

Abstract
The DNA architectural protein Xis regulates the construction of higher-order nucleoprotein intasomes that integrate and excise the genome of phage lambda from the Escherichia coli chromosome. Xis modulates the directionality of site-specific recombination by stimulating phage excision 106-fold, while simultaneously inhibiting phage reintegration. Control is exerted by cooperatively assembling onto a ≈35-bp DNA regulatory element, which it distorts to preferentially stabilize an excisive intasome. Here, we report the 2.6-Å crystal structure of the complex between three cooperatively bound Xis proteins and a 33-bp DNA containing the regulatory element. Xis binds DNA in a head-to-tail orientation to generate a micronucleoprotein filament. Although each protomer is anchored to the duplex by a similar set of nonbase specific contacts, malleable protein–DNA interactions enable binding to sites that differ in nucleotide sequence. Proteins at the ends of the duplex sequence specifically recognize similar binding sites and participate in cooperative binding via protein–protein interactions with a bridging Xis protomer that is bound in a less specific manner. Formation of this polymer introduces ≈72° of curvature into the DNA with slight positive writhe, which functions to connect disparate segments of DNA bridged by integrase within the excisive intasome.
Keywords: DNA bending, recombination directionality factors, site-specific DNA recombination, x-ray structure
Mobile genetic elements such as bacteriophages, conjugative transposons, and pathogenicity islands promote the lateral exchange of foreign DNA, enabling bacteria to acquire metabolic, pathogenic, and antibiotic resistance determinants. To prevent potentially catastrophic changes in the genome, these DNA rearrangements are often tightly controlled by regulatory factors that function together with the recombinase. The integration and excision reactions of phage λ, which are controlled by the phage-encoded Xis protein, serve as a paradigm for studies of regulated site-specific recombination (1). Upon infection, specific DNA attachment sites located on the circularized phage genome (attP) and bacterial chromosome (attB) recombine to generate the integrated prophage with flanking hybrid sites (attL and attR) (Fig. 1A). Cellular DNA damage initiates a series of events that result in prophage excision to regenerate attP on the episomal phage genome and attB on the chromosome. Although the DNA strand transfer steps in each reaction are catalyzed by the phage-encoded tyrosine recombinase integrase (Int) protein, and are mechanistically similar, directionality control is achieved by guiding the assembly of distinct higher-order nucleoprotein structures called intasomes. Viral integration occurs within an integrative intasome containing Int and the Escherichia coli-encoded integration host factor (IHF) (2, 3), whereas excision is performed within an alternative excisive intasome complex containing Int, Xis, IHF, and the factor for inversion stimulation (4–7).
Fig. 1.

Integrative and excisive recombination of phage λ. (A) Schematic representation of the phage λ site-specific recombination reactions. The supercoiled phage genome inserts into the E. coli chromosome by recombination between the attP (phage) and attB (bacterial) sites to generate the attL and attR sites that are substrates for excisive recombination. Proteins required for integration and excision are indicated: Int, integration host factor (IHF), and factor for inversion stimulation (Fis). Excise (Xis) is required for excision and inhibits integration. Filled symbols, binding sites used; open symbols, binding sites not used during the integration or excision reactions (1). (B) Xis binding sequence used to form crystals with red nucleotides depicting 5-bromo-uracil substitutions used for phasing. The dot between nucleotides 15 and 16 represents a nick in the DNA present after annealing the three oligonucleotides. The nucleotide numbers in parentheses are relative to the center of the attR core site, and the lines represent the conventional binding sites based on in vitro footprinting data and DNA sequence relationships (40).
Xis is the master regulator of λ recombination, stimulating phage excision in vivo >106-fold, while simultaneously inhibiting phage reintegration (8–10). These dual and opposing effects are elicited by its cooperative binding to a ≈35-bp DNA segment that contains two conserved sequence elements, X1 and X2 (Fig. 1B). DNA bending by Xis promotes formation of the excisive intasome, but antagonizes formation of an integrative intasome (8, 11). In addition, Xis stabilizes the synaptic complex by directly interacting with Int bound to the arms of the phage. Previously, the NMR structure of Xis (12, 13) and the x-ray structure of the complex between a single Xis protein and DNA have been reported (14). Although this work revealed how Xis recognizes a single binding site, the mechanism through which it cooperatively binds and distorts the attR regulatory element has remained enigmatic. Here, we show that three Xis monomers work in concert to substantially bend DNA by forming a micronucleoprotein filament. The mechanistic implications of the structure on the site-specific recombination reactions of phage λ and the functions of recombination directionality factors in other systems are discussed.
Results and Discussion
Three Xis Proteins Cooperatively Bind to the attR Regulatory Element.
Traditionally, two Xis proteins have been thought to bind to two related 13-bp sequences called X1 and X2 (Fig. 1B). However, the small size of the Xis protein and the 7-bp separation between the X1 and X2 sites has made it difficult to envision how cooperative binding could be achieved. Moreover, the sites are arranged in a head-to-tail configuration that would appear to necessitate the formation of an unusual asymmetric protein–DNA complex. To determine the number of Xis proteins that bind the regulatory element, we performed EMSAs. As observed (8, 13), Xis forms a single low mobility complex over the Xis binding region embedded in a 263-bp fragment from attR (Fig. 2A). As this DNA fragment contains at least two binding sites for Xis, the results suggest that binding is highly cooperative. Unexpectedly, we find that cooperative binding by Xis is compromised on short DNA fragments. The gel in Fig. 2B reveals three distinct Xis-dependent bands over a broad range of Xis levels on a 54-bp fragment covering the Xis binding region [see supporting information (SI) Fig. 6]. The number of Xis proteins in the slowest mobility complex (labeled 3X in Fig. 2B) was addressed by using a fluoroscein-labeled DNA fragment and 32P-labeled Xis. The Xis used for this experiment had the C-terminal 17-aa residues that are disordered in solution replaced with a kinase tag (Δ55XisHMK). We have shown previously that Δ55Xis binds attR in a cooperative manner that is nearly indistinguishable from the full-length protein (13). Quantifying the fluorescence and radioactivity in the complex gave a molar ratio of 3.6 ± 0.9 Xis molecules per DNA substrate. These experiments suggest that at least three Xis protomers are present in the attR regulatory complex.
Fig. 2.

A trimer of Xis cooperatively binds DNA. (A) Cooperative Xis binding to long attR DNA fragments. Increasing concentrations of Xis were incubated with a 32P-labeled attR fragment (−220 to +43) and electrophoresed in a native polyacrylamide gel. (B) Xis binding to short attR DNA fragments (−104 to −51) exhibits reduced cooperativity. 1X, 2X, and 3X represent DNA complexes postulated to contain one, two, and three Xis monomers, respectively. (C) Cross-linking of Xis–attR complexes. 32P-labeled XisHMK was incubated with fluorescein-labeled attR DNA (−104 to −54), subjected to cross-linking with BS3, and electrophoresed in a native gel as in B. Xis complexes representing 2X and 3X were extracted from the native gel and subjected to SDS/PAGE. The number of cross-linked Xis monomers in each product band is based on their apparent molecular mass. (D) Cross-linking of Δ55Xis–attR complexes. Protocol was the same as in C except that 32P-labeled Δ55XisHMK was subjected to cross-linking with BS3 (11-Å spacer) or DSG (8-Å spacer), and the cross-linked products were extracted from the dominant slowest migrating complex. (E) Binding of Int1–64 and two Xis protomers to attR fragments missing the X2 site. Xis (0.53 μM) and HisInt1–64 (3 μM) were incubated with a 32P-labeled 45-bp probe (−121 to −77) containing the P2 and X1 binding sites as designated and electrophoresed in a native polyacrylamide gel. The identities of each of the bands based on their electrophoretic mobilities are denoted. The sequences of the oligonucleotide substrates are given in SI Fig. 6.
Protein cross-linking experiments were performed to independently define the number of Xis protomers in the attR complex. 32P-labeled full-length XisHMK or Δ55XisHMK were incubated with the fluoroscein-labeled attR substrate and subjected to cross-linking with amine-specific cross-linkers. After purification of the Xis–DNA complexes on native gels, the cross-linked Xis molecules were displayed on SDS/PAGE. The complex that is predicted to contain two Xis protomers from the native gel (2X in Fig. 2B) contains a single product after cross-linking with bis(sulfosuccinimidyl)suberate (BS3) (11-Å spacer) whose migration is consistent with two linked Xis monomers (Fig. 2C, lane 2), and the slowest mobility band on the native gel (3X in Fig. 2B) contains products consistent with cross-linked trimers and dimers (Fig. 2C, lane 3). The slow mobility complexes formed with Δ55Xis and cross-linked with BS3 or disuccinimidyl glutarate (DSG) (8-Å spacer) also contained linked trimer and dimer products (Fig. 2D). The less efficient cross-linking obtained with Δ55Xis compared with that of full-length Xis probably reflects the absence of the lysine-rich and unstructured C terminus on the truncated derivative. Combined, the cross-linking and EMSA stoichiometry data indicate that three proximally positioned Xis proteins cooperatively bind the attR regulatory element.
EMSA experiments using truncated attR DNA segments suggest that the third Xis protomer binds between the X1 and X2 sites. In Fig. 2E, the DNA probe contained the P2 Int binding site and sequences over the Xis regulatory unit up to the beginning of the X2 sequence (−121 to −77) (SI Fig. 6). For this experiment we also used the N-terminal His-tagged domain of Int (HisInt1–64) that contains the DNA-binding and Xis cooperativity determinants (15–17). As shown in lane 2 of Fig. 2E, a weak complex is formed in the presence of HisInt1–64 alone, which corresponds to a monomer of HisInt1–64 bound to P2. In the absence of HisInt1–64, WT Xis forms two weak complexes on this substrate (Fig. 2E, lane 3), whereas addition of both Xis and HisInt1–64 results in the formation of two prominent novel complexes (Fig. 2E, lane 4). The migration of the faster band is consistent with the presence of one Xis monomer and one HisInt1–64 monomer, whereas the slower migrating band is consistent with the presence of two Xis monomers and one HisInt1–64 monomer. This result supports a model in which Xis bound to X1 recruits an additional Xis protomer in the presence of Int, even though the X2 site is missing from the probe, and strongly implies that in the trimeric Xis–DNA complex, sites X1 and X2, and the intervening DNA, are occupied by Xis.
The Structure of Three Xis Proteins Bound to the X1–X2 DNA Segment Reveals a Micronucleoprotein Filament.
To gain a high-resolution view of the cooperative Xis–DNA complex, we solved the cocrystal structure of Δ55Xis bound to a 33-bp DNA duplex containing sites X1 and X2 (the Xis–DNAX1–X2 complex). The structure was refined to 2.6-Å with Rwork and Rfree values of 19.9% and 24.6%, respectively (SI Table 1). The asymmetric unit of the Xis–DNAX1–X2 complex contains three proteins bound to the DNA molecule. The complex resembles a nucleoprotein filament as the proteins are positioned in a head-to-tail arrangement along one face of the duplex forming a continuous interface that buries 5,100 Å of solvent-exposed surface area (Fig. 3A). The proteins adopt nearly identical “winged” helix structures that are very similar to the structure of Xis in the DNA free state and its conformation when bound to a single DNA binding site (described in Fig. 3A legend). However, in this complex, unique protein–protein interactions function to cumulatively bend the duplex by ≈72°. A summary of the Xis–DNA contacts observed in the structure is presented in SI Fig. 7.
Fig. 3.

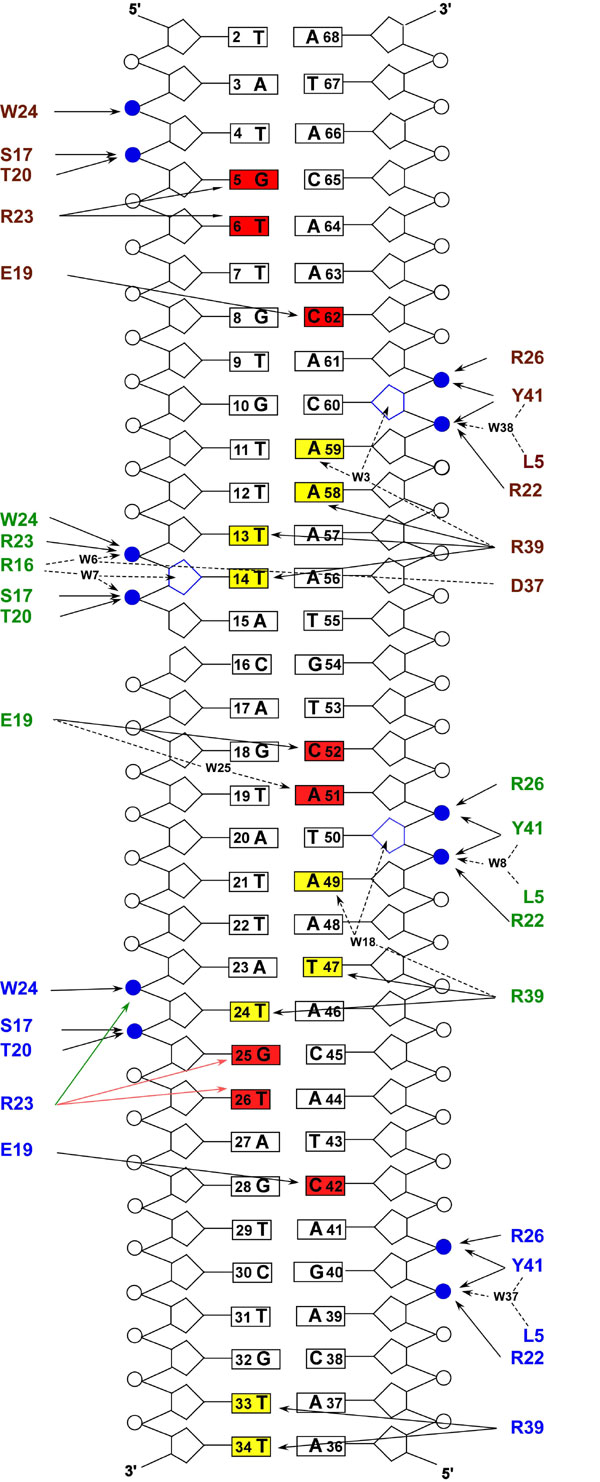
The structural basis of cooperative binding is revealed by crystallography. (A) X-ray crystal structure of Xis bound to the Xis binding region. Xis monomers bound to the X1, X1.5, and X2 sites are colored dark salmon, green, and blue, respectively. The nick present in the top strand of the DNA duplex is a result of annealing three complementary DNA strands (Fig. 1B). The proteins adopt nearly identical “winged” helix structures (the backbone coordinates of residues Tyr-2–Val-50 in each protomer can be superimposed with a rmsd of 0.22 Å). The secondary structural elements in this fold are arranged in the following configuration: β1-α1-L-α2-B1-β2-β3-W-β4-B2-β5, where L, W, and B are the loop, wing, and bulge structures, respectively. Residues within each secondary structural element are as follows: β1 (Tyr-2–Thr-4), α1 (Leu-5–Arg-11), L (Gln-12–Ser-17), α2 (Leu-18–Arg-26), B1 (Glu-27–Arg-29), β2 (Ile-30–Phe-31), β3 (Val-35–Asp-37), W (Gly-38–Arg-39), β4 (Glu-40–His-44), B2 (Glu-45–Ala-47), and β5 (Val-48–Lys-49). (B and C) Residues within Xis–DNA interfaces involved in hydrogen bonding. (B) Shown is the interface between DNA and the Xis monomer bound at X1. (C) Shown is the interface between DNA and the Xis monomer bound at X1.5.
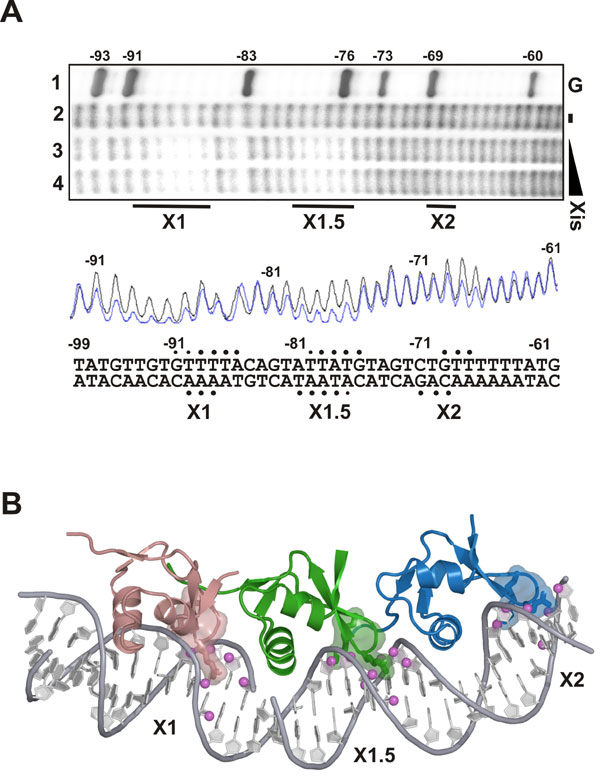
The three tandemly arranged proteins are positioned over their respective binding sites in a similar manner, with each inserting its α2 helix and wing motif into the major and minor grooves, respectively. Base-specific recognition is primarily achieved by Xis interactions with the terminal X1 and X2 sites that share nearly identical nucleotide sequences. In contrast, the third protomer is bound in a largely nonspecific manner to a central site that differs substantially in nucleotide sequence, hereafter referred to as site X1.5. Sites X1 and X2 are recognized by major groove contacts from the side chains of Glu-19 and Arg-23 in the α2 helix. At each interface, the side chain of Glu-19 forms a salt bridge with the guanidino group of Arg-26, which positions it to accept a hydrogen bond from the N4 amine of a conserved cytosine base (C42 and C62 in sites X1 and X2, respectively) (Fig. 3B). In addition, the guanidino group of Arg-23 donates hydrogen bonds to conserved G5-T6 (site X1) and G25-T26 (site X2) base steps. Locking the recognition helix into place is a network of contacts to the surrounding phosphodiester backbone that originate from residues within the turn preceding helix α2, the β3/β4 wing, and the recognition helix (SI Fig. 7). All of the nonspecific and specific interactions at sites X1 and X2 are compatible with extensive mutagenesis data describing the Xis–DNA interaction (13, 18). Moreover, DNA regions protected from hydroxyl radical cleavage by Xis binding to long (203 bp) DNA fragments in solution correspond well to the locations of the Xis wings at all three binding sites in the crystal (SI Fig. 8).
In the crystal structure, pliant interfacial side chains enable Xis to nonspecifically bind the central X1.5 site. In the major groove of site X1.5, an A-C base step replaces the conserved G-T base step that is sequence-specifically recognized in sites X1 and X2. To accommodate this alteration, the side chain of Arg-23 is rotated away from the major groove to form a salt bridge with the phosphate of T14 (Fig. 3C). Interestingly, Arg-23 can readily toggle between specific and nonspecific binding conformations, because model building of the electron density at site X2 reveals that it contacts the G-T base step only ≈25% of the time in the crystal, whereas the rest of the time it adopts the phosphate contacting conformation observed at site X1.5. Structural plasticity in the protein–DNA interface extends into the minor groove where contacts primarily originate from the side chain of Arg-39 located in the wing. Hydrogen bonds to the O2 atoms of T14, T24, and T34 are conserved at all three sites, but additional hydrogen bonds to thymines and adenines are formed at each site, depending on the sequence. Although radical changes in the nucleotide sequence of X1.5 can be made without adversely affecting filament formation (data not shown), the DNA sequence within X1.5 contains an appropriately positioned cytosine base that enables a lone sequence-specific contact from the side chain of Glu-19 (Fig. 3C).
Malleable Xis–Xis Interactions Promote Cooperative Filament Assembly and DNA Bending.
Cooperative binding is mediated by the Xis protein bound to site X1.5, which contains two protein-binding surfaces that participate in reciprocal interactions with adjacent protomers. It uses residues within the turn that connects helices α1 and α2 (the α1–α2 turn) to contact the wing of the protomer bound to site X1 (Fig. 4A; X1–X1.5 interface), whereas residues within its wing contact the α1–α2 turn of Xis bound at site X2 (Fig. 4B; X1.5–X2 interface). Electrostatic interactions predominate within each interface and exclude a similar amount of solvent accessible surface area (the X1–X1.5 and X1.5–X2 interfaces bury 700 and 600 Å, respectively). Although similar sets of amino acids are involved, the interprotomer interactions at each interface are strikingly different, because the protomers bound to sites X1–X1.5 are rotated with respect to one another by 15°, whereas those bound at the X1.5–X2 interface are positioned at a 31° angle. As a result, only a single set of hydrogen bonding and electrostatic contacts are conserved within each interface; the side chain of Arg-16 originating from the α1–α2 turn contacts the carboxyl side chains of Asp-37 and Glu-40 in the neighboring protomer (Fig. 4 A and B). Consistent with the importance of these interactions implied by the structure, mutants containing alanine substitutions at either Arg-16 and Asp-37 no longer cooperatively bind DNA and only form nonspecific complexes (Fig. 4 C and D) (13). In addition, an Xis protein containing a Glu-40–Ala mutation has been reported to exhibit increased nonspecific binding (18).
Fig. 4.

Protein–protein interactions stabilize a nucleoprotein filament. (A and B) Cross-eyed stereoviews of residues within Xis–Xis interfaces involved in hydrogen bonding or ionic interactions in the Xis cooperative complex. (A) The interface between the Xis monomers bound at X1 (dark salmon) and X1.5 (green). (B) The interface between Xis monomers bound at X1.5 (green) and X2 (blue). (C–F) The effects of mutations in the Xis protein interfaces. Each panel shows a titration of the Xis mutant on the 263 attR fragment (−220 to +43) used in Fig. 2A. The identity of the Xis mutant is indicated at the top: R16A (C), D37A (D), R13E (E), and a double R13A and R14A mutant (F). The lanes designated WT contained reactions using 116 nM WT Xis; − designates that no Xis was added. Data shown for the R16A mutant are modified from figure 5C of ref. 13.
The remaining interprotomer interactions are unique, but generally involve the same amino acids. For example, in both interfaces the side chain of Arg-14 interacts with residues in an adjacent protomer. When it is located in the X2-bound Xis protein, Arg-14 forms a salt bridge to the Glu-40 side chain of Xis bound to site X1.5. In contrast, when Arg-14 originates from the X1.5-bound protomer, it contacts the side chain of Glu-7 and backbone atoms of the protein at site X1. The side chain of Arg-13 is involved in direct interactions only at the X1–X1.5 interface, where it is positioned to form multiple contacts with Asp-37. These interactions appear to play a less important role in stabilizing the complex because R13A and R14A mutants of Xis have comparatively milder binding phenotypes as compared with alanine mutants that alter Arg-16 and Asp-37 (data not shown). However, single R13E (Fig. 4E) and double R13A, R14A (Fig. 4F) mutants exhibit severe reductions in cooperative binding, demonstrating that these residues also participate in stabilizing the attR complex. Related, but nonidentical, contacts between interfaces within protein filaments that polymerize on DNA have been observed previously (e.g., refs. 19 and 20).
Although Xis only modestly distorts DNA when bound to a single site (14), interprotomer interactions within the filament stabilize in-phase DNA distortions that cumulatively bend the duplex by 72°. At each binding site a series of modest positive roll angle changes in base steps proximal to the recognition helix narrows the major groove by as much as 2.5 Å (8.5 Å in the complex versus 11 Å in typical B-form DNA). Further bending toward the protein occurs at the adjacent minor groove interfaces, which are also compacted to accommodate insertion of Arg-39 for direct and water-mediated hydrogen bonding. Contacts from adjacent Xis protomers in the filament are required for maximal bending. At sites X1 and X1.5, which exhibit the largest degree of bending (38° and 24°, respectively), interactions originating from a downstream positioned Xis protomer facilitate minor groove compression by neutralizing anionic phosphates and stabilizing the positioning of the wing (Fig. 4 A and B). The absence of these contacts at the terminal X2 interface and in a complex that contained a single Xis bound to DNA (14) may explain why the DNA in these structures is only modestly bent. Although there is a nick in the DNA used to form the crystal, a comparison of bending between sites X1 and X1.5, where the nick resides, indicates that its contribution to the overall bending of the DNA is minimal.
Models of the excisive intasome require Xis to juxtapose distally positioned Int arm- and core-binding sites by substantially altering the trajectory of DNA. The curvature introduced by the three contiguous Xis protomers in the Xis–DNAX1–X2 crystal structure demonstrates how Xis performs this critical architectural function. We attempted to dock the Xis–DNAX1–X2 crystal structure onto a recent structure of an Int tetramer bound to antiparallel discontinuous DNA segments representing the P and P′ arm binding sites plus core binding sites in a Holliday junction configuration (21). The resulting connection of the P arm and core Int binding sites generated a structure for the intact excisive intasome that is incompatible with the previously established temporal order of DNA exchanges that occur during the excision reaction (22, 23). Thus, additional structural information with respect to the configuration of Int arm sites will be required to generate a structural model for the excisive intasome.
Nucleoprotein Filaments.
Xis is the founding member of a diverse family of proteins that function to control reactions that rearrange DNA, called recombination directionality factors (RDFs) (24). Many of the RDFs share two common features with Xis: they adopt a similar winged-helix fold and cooperatively bind DNA as oligomers to produce extensive footprints (25–30). For example, the small Xis protein encoded by mycobacteriophage L5 is predicted to bind to four related sequence motifs spaced ≈10 bp apart to form a stable and bent DNA complex (27). The low molecular weight Xis protein from the Tn916 transposon also exhibits similar binding behavior and produces large footprints that contain hypersensitive cleavage sites characteristic of wrapped or bent DNA (31, 32). It seems likely that many of these proteins also assemble into nucleoprotein filaments, which function to stabilize recombinogenic higher-order structures. In the λ Xis microfilament, the bends introduced by each Xis protein are not coplanar, resulting in positive writhe that is readily visualized when an extended filament is modeled (Fig. 5). The DNA encircles the Xis protomers in the filament, possibly explaining why DNA bound by RDFs frequently exhibit periodic DNase I hypersensitivity.
Fig. 5.

Structure-based model of an extended Xis–DNA filament. Units of the Xis–DNAX1–X2 crystal structure were stacked end-to-end by superimposing site X1 over X1.5 to assemble a pseudo-continuous helix with a pitch of ≈22 nm. The model is shown perpendicular (Left) and parallel (Right) to the superhelical axis. Proteins are blue; DNA is orange and red.
Nucleoprotein filaments play important roles in other types of reactions. The most intensively studied filaments are those assembled by the RecA/RAD51 family of proteins, which catalyze homologous DNA pairing and strand exchange (33). Polymerization of DnaA within the oriC DNA region is believed to control open complex formation during initiation of replication (34). Formation of H-NS filaments containing one or two DNA duplex segments has been shown to silence transcription at promoters (35). In addition, localized filaments of nucleoid proteins are believed to contribute to compaction of bacterial chromosomes (36). Some of these protein filaments have been visualized by x-ray crystallography at varying resolutions [e.g., RecA (37), RAD51 (19), and DnaA (31)], but in most cases, the DNA was not present during crystal growth or not visible in the electron density maps. Although the details of the molecular interactions involved in each system will no doubt vary, the present structure of the Xis–DNAX1–X2 complex reveals how pliant protein–protein and protein–DNA interactions contribute to polymerization along DNA.
Materials and Methods
DNA Binding, Stoichiometry, and Cross-Linking Assays.
Xis–DNA binding reactions and EMSAs using WT and mutant derivatives of Xis were performed as described (13). Full-length XisHMK contains the sequence coding for MRRASL added by PCR to the N terminus of otherwise WT Xis, and Δ55XisHMK contains the sequence coding for ARRASL(TAA) added after residue 55 of Xis containing Cys-28 replaced with alanine. HisInt1–64 that was used in Fig. 2E contains the first 64 residues of Int with a six-histidine tag at the N terminus. DNA probes were generated by PCR where one or both primers were 5′ 32P-end-labeled; Fig. 2E shows a synthetic duplex oligonucleotide probe representing the attR sequence from −121 to −77 (SI Fig. 6). HPLC-purified fluorescein (6-FAM)-labeled 51-bp oligonucleotides representing the attR sequence between −104 to −54 (SI Fig. 6) were purchased from Sigma-Genosys (St. Louis, MO). Stoichiometry experiments using 32P-labeled Δ55XisHMK and the 6-FAM duplex oligonucleotides were performed essentially as described (38). Xis cross-linking reactions were performed in a similar manner to those described in ref. 39. Binding reactions were assembled on the 6-FAM 51-bp attR oligonucleotides with 525 nM 32P-labeled XisHMK or Δ55XisHMK. The reactions were then incubated with 3.4 mM BS3 or DSG for 60 min at 37°C, quenched with the addition of 20 mM glycine, and applied to an 8% native polyacrylamide gel. Xis–DNA complexes were localized by fluoroimaging and phosphorimaging, relevant bands were excised, and Xis proteins were recovered and subjected to SDS/PAGE following the protocols described (39).
Crystallization, Data Collection, and Structure Determination.
Details describing complex crystallization, data collection, and structure determination are provided in SI Text. A representative portion of the experimental electron density map after solvent flattening is shown in SI Fig. 9.
Supplementary Material
Acknowledgments
We thank Michael Sawaya, Carlos Lopez, Corie Ralston, and the ALS beamline 8.2.2 staff for useful discussions. This work was supported by National Institutes of Health Grants GM57487 (to R.T.C.) and GM38509 (to R.C.J.).
Abbreviations
- Int
integrase
- BS3
bis(sulfosuccinimidyl)suberatel
- DSG
disuccinimidyl glutarate.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2IEF).
This article contains supporting information online at www.pnas.org/cgi/content/full/0607820104/DC1.
References
- 1.Azaro MA, Landy A. In: Mobile II. Craig NL, Craigie R, Gellert M, Lambowitz AM, editors. Am Soc Microbiol: Washington, DC; 2002. pp. 118–148. [Google Scholar]
- 2.Miller HI, Kikuchi Y, Nash HA, Weisberg RA, Friedman DI. Cold Spring Harb Symp Quant Biol. 1979;43:1121–1126. doi: 10.1101/sqb.1979.043.01.125. [DOI] [PubMed] [Google Scholar]
- 3.Nash HA, Robertson CA. J Biol Chem. 1981;256:9246–9253. [PubMed] [Google Scholar]
- 4.Abremski K, Gottesman S. J Biol Chem. 1982;257:9658–9662. [PubMed] [Google Scholar]
- 5.Guarneros G, Echols H. J Mol Biol. 1970;47:565–574. doi: 10.1016/0022-2836(70)90323-2. [DOI] [PubMed] [Google Scholar]
- 6.Ball CA, Johnson RC. J Bacteriol. 1991;173:4027–4031. doi: 10.1128/jb.173.13.4027-4031.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson JF, Moitoso de Vargas L, Koch C, Kahmann R, Landy A. Cell. 1987;50:901–908. doi: 10.1016/0092-8674(87)90516-2. [DOI] [PubMed] [Google Scholar]
- 8.Bushman W, Yin S, Thio LL, Landy A. Cell. 1984;39:699–706. doi: 10.1016/0092-8674(84)90477-x. [DOI] [PubMed] [Google Scholar]
- 9.Franz B, Landy A. EMBO J. 1995;14:397–406. doi: 10.1002/j.1460-2075.1995.tb07014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moitoso de Vargas L, Landy A. Proc Natl Acad Sci USA. 1991;88:588–592. doi: 10.1073/pnas.88.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson JF, Landy A. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rogov VV, Lucke C, Muresanu L, Wienk H, Kleinhaus I, Werner K, Lohr F, Pristovsek P, Ruterjans H. Eur J Biochem. 2003;270:4846–4858. doi: 10.1111/j.1432-1033.2003.03884.x. [DOI] [PubMed] [Google Scholar]
- 13.Sam MD, Papagiannis C, Connolly KM, Corselli L, Iwahara J, Lee J, Phillips M, Wojciak JM, Johnson RC, Clubb RT. J Mol Biol. 2002;324:791–805. doi: 10.1016/s0022-2836(02)01150-6. [DOI] [PubMed] [Google Scholar]
- 14.Sam MD, Cascio D, Johnson RC, Clubb RT. J Mol Biol. 2004;338:229–240. doi: 10.1016/j.jmb.2004.02.053. [DOI] [PubMed] [Google Scholar]
- 15.Warren D, Sam MD, Manley K, Sarkar D, Lee SY, Abbani M, Wojciak JM, Clubb RT, Landy A. Proc Natl Acad Sci USA. 2003;100:8176–8181. doi: 10.1073/pnas.1033041100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarkar D, Azaro MA, Aihara H, Papagiannis CV, Tirumalai R, Nunes-Duby SE, Johnson RC, Ellenberger T, Landy A. J Mol Biol. 2002;324:775–789. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- 17.Cho EH, Gumport RI, Gardner JF. J Bacteriol. 2002;184:5200–5203. doi: 10.1128/JB.184.18.5200-5203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho EH, Alcaraz R, Jr, Gumport RI, Gardner JF. J Bacteriol. 2000;182:5807–5812. doi: 10.1128/jb.182.20.5807-5812.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, Rice PA. Nat Struct Mol Biol. 2004;11:791–796. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 20.Kim CA, Phillips ML, Kim W, Gingery M, Tran HH, Robinson MA, Faham S, Bowie JU. EMBO J. 2001;20:4173–4182. doi: 10.1093/emboj/20.15.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. Nature. 2005;435:1059–1066. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nunes-Düby SE, Matsumoto L, Landy A. Cell. 1987;50:779–788. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 23.Kitts PA, Nash HA. J Mol Biol. 1988;204:95–107. doi: 10.1016/0022-2836(88)90602-x. [DOI] [PubMed] [Google Scholar]
- 24.Lewis JA, Hatfull GF. Nucleic Acids Res. 2001;29:2205–2216. doi: 10.1093/nar/29.11.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu A, Haggard-Ljungquist E. J Bacteriol. 1993;175:7848–7855. doi: 10.1128/jb.175.24.7848-7855.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esposito D, Scocca JJ. J Biol Chem. 1997;272:8660–8670. doi: 10.1074/jbc.272.13.8660. [DOI] [PubMed] [Google Scholar]
- 27.Lewis JA, Hatfull GF. J Mol Biol. 2003;326:805–821. doi: 10.1016/s0022-2836(02)01475-4. [DOI] [PubMed] [Google Scholar]
- 28.Abbani M, Iwahara M, Clubb RT. J Mol Biol. 2005;347:11–25. doi: 10.1016/j.jmb.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 29.Esposito D, Scocca JJ. Mol Microbiol. 1994;13:685–695. doi: 10.1111/j.1365-2958.1994.tb00462.x. [DOI] [PubMed] [Google Scholar]
- 30.Eriksson JM, Haggard-Ljungquist E. J Bacteriol. 2000;182:6714–6723. doi: 10.1128/jb.182.23.6714-6723.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Connolly KM, Iwahara M, Clubb RT. J Bacteriol. 2002;184:2088–2099. doi: 10.1128/JB.184.8.2088-2099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rudy CK, Scott JR, Churchward G. J Bacteriol. 1997;179:2567–2572. doi: 10.1128/jb.179.8.2567-2572.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.West SC. Nat Rev Mol Cell Biol. 2003;4:435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 34.Erzberger JP, Mott ML, Berger JM. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- 35.Dame RT. Mol Microbiol. 2005;56:858–870. doi: 10.1111/j.1365-2958.2005.04598.x. [DOI] [PubMed] [Google Scholar]
- 36.Luijsterburg MS, Noom MC, Wuite GJ, Dame RT. J Struct Biol. 2006;156:262–272. doi: 10.1016/j.jsb.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Story RM, Weber IT, Steitz TA. Nature. 1992;355:318–325. doi: 10.1038/355318a0. [DOI] [PubMed] [Google Scholar]
- 38.Sanders ER, Johnson RC. J Mol Biol. 2004;340:753–766. doi: 10.1016/j.jmb.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 39.Dhar G, Sanders ER, Johnson RC. Cell. 2004;119:33–45. doi: 10.1016/j.cell.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 40.Yin S, Bushman W, Landy A. Proc Natl Acad Sci USA. 1985;82:1040–1044. doi: 10.1073/pnas.82.4.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}