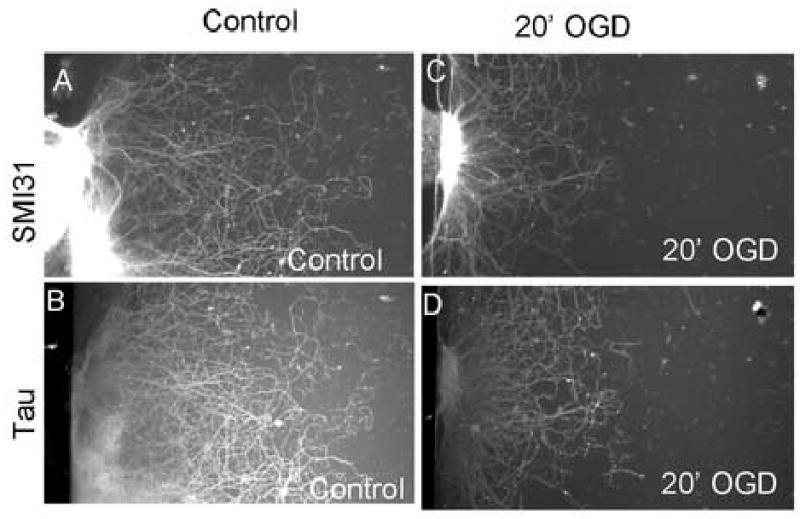
SMI31 (A and C) and anti-tau (B and D) immunofluorescence of axons in chamber cultures demonstrated substantial loss of axonal neurofilaments after 20′ of OGD and 24 hours of recovery (figures C and D) as compared with control, sham wash axons (figures A and B).
SMI31 recognizes phosphorylated neurofilaments. An increase in non-phosphorylated neurofilaments may indicate the early stages of injury (Meller et al., 1994). Immunoreactivity for non-phosphorylated neurofilaments recognized by SMI32 was not detected in OGD treated axons at 1, 6 and 24 hours after the initial injury (data not shown). This suggests a loss of neurofilaments rather than alteration in their phosphorylation state. Immunoreactivity for axonal tau illustrated a similar decrease in axon length (Figure 3 and Table 1). These results support the hypothesis that OGD leads to a loss of axonal structure.
It was interesting that sustained OGD did not result in increased axon injury with longer durations. One possible explanation might be that the exchange of oxygen- and glucose-free medium was not sufficiently complete within the axon compartment of the cell culture chamber array. To test this with a method less sensitive to small differences in retained medium, we applied “chemical ischemia” with 1 mM NaCN and 10 mM 2-deoxyglucose (2-DG). Results were similar to those observed with OGD, with comparable injury for all three structural parameters tested over a similar time-course (Table 1). Increasing durations of chemical anoxia did not significantly increase axonal injury. There was never more than a 61% reduction in axon length, even with two hours of chemical ischemia. This suggests that results did not depend upon the model of energy deprivation applied.
The neuronal cell bodies inside the chamber were not injured by transient OGD. YFP expressing neurons maintained normal fluorescence intensity and morphology (Figure 2). Moreover, there was no loss of neuronal viability as assessed by propidium iodide uptake. In neuronal cultures treated with OGD in the axonal compartment, or with normoxic wash conditions, at 24 hrs PI uptake was observed in 4 +/− 4.5% of neurons (normalized to cultures exposed to 500 μM NMDA to kill all neurons).
OGD induced injury to isolated axons is not excitotoxic
Since many modes of injury to white matter are excitotoxic, we tested the role of glutamate receptors in the vulnerability of isolated axons. We blocked NMDA and non-NMDA receptors by co-application of MK-801 (10 μM) and NBQX (30 μM) to the axonal chamber during the OGD insult. These antagonists had no effect on axon lengths (Table 1). Similarly, the GluR agonists kainate (100 μM) and NMDA (100 μM) applied for two hours had no effect upon axon length immediately (data not shown) or twenty-four hours later (Table 1). This suggests that intense activation of ionotropic glutamate receptors is not sufficient to cause injury in isolated, unmyelinated cortical axons.
OGD induced injury to isolated axons is dependent upon tetrodotoxin (TTX)-sensitive voltage-gated sodium channels
Axons are rich in voltage-gated sodium channels, which have been implicated in many models of axonal injury. Application of the voltage-gated sodium channel blocker, 10 μM TTX, during 20′ OGD insult prevented reduction in axon lengths (Table 1). Activation of voltage-sensitive sodium channels with veratridine (10 μM) for 20′ led to irreversible injury of axons (Table 1).
OGD induced injury to isolated axons is dependent upon extracellular calcium and calpain activation
Removal of extracellular calcium from the OGD media prevented injury (Table 1). Elevation in intra-axonal calcium may activate calcium-dependent proteases, such as calpain, and over-stimulation of this enzyme may lead to structural damage. Co-application of a calpain inhibitor, ALLM (1 μM), also prevented structural loss of axons in response to OGD (Table 1). These results suggest that an influx of calcium during axonal energy deprivation triggers calpain activation to pathological levels leading to axonal injury.
DISCUSSION
We report here that cortical axons in compartmented cultures are structurally damaged by transient deprivation of oxygen and glucose. Axons are not protected by ionotropic glutamate receptor antagonists, and are not vulnerable to direct application of excitotoxic agonists. OGD-induced axonal injury is dependent upon activation of TTX-sensitive voltage-gated sodium channels, the presence of extracellular calcium, and calpain activation.
Compartmented culture system for examination of axon vulnerability
In vitro models are valuable for examining the response of axons to energy deprivation, without the influences of alterations in local blood flow or neurochemical environment which occur in vivo. These experiments make use of a culture system for separation of neuronal axons from their cell bodies and dendrites. Using primary murine cortical cultures, we previously observed that cortical dendrites and axons are differentially injured when cultures are exposed to glutamate receptor agonists or toxins (Hasbani et al., 1998). Dissociated cultures allow microscopic identification of axons by morphological or immunocytochemical features, but it is not possible to determine whether axon injury occurs as a secondary consequence of damage to neuronal cell bodies. Axons can be grown in chambered systems such as that described by Campenot for rat sympathetic neurons (Campenot, 1982). Commercially available Campenot designs are poorly suited for rodent cortical neurons, because their Teflon barriers of several millimeters exceed the length of most cultured cortical axons. The use of a glass coverslip barrier reduced this distance to less than 200 μM, while still providing pharmacological separation of the two compartments (Ivins et al., 1998). We modified the method of Ivins et al (1998) by plating embryonic cortical tissue as micro-explants of several hundred cells rather than fully dissociated cultures, which resulted in much longer neurite extension on our poly-lysine / laminin-coated polystyrene substrate.
Several potential limitations of the culture model should be recognized. In our experiments, there was considerable culture-to-culture variability, which was due in part to variable neurite penetration through the silicone grease used to seal the glass coverslip edge to the polystyrene culture surface. More consistent axon penetration could likely be obtained by use of patterned substrates or microfluidic chamber designs (Taylor et al., 2006), or a more biocompatible sealing material. The culture system uses tissue derived from neonatal animals, and deliberately excludes important interactions of axons with myelin and glial cells normally present in white matter. In the current experiments we did not include other cell types since the specific goal was to examine vulnerability of isolated axons. The compartmented culture system provides a complementary approach to in situ models for axon injury, such as acute tissue slices and isolated nerve preparations.
Axons are vulnerable to brief oxygen-glucose deprivation
Axon integrity was assessed under fluorescence microscopy and provided similar results with fixed cultures immunolabeled for phosphorylated neurofilaments or tau, or living cultures prepared from transgenic mice expressing YFP (Feng et al., 2000). We observed loss of axon structure and length by these parameters following transient OGD exposures as brief as 20 minutes. This is less than half of the duration needed to produce acute morphological damage in neuronal cell bodies and dendrites (Hasbani et al., 2001), or delayed neuronal death (Goldberg and Choi, 1993) under identical experimental conditions. OGD induced injury of axons was not immediately lethal to neuronal cell bodies. Transgenic neurons maintained their expression of YFP and wildtype neurons did not take up propidium iodide throughout the experimental time-course. We did not examine neuronal survival more than 24 hours after axon injury.
It is interesting that extending the duration of OGD did not increase the injury seen to axons. We considered several possible reasons. First, local surface tension or liquid flow might have prevented solution exchange in the area of proximal axons near the coverslip barrier. However, pharmacological application of veratridine (Table I) or hydrogen peroxide caused axonal injury right up to the coverslip (data not shown). This suggests that proximal axons were not excluded from media exchange. Second, oxygen or glucose might have diffused from the cell body compartment to supply proximal axons on the other side. We observed a similar extent of injury when energy deprivation was produced by chemical application of cyanide and 2-deoxyglucose in the axon compartment, which suggests that the observed pattern was not related to local oxygen or glucose diffusion. A third possibility is that the most proximal axon segments receive metabolic or trophic support from the nearby cell body.
Ionic mechanisms of axon vulnerability
Hypoxic injury of isolated axons was prevented by blockade of voltage-gated sodium channels with TTX (Table 1). Tetrodotoxin-sensitive sodium channels contribute to injury of myelinated axons in optic nerve anoxia (Waxman et al., 1994; Garthwaite et al., 1999; Jiang and Stys, 2000) and spinal cord contusion (Wolf et al., 2001) models. A role for TTX-sensitive sodium channels in hypoxic injury has not previously been established for non-myelinated axons. Sodium channels in non-myelinated axons are expressed diffusely rather than clustered at nodes of Ranvier, but the overall channel numbers may not be very different (Utzschneider et al., 1993). Persistent activation of TTX-sensitive sodium channels is critical for late calcium disregulation in isolated axons following stretch injury (Wolf et al., 2001).
In isolated optic nerves exposed to anoxia, action potential loss (Stys et al., 1990) and ultrastructural breakdown (Waxman et al., 1993) are highly dependent on the presence of extracellular calcium during injury. Similarly, we found that omission of calcium from the OGD media prevented the injury of isolated axons (Table I).
One putative action of this rise in [Ca2+]i in axons may be the activation of calcium-dependent proteases such as calpain. In our experiments, application of the calpain inhibitor ALLM prevented axon structural loss following brief OGD, supporting a role of calpain or other intracellular protease. The calpain inhibitor used in this study may also inhibit other neutral cysteine proteases (Sasaki et al., 1990). Jiang and Stys observed that a calpain-specific breakdown product appeared in optic nerve axons following anoxia; however blockade of calpain activation did not preserve axon function (Jiang and Stys, 2000). Observed differences may be related to the duration of study (one hour in optic nerves vs 24 hrs in cultured axons) or experimental endpoints (compound action potential in optic nerves vs axon morphology in culture).
Isolated axons are not vulnerable to excitotoxic mechanisms
Our study was prompted by the observation that glutamate receptor blockade protects axons in several white matter injury models. Therefore we examined the hypothesis that axons are directly vulnerable to glutamate receptor activation during energy deprivation. However, we observed that isolated cortical axons were not injured by prolonged AMPA/KA or NMDA receptor activation, which would be highly toxic to neuronal cell bodies or dendrites, and AMPA/KA or NMDA antagonists did not protect axons from OGD. We conclude that ionotropic glutamate receptor activation does not directly contribute to anoxic injury of unmyelinated axons in primary murine cortical culture.
The AMPA/KA receptor blocker, NBQX, protected cortical axons from OGD-induced injury in an acute white matter preparation of the corpus callosum (Tekkok and Goldberg, 2001). Since this effect does not appear to mediated by axonal glutamate receptors and does not require the presence of neuronal cell bodies, it is likely to be mediated indirectly, by activation of AMPA/KA receptors on cells resident in white matter, glia. While we cannot rule out that myelinated axons in vivo may express glutamate receptors that would change their vulnerability to glutamate, the selective vulnerability of glial cells to excitotoxicity presents an intriguing hypothesis. Both astrocytes (David et al., 1996) and oligodendrocytes (Matute et al., 1997; McDonald et al., 1998) are directly vulnerable to AMPA/KA receptor overactivation in vitro, although oligodendrocytes are much more sensitive. Recent studies suggest that oligodendrocytes and their myelin are also vulnerable to NMDA receptor mediated excitotoxicity (Karadottir et al., 2005; Salter and Fern, 2005; Micu et al., 2006). Activation of glutamate receptors on white matter glia might trigger axonal injury by loss of glial cell bodies or myelin, or by glial dysfunction (for example, loss of ion buffering or lactate transport) or by secondary release of toxic mediators.
Summary and conclusions
Murine cortical axons isolated in a compartmented culture chamber are highly vulnerable to local oxygen-glucose deprivation. This injury is dependent upon sodium, calcium and calpain activation but not upon glutamate receptor stimulation. This simple system provides an opportunity to study injury mechanisms in axons and cell-cell interactions relevant to hypoxic-ischemic, traumatic, and inflammatory etiologies of white matter injury.
Acknowledgments
We thank Olga Strots for technical assistance and M. Joshua Hasbani for early development of axon studies. This work was supported by grants (to MPG) from the American Heart Association, Juvenile Diabetes Research Foundation, and NIH NS36265 and NS32636.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences. Curr Opin Neurol. 2001;14:271–278. doi: 10.1097/00019052-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Brown AM, Wender R, Ransom BR. Metabolic substrates other than glucose support axon function in central white matter. J Neurosci Res. 2001;66:839–843. doi: 10.1002/jnr.10081. [DOI] [PubMed] [Google Scholar]
- Campenot RB. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev Biol. 1982;93:13–21. doi: 10.1016/0012-1606(82)90233-0. [DOI] [PubMed] [Google Scholar]
- David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar D, Yam P, McCulloch J. Drug development for stroke: importance of protecting cerebral white matter. Eur J Pharmacol. 1999;375:41–50. doi: 10.1016/s0014-2999(99)00280-0. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R, Ransom BR, Waxman SG. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol. 1995;74(1):369–77. doi: 10.1152/jn.1995.74.1.369. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Patneau DK, Mayer ML, Vaccarino FM. Excitatory Amino-Acid Receptors in Glial Progenitor Cells - Molecular and Functional-Properties. Glia. 1994;11:94–101. doi: 10.1002/glia.440110204. [DOI] [PubMed] [Google Scholar]
- Garthwaite G, Brown G, Batchelor AM, Goodwin DA, Garthwaite J. Mechanisms of ischaemic damage to central white matter axons: a quantitative histological analysis using rat optic nerve. Neuroscience. 1999;94:1219–1230. doi: 10.1016/s0306-4522(99)00389-9. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbani MJ, Hyrc KL, Faddis BT, Romano C, Goldberg MP. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Experimental Neurology. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- Hasbani MJ, Viquez NM, Goldberg MP. NMDA receptors mediate hypoxic spine loss in cultured neurons. Neuroreport. 2001;12:2731–2735. doi: 10.1097/00001756-200108280-00028. [DOI] [PubMed] [Google Scholar]
- Ivins KJ, Bui ET, Cotman CW. Beta-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis. 1998;5:365–378. doi: 10.1006/nbdi.1998.0228. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Stys PK. Calpain inhibitors confer biochemical, but not electrophysiological, protection against anoxia in rat optic nerves. J Neurochem. 2000;74:2101–2107. doi: 10.1046/j.1471-4159.2000.0742101.x. [DOI] [PubMed] [Google Scholar]
- Kanellopoulos GK, Xu XM, Hsu CY, Lu X, Sundt TM, Kouchoukos NT. White matter injury in spinal cord ischemia: protection by AMPA/kainate glutamate receptor antagonism. Stroke. 2000;31:1945–1952. doi: 10.1161/01.str.31.8.1945. [DOI] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jiang Q, Stys PK. Important role of reverse Na(+)-Ca(2+) exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84:1116–1119. doi: 10.1152/jn.2000.84.2.1116. [DOI] [PubMed] [Google Scholar]
- Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci U S A. 1997;94:8830–8835. doi: 10.1073/pnas.94.16.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken E, Fowler J, Dewar D, Morrison S, McCulloch J. Grey matter and white matter ischemic damage is reduced by thecompetitive AMPA receptor antagonist, SPD 502. J Cereb Blood Flow Metab. 2002;22:1090–1097. doi: 10.1097/00004647-200209000-00006. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- Meller D, Eysel UT, Schmidt-Kastner R. Transient immunohistochemical labelling of rat retinal axons during Wallerian degeneration by a monoclonal antibody to neurofilaments. Brain Res. 1994;648:162–166. doi: 10.1016/0006-8993(94)91917-8. [DOI] [PubMed] [Google Scholar]
- Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, Mcrory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, Mcrory JE, Trapp BD, Yin XH, Wang WL, Woulfe J, Stys PK. Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron. 2003;40:53–63. doi: 10.1016/j.neuron.2003.08.016. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Zamponi GW, Barr W, Kiedrowski L, Stys PK. Protection of ischemic rat spinal cord white matter: Dual action of KB-R7943 on Na+/Ca2+ exchange and L-type Ca2+ channels. Neuropharmacology. 2005;48:566–575. doi: 10.1016/j.neuropharm.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Li Y, Ali S, Altiok N, Back SA, Volpe JJ. Intracellular redox state determines whether nitric oxide is toxic or protective to rat oligodendrocytes in culture. J Neurochem. 1999;73:476–484. doi: 10.1046/j.1471-4159.1999.0730476.x. [DOI] [PubMed] [Google Scholar]
- Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kishi M, Saito M, Tanaka T, Higuchi N, Kominami E, Katunuma N, Murachi T. 90 A.D. Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J Enzyme Inhib. 3:195–201. doi: 10.3109/14756369009035837. [DOI] [PubMed] [Google Scholar]
- Stys PK. General mechanisms of axonal damage and its prevention. Journal of the Neurological Sciences. 2005;233:3–13. doi: 10.1016/j.jns.2005.03.031. [DOI] [PubMed] [Google Scholar]
- Stys PK, Lopachin RM. Mechanisms of calcium and sodium fluxes in anoxic myelinated central nervous system axons. Neuroscience. 1998;82:21–32. doi: 10.1016/s0306-4522(97)00230-3. [DOI] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG, Davis PK. Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci U S A. 1990;87:4212–4216. doi: 10.1073/pnas.87.11.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Rhee S, Jeon N. Microfluidic chambers for cell migration and neuroscience rese. Methods Mol Biol. 2006;321:167–77. doi: 10.1385/1-59259-997-4:167. [DOI] [PubMed] [Google Scholar]
- Tekkok SB, Goldberg MP. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DA, Thio C, Sontheimer H, Ritchie JM, Waxman SG, Kocsis JD. Action-Potential Conduction and Sodium-Channel Content in the Optic-Nerve of the Myelin-Deficient Rat. Proceedings of the Royal Society of London Series B-Biological Sciences. 1993;254:245–250. doi: 10.1098/rspb.1993.0153. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Waxman SG, R B, S PK. 91 A.D. Non-synaptic mechanisms of Ca(2+)-mediated injury in CNS white matter. Trends Neurosci. 14:461–468. doi: 10.1016/0166-2236(91)90046-w. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Black JA, Ransom BR, Stys PK. Protection of the axonal cytoskeleton in anoxic optic nerve by decreased extracellular calcium. Brain Res. 1993;614:137–145. doi: 10.1016/0006-8993(93)91027-p. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Black JA, Ransom BR, Stys PK. Anoxic injury of rat optic nerve: ultrastructural evidence for coupling between Na+ influx and Ca(2+)-mediated injury in myelinated CNS axons. Brain Res. 1994;644:197–204. doi: 10.1016/0006-8993(94)91680-2. [DOI] [PubMed] [Google Scholar]
- Wilke S, Thomas R, Allcock N, Fern R. Mechanism of acute ischemic injury of oligodendroglia in early myelinating white matter: the importance of astrocyte injury and glutamate release. J Neuropathol Exp Neurol. 2004;63:872–81. doi: 10.1093/jnen/63.8.872. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka A, Hardy M, Younkin DP, Grinspan JB, Stern JL, Pleasure D. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors mediate excitotoxicity in the oligodendroglial lineage. J Neurochem. 1995;64:2442–2448. doi: 10.1046/j.1471-4159.1995.64062442.x. [DOI] [PubMed] [Google Scholar]