

Abstract
The discovery of quantitative trait loci (QTL) in model organisms has relied heavily on the ability to perform controlled breeding to generate genotypic and phenotypic diversity. Recently, we and others have demonstrated the use of an existing set of diverse inbred mice (referred to here as the mouse diversity panel, MDP) as a QTL mapping population. The use of the MDP population has many advantages relative to traditional F2 mapping populations, including increased phenotypic diversity, a higher recombination frequency, and the ability to collect genotype and phenotype data in community databases. However, these methods are complicated by population structure inherent in the MDP and the lack of an analytical framework to assess statistical power. To address these issues, we measured gene expression levels in hypothalamus across the MDP. We then mapped these phenotypes as quantitative traits with our association algorithm, resulting in a large set of expression QTL (eQTL). We utilized these eQTL, and specifically cis-eQTL, to develop a novel nonparametric method for association analysis in structured populations like the MDP. These eQTL data confirmed that the MDP is a suitable mapping population for QTL discovery and that eQTL results can serve as a gold standard for relative measures of statistical power.
THE use of modern genetics in model organisms relies heavily on the use of controlled breeding. Experimentally derived crosses enable researchers to generate both genotypic and phenotypic diversity, and the resulting populations are analyzed to identify genomic loci that underlie disease and traits. Data from these experimental designs are analyzed using what are now classical methods of linkage analysis (Lander and Botstein 1989; Haley and Knott 1992). In rodent models over the past 15–20 years, these methods have been used to identify thousands of quantitative trait loci (QTL) across a wide range of phenotypes (Flint et al. 2005).
Recently, the increasing availability of high-throughput genotyping technologies has enabled the use of genomewide association analyses for QTL discovery and, in some cases, in alternate mapping populations. For example, several research groups have investigated the use of a panel of diverse inbred strains of mice (Bogue and Grubb 2004) (collectively referred to here as the mouse diversity panel, MDP) for these QTL mapping studies (Grupe et al. 2001; Liao et al. 2004; Pletcher et al. 2004; Cervino et al. 2005). In contrast to mapping populations derived from controlled crosses, the strains of the MDP have been derived over the past century of semistructured breeding and inbreeding (Beck et al. 2000). We recently introduced an algorithm for association analysis in the MDP based on the local inferred haplotype pattern, an approach we termed “haplotype association mapping” (HAM) (Pletcher et al. 2004; McClurg et al. 2006).
The MDP is an attractive choice as a mapping population for several reasons. Because these mouse strains were derived over the past century from crossing different mouse populations, the MDP has a greater genetic and phenotypic diversity than is found in a typical F2 population derived from two parental strains. Since these mice are inbred, genotype data can be collected in community databases and applied to all mapping studies in the MDP. Finally, higher recombination rates and dense genotype maps result in more precisely defined QTL regions, facilitating the refinement of QTL to quantitative trait genes (QTG).
Nevertheless, there are also significant challenges to performing QTL studies in the MDP population. First, because we are limited to ∼30–50 strains in the MDP for which we have dense genotype data, the question exists of whether there is sufficient statistical power to detect QTL (Chesler et al. 2001; Darvasi 2001). Moreover, no analytical method for analyzing power has yet been developed. Second, the uncontrolled breeding process from which the MDP was derived can lead to spurious associations to background genetic structure if this population structure is not accounted for. Although the Collaborative Cross effort (Churchill et al. 2004) will eventually address both these drawbacks experimentally, this mapping population is not yet available. In the mean time, QTL mapping in the MDP requires that these effects be accounted for analytically.
Ideally, computation of power and optimization of the association algorithm would utilize a set of positive controls where the QTG underlying a phenotypic trait has been unequivocally determined. However, in the case of genetic association analysis, particularly in the case of complex traits, the availability of these positive controls is severely limited (Flint et al. 2005). Consequently, estimates of statistical power for QTL mapping studies typically rely on simulated genotype and phenotype data that are based on parametric assumptions. Although these simulation studies produce useful estimates of statistical power, the parametric assumptions of normality are usually not satisfied in the typical real-world study. This limitation is especially clear when using association mapping in the MDP. Therefore, the degree to which these parametric estimates reflect the true statistical power in this population is unclear.
In this article, instead of making parametric assumptions on simulated data, we used real experimental data and made assumptions on the identity of the true positives. The success of this approach clearly rested on the ability to identify a set of high-confidence true positives. Here, we utilized expression QTL (eQTL) data that maps gene expression measurements across the MDP as phenotypes for association analysis. cis-eQTL are commonly considered to be a highly enriched set of true positives with a low false-positive rate (Chesler et al. 2006). Although we did not know the identity of all genes that are truly cis-regulated (and hence could not calculate absolute measures of power), we used the presence of cis-eQTL as a relative measure of statistical power. We applied this approach to the development of an algorithm to account for the inherent population structure in the MDP. Furthermore, we assessed the ability of two-factor ANOVA models to improve the algorithm's sensitivity.
Finally, since phenotype data measured in the MPD population are becoming increasingly available (Bogue and Grubb 2004), we also created a web site where users can submit phenotypes for analysis using the HAM algorithm (at http://snpster.gnf.org).
METHODS
Sample preparation:
All mice were maintained on a 12-hr light/dark cycle at least 1 week before collecting hypothalami and individually housed with food and water available ad libitum. At 25 weeks of age, mice were sacrificed under light anesthesia at Zeitgerber time (ZT) 6 (ZT 0 defined as lights on) 2 hr after food deprivation. Animals were killed by cervical dislocation and hypothalami were taken by making two coronal cuts each just posterior to the optic chiasm and the pituitary stalk. A pair of sagittal cuts was made 1.5 mm from the midline. A final horizontal cut was made 1 mm dorsal to the floor of the hypothalamus. Tissues were snap frozen in liquid nitrogen and stored at −80° for subsequent analysis.
Gene expression data:
Hypothalamus samples were isolated from male mice of 32 strains (n = 3 except as noted below): 129S1/SvImJ*, A/J*, AKR/J*, BALB/cByJ*, BTBRT+tf/J*, BUB/BnJ*, C3H/HeJ, C57BL/6J*, C57BLKS/J, C57L/J, CAST/EiJ (n = 2), CBA/J, CZECHII/EiJ, DBA/2J*, FVB/NJ, I/LnJ, JF1/Ms, MA/MyJ, MOLF/EiJ, MSM/Ms (n = 2), NOD/LtJ (n = 1), NZB/BlNJ*, NZW/LacJ*, PERA/EiJ, PL/J (n = 2), RIIIS/J*, SEA/GnJ, SJL/J, SM/J*, SPRET/EiJ, SWR/J, and WSB/EiJ (n = 2). RNA from male replicates was pooled prior to amplification and subsequently hybridized to a single chip per strain. Hypothalamus samples from female mice of 12 strains (indicated with “*” above) were also isolated (n = 1). Gene expression analysis was performed according to standard procedures (Su et al. 2004). Briefly, RNA was isolated from frozen tissue using Trizol followed by cleanup with the RNeasy kit. RNA was amplified and labeled using the Affymetrix one-cycle target labeling kit. Samples were hybridized to GNF1M whole-genome mouse arrays (Su et al. 2004), and data were processed using the gcRMA algorithm (Wu et al. 2004). Raw data were deposited in GEO (http://ncbi.nih.gov/geo) under series accession no. GSE5961.
In this study, data were filtered to remove probe sets whose expression was either undetectable (maximum expression across strains <200) or invariant across strains (ratio of maximum expression to minimum expression across strains <3). Results were qualitatively the same when no filtering for differential expression was performed. In addition, the results described in this article were generated using the default cumulative distribution function (CDF) file. Although summarization algorithms are designed to be robust to single-probe outliers, the presence of SNPs in the probe sequence could theoretically lead to spurious detection of cis-eQTL. An analysis performed after removing all probes overlapping a SNP in dbSNP from the CDF file resulted in qualitatively similar results (supplemental Figure 1 at http://www.genetics.org/supplemental/).
Haplotype association mapping:
The original HAM method using inferred haplotypes has been previously described (Pletcher et al. 2004; McClurg et al. 2006). Briefly, the HAM approach inferred the haplotype structure from a set of contiguous genotype calls across the MDP and used these inferred haplotype groups as the independent factor in a one-factor ANOVA. For each phenotype, the F-statistic was calculated to quantify the between-group variance relative to within-group variance. The background distribution used to estimate the significance of the test statistic was either computed parametrically (the F-distribution) or simulated nonparametrically using 1E6 bootstraps of the phenotype vector. The resulting P-value was then transformed using a −log10 transformation to produce an association score.
SNP genotype data were collected primarily from three sources: GNF (Wiltshire et al. 2003), Rosetta/Merck (Cervino et al. 2005), and the Broad Institute (http://www.broad.mit.edu/∼mjdaly/mousehapmap). All SNP locations were mapped to NCBIM33. In total, the genotype data set contained allele calls for ∼157,000 SNPs across 46 strains in the MDP, resulting in a median inter-SNP distance of 6.02 kb across autosomes.
Weighted bootstrap:
We first calculated the genetic similarity matrix, as defined previously (McClurg et al. 2006). Briefly, this matrix calculated the pairwise similarity between two strains as the number of genotype calls in common divided by the total number of genotypes called in both strains. This matrix was “weighted” by raising all ratios to the exponent of the weight factor. A weight exponent of zero corresponded to a genetic similarity matrix of all ones.
As described above in the discussion of HAM, the significance of the calculated test statistic was estimated nonparametrically by simulating the null using bootstrapped phenotype values. A bootstrapped phenotype vector was created by choosing a replacement phenotype value for each strain in the phenotype vector. In the unweighted case, all strains were equally likely to be selected as a replacement for a given strain. In our weighted bootstrap, the probability of choosing the phenotype of a particular strain A as a replacement for strain B was proportional to the value in the genetic similarity matrix. When the weight exponent (described above) was set to zero, this corresponded to the unweighted case (since all values in the matrix are 1). As the weight exponent was increased, the likelihood also was increased of choosing a substitution strain in the null distribution that was genetically (and hence phenotypically) similar. The net effect was that the null distribution was increased for strong associations that are due to population structure, thereby selectively decreasing their significance and association score. This weighted bootstrap had a lesser negative effect on association scores that are not due to population structure.
Cis-eQTL enrichment calculation:
The cis-eQTL enrichment score was calculated as the ratio of peak density in the cis-eQTL band relative to the overall peak density. We first applied a prefilter to remove multiple adjacent associations that reflect the same strain distribution pattern. First, the top genomewide association was identified, and all association scores within 500 kb were removed. The second-highest association score was similarly identified and isolated, and this process was iteratively applied for all peaks. After prefiltering, we had at most one representative for each 1-Mb interval for each probe set.
Cis-eQTL were defined as associations between a gene expression vector and a genomic locus within 500 kb of the gene's genomic location. At a given threshold, the percentage of cis-eQTL of all eQTL peaks was calculated. Since the defined cis-region was ∼1/2600 of the overall genome size (∼2600 Mb), the cis-eQTL enrichment was the percentage of cis-eQTL × 2600.
Trans-eQTL band calculation:
Trans-eQTL bands were detected by tabulating the number of genes whose expression was associated with a single genetic locus. The significance of each trans-eQTL band was calculated by comparing it to a background distribution of trans-eQTL sizes. This background distribution was generated by creating 1000 random bootstrap samples of the eQTL matrix.
Two-factor ANOVA model:
The two-factor ANOVA model incorporated two main effects (haplotype and sex) and an interaction effect. For a genomic locus with three haplotype groups, the full model F was:
 |
The Xijkn were indicator variables with values dependent on sex and haplotype. The error terms were independent and normally distributed. To test the significance of the haplotype effect, the full model was compared to a reduced haplotype model Rhap in which the haplotype main effect terms were removed. A general linear test statistic was used to compare the variance explained by the full and reduced model:
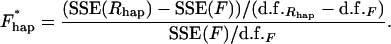 |
The d.f. terms were the degrees of freedom of the two models involved. Finally the significance of this statistic was assessed nonparametrically using our weighted bootstrap procedure to create a background distribution of 1E6 random statistics. As in the one-factor case, the resulting P-value was transformed via −log10 to give an association score.
RESULTS
eQTL data:
Hypothalamus tissue was harvested from 32 strains of male mice from the MDP. Total RNA was isolated from frozen tissue and hybridized to a custom Affymetrix whole-genome mouse chip (GNF1M) (Su et al. 2004), and data were analyzed using the gcRMA algorithm (Wu et al. 2004). The 36,182 probe sets represented on the chip were filtered to remove probe sets whose expression was either undetectable or invariant across strains. For each of the remaining 3725 probe sets, the expression pattern across strains was used as an input phenotype for the HAM algorithm.
Although each HAM analysis is performed completely independently of any knowledge of the gene's position in the genome, a strong association is commonly observed between gene expression across the MDP and the haplotype pattern near the gene locus. This pattern is referred to as the “cis-eQTL band” and is a robust pattern observed in many eQTL studies to date in a range of organisms and tissues (for example, Schadt et al. 2003; Brem and Kruglyak 2005; Bystrykh et al. 2005; Chesler et al. 2005; Cheung et al. 2005; Hubner et al. 2005; Stranger et al. 2005). Mechanistically, it is commonly hypothesized that cis-eQTL are explained by a regulatory polymorphism that alters the ability of a transcription factor or enhancer to activate transcription, although allelic changes in mRNA stability and local DNA structure are also consistent with cis-regulation. Regardless of the specific mechanism underlying the observation, it is commonly assumed that there is a very low false-positive rate among cis-eQTL (Chesler et al. 2006). Therefore, in this study we use cis-eQTL as a large set of surrogate true positives and conclude that methods that identify more cis-eQTL are relatively more powerful.
We quantified the strength of the cis-eQTL signal by calculating an enrichment score, defined generally as the ratio of the eQTL peak density along the cis-eQTL band to the background genomewide peak density. We defined cis-eQTL to be any association within 500 kb of the gene location (although evidence of cis-eQTL enrichment extends as far as 5 Mb; see supplemental Figure 2 at http://www.genetics.org/supplemental/).
Parametric vs. nonparametric methods:
In this article, we considered three variants of the original HAM method that differ in their approach to determining statistical significance. We first applied a parametric HAM method of measuring genetic association (McClurg et al. 2006). Briefly, this method used an inferred local haplotype based on a genotype calls over three contiguous SNPs. An F-test statistic was calculated on the basis of the inferred haplotype grouping. Using the parametric version of HAM, significance of the F-test statistic was calculated from the theoretical background distribution, the F-distribution. This procedure was repeated for all three-SNP windows in the genotype data.
Using parametric HAM, we examined the top 10,000 associations (nominal P < 1E-12) over all probe sets and SNP positions and found 19 associations within the ±500-kb window defining the cis-eQTL band (Figure 1A). Compared to the size of the cis-eQTL band relative to the size of the mouse genome (1 Mb/∼2600 Mb), these results indicated a cis-eQTL enrichment factor of 4.94. Although the enrichment was modest, the results were statistically significant (P < 0.01) on the basis of eQTL analysis using 100 random permutations of the expression data. None of these random simulations produced a cis-eQTL enrichment that exceeded the observed value, and the maximum enrichment observed in the background set was 2.08.
Figure 1.—

eQTL plots for hypothalamus. eQTL plots were generated using three haplotype association mapping (HAM) methods: (A) parametric analysis, (B) nonparametric analysis, and (C) weighted nonparametric analysis. In all plots, the x-axis represents the genomic SNP axis and the y-axis represents the genomic probe set axis. Each spot represents an association between the expression of a gene and the strain distribution pattern at a SNP location. Alternating colors indicate chromosome boundaries on the x-axis. In each plot, the top 10,000 eQTL associations are shown.
We next performed eQTL analysis using a nonparametric HAM version of our association method (McClurg et al. 2006). As before, an F-statistic was calculated on the basis of the inferred haplotype pattern. Instead of calculating a P-value using a theoretical background F-distribution, we simulated the true background distribution using 1E6 bootstraps of the input phenotype. The eQTL map using nonparametric HAM is shown in Figure 1B. Of the top 10,000 associations (10,384 with ties, nominal P < 1E-6), 80 were within the cis-eQTL band, corresponding to a cis-eQTL enrichment factor of 20.03. Again, relative to 100 randomized eQTL simulations, this enrichment score was highly significant (P < 0.01). To confirm that this trend is robust, we also examined cis-eQTL enrichment ratios using several other thresholds (Figure 2).
Figure 2.—

Comparison of parametric and nonparametric HAM methods. The chart displays the cis-eQTL enrichment as a function of eQTL rank for each of three different HAM variants.
On the basis of its higher cis-eQTL enrichment factor, nonparametric HAM was more sensitive to detecting cis-eQTL associations and therefore was considered to be a more powerful method. Nevertheless, it was apparent from Figure 1, A and B, that although the cis-eQTL band is statistically significant, it was not the most prominent signal in these eQTL maps. In both eQTL maps, a large number of horizontal bands were also clearly visible. Inspection of these horizontal bands revealed that this pattern reflects the case where the expression of one gene across strains was associated with the background population structure present in the MDP. Unlike F2 mapping populations in which parental haplotypes are shared with progeny in equal proportions, the MDP contains clusters of strains that are more related to each other than to the other strains in the MDP. Clustering the global patterns of gene expression in hypothalamus revealed a grouping of strains that closely matches the known ancestry of the MDP (Figure 3). In the eQTL context, nonspecific associations to this background population structure manifested themselves as horizontal bands.
Figure 3.—

Clustering of gene expression data. The clustering dendrogram displays the relationship of global gene expression patterns between strains. Coloring of the strain names reflects clusters derived from clustering of genotype data. The clear relationship between global gene expression patterns and genomewide genetic similarity underscores the need to account for population structure in association analyses.
Accounting for population structure:
Here, we introduced a weighted HAM method to account for the population structure inherent in the MDP. To compute the association between a phenotype and an inferred haplotype pattern, the F-statistic was computed as usual. Next, significance was calculated using a weighted bootstrap procedure to simulate the null distribution. In nonparametric HAM, 1E6 bootstraps were performed in which a randomly chosen phenotype value was sampled from the vector of all phenotype values for each strain in the analysis. In the case of nonparametric HAM, the phenotype values from all strains were equally likely to be sampled as the replacement. Here, weighted HAM utilized the genomewide genetic similarity to weight the sampling procedure. In this procedure, genetically similar strains were more likely to be selected as a replacement in the background distribution (details in methods). This adjustment had the overall effect of increasing the distribution of association scores in the null distribution, thereby selectively decreasing the significance of nonspecific associations.
The effect of the genomewide genetic similarity correction can be adjusted using an empirical weighting exponent. An analysis with the weight exponent of zero exactly corresponded to an unweighted nonparametric HAM analysis (Figure 1B). Figure 4 displays the cis-eQTL enrichment as a function of the weight exponent. For all subsequent studies of the weighted HAM method, we used a weighting exponent equal to three (Figure 1C), chosen as the lowest weighting exponent that visibly reduces nonspecific background association. Using the weighted nonparametric HAM approach, we observed 190 cis-eQTL among the top 10,000 associations (10,030 with ties, nominal P < 0.0004), corresponding to a cis-eQTL enrichment factor of 49.25. Again, the improved cis-eQTL enrichment was evident over a wide range of eQTL ranks (Figure 2) and was highly statistically significant relative to randomized eQTL simulations (P < 0.01).
Figure 4.—

Weight exponent analysis. To optimize the choice of the weight exponent, we calculated cis-eQTL enrichment using a range of weight powers and eQTL ranks. We chose a weight exponent of three for all subsequent studies.
Trans-eQTL band enrichment:
The diagonal cis-eQTL band was the most striking pattern in our hypothalamus eQTL data (Figure 1C) and was composed of individual cis-eQTL peaks. However, genetic associations to a gene's expression pattern that do not map back to the genomic position of the gene itself are called “trans-eQTL peaks,” and we also observed “trans-eQTL bands” that form vertical lines in the eQTL plot. These patterns resulted from the case where the expression patterns of multiple genes all associated to the haplotype pattern at a single genetic locus. These patterns have also been observed in previous eQTL studies (Bystrykh et al. 2005; Chesler et al. 2005) and are presumed to reflect the situation where an upstream transcriptional regulator at the QTL locus affects the expression of multiple downstream target genes. As another distinctly nonrandom pattern that was likely to be enriched in true-positive associations, the occurrence of trans-eQTL bands was also used as a metric to evaluate relative statistical power.
To determine the threshold of significance for the number of genes in a trans-eQTL band, we performed 1000 bootstrap samples of the top 10,000 association scores while randomizing genomic position. This analysis indicated that trans-eQTL bands with greater than nine genes were statistically significant (α = 0.05). The results comparing the parametric, nonparametric, and weighted HAM methods are summarized in Table 1. This analysis showed that the weighted HAM analysis resulted in the most significant trans-eQTL bands among the three methods. Moreover, when comparing the size and significance of the largest trans-eQTL band from each method, the weighted HAM method also showed the best performance. Overall, analysis of trans-eQTL bands corroborated the findings of the cis-eQTL enrichment.
TABLE 1.
Trans-eQTL bands
| No. significant (α = 0.05) | Largest trans-eQTL band
|
||
|---|---|---|---|
| Size | P-value | ||
| Parametric | 0 | 7 | NS |
| Nonparametric | 1 | 10 | 0.03 |
| Weighted | 25 | 28 | <0.001 |
Two-factor ANOVA model:
Since phenotype data for association analysis are commonly collected for both males and females separately, we next investigated the use of a two-factor ANOVA model to simultaneously model haplotype effects and sex effects. Although the application of a two-factor ANOVA model requires simply obtaining and phenotyping both male and female mice of each strain, the validation of the model using the concept of cis-eQTL enrichment required collection of gene expression data across strains for both sexes. Therefore, we chose a 12-strain subset of the samples used in the hypothalamus study and performed an eQTL analysis. Clearly, with only 12 strains, statistical power was drastically reduced. Over the top 500 associations, the cis-eQTL enrichment was reduced from 362 (corresponding to 27 cis-eQTL) in the full 32-strain analysis to 26 (5 cis-eQTL) in the 12-strain subset. However, over 1000 randomized simulations, no computed cis-eQTL enrichment scores exceeded 16 (P < 0.001), and an enrichment threshold corresponding to α = 0.05 was only ∼5-fold cis-eQTL enrichment. These simulations indicated that the cis-eQTL band in the 12-strain subset was still highly statistically significant. (trans-eQTL analysis did not reveal a statistically significant signal in the 12-strain analysis.)
We then obtained hypothalamus samples from female mice for each strain in the 12-strain subset. Total RNA was isolated and gene expression was measured using our custom whole-genome Affymetrix array. Over the top 500 associations, the cis-eQTL enrichment in the female samples was 41.6 (8 cis-eQTL), comparable to the score observed in the male samples. Next, we considered a two-factor ANOVA analysis over sex and haplotype. This model identified 27 of the top 500 eQTL peaks as cis-eQTL, corresponding to a cis-eQTL enrichment ratio of 140.4. This comparison between the three eQTL runs clearly showed that the two-factor ANOVA model demonstrates much stronger enrichment of the cis-eQTL band (Figure 5). To confirm that this effect was not simply due to an increase in the number of animals phenotyped, we also performed a one-factor ANOVA analysis in which the sex factor was disregarded. This analysis showed cis-eQTL enrichment comparable to that in the individual sex-restricted one-factor analyses.
Figure 5.—

Two-factor association analysis. Strain distribution pattern and sex were treated as independent factors in a two-factor HAM analysis. An eQTL analysis was performed using hypothalamus over 12 strains. For comparison, the cis-eQTL enrichment ratios are shown for the corresponding one-factor analyses.
DISCUSSION
The prospect of using the MDP for genomewide association mapping holds many potential advantages relative to more traditional mapping populations. In particular, the high recombination frequency between strains, coupled with dense genotyping data sets, leads to relatively precisely defined QTL with fewer candidate genes. Furthermore, because these strains are inbred, community efforts for genotype and phenotype data significantly decrease the cost of performing an association analysis for a phenotype of interest. Nevertheless, the two important liabilities of using the MDP as a mapping population are the lack of a framework to assess statistical power and the background population structure. Here, we have introduced two approaches that address these issues.
Estimation of statistical power is an important aspect of characterizing any QTL mapping strategy. Several seminal studies of statistical power have been published that aid the interpretation of QTL results from F2 mapping studies (Soller et al. 1976; Darvasi and Soller 1997). However, these power estimates typically employ parametric assumptions on the structure of the data. In the context of F2 mapping populations, these statistical assumptions are usually reasonable due to the defined structure resulting from controlled breeding. However, the MDP is fundamentally different in a variety of ways. Most notably, this population is inbred with no heterozygous alleles, and the pedigree is unbalanced and largely uncertain.
Since the theoretical framework to compute absolute statistical power has not yet been worked out for the MDP, here we used cis-eQTL enrichment as a measure of relative power. Instead of assumptions on the structure of the data, this approach made assumptions on the identity of true positives. Here, we generated the first eQTL data set in the MDP population. The results were qualitatively the same as previously observed in mouse and in other organisms. In the eQTL context, cis-eQTL can be considered a reliable set of true positives (Chesler et al. 2006), even after correcting for extensive multiple testing across genomic loci and gene expression probes (data not shown). Although it is not known how many genes are truly cis regulated in the MDP (and hence we cannot calculate absolute power), it is likely that an algorithm that enriches for cis-eQTL is relatively more powerful. Furthermore, although the expression traits underlying cis-eQTL are likely to result from alleles with very high genetic effect sizes, the algorithmic optimizations and conclusions drawn from these studies will likely extrapolate to analysis of complex traits.
We first applied this power assessment metric to compare parametric and nonparametric calculation of association significance. Both methods utilized the same test statistic, but the former compared the computed test statistic against a theoretical background distribution whereas the latter simulated the background distribution using extensive bootstrapping. The nonparametric HAM method showed a substantially higher cis-eQTL enrichment, which indicated a higher statistical power relative to the parametric HAM. These results corroborated our previous findings that used F2-derived QTL for a well-studied complex trait as a gold standard (McClurg et al. 2006). Moreover, these results underscored that phenotypic data in the MDP often do not follow typical parametric assumptions of normality.
We next applied the cis-eQTL enrichment metric to assess methods for correcting population structure effects inherent in the MDP. Previously, we accounted for these effects by altering the standard F-statistic using a weight factor reflecting genomewide genetic similarity (McClurg et al. 2006). However, this method had an unknown effect on the test statistic's null distribution. The challenges of association mapping in structured populations have also been previously addressed in larger population sizes by identifying unstructured subpopulations (Pritchard et al. 2000a,b). Others have recently proposed an alternate method for accounting for population structure in the context of a mouse heterogeneous stock population using a “bootstrap posterior probability” (Valdar et al. 2006). However, because of the limited population size in the MDP, these previous efforts cannot be directly adapted to this mapping population. Recently, another method was introduced in the context of association mapping in an Arabidopsis population similar to the MDP (Toomajian et al. 2006). As these authors alluded to, their algorithm could be applicable to other structured mapping populations (including the MDP).
Here, we have introduced an alternate method to generate a more appropriate null distribution of the F-statistic in the MDP, thereby reducing the significance of P-values for nonspecific associations. Since some strains in the MDP are clearly more related to each other than to the rest of the strain set, our weighted bootstrap procedure utilized genomewide genetic similarity when selecting replacement phenotype values. This modification represented a different null hypothesis being tested. The unweighted nonparametric method sought to reject the null hypothesis that the association at a given locus was stronger than a random bootstrap of strain labels. In contrast, the weighted HAM method tested the null hypothesis that the association at a given locus was no stronger than the association to the global genome structure. To generate this modified null distribution, strains sharing a high degree of genetic similarity were more likely to be sampled when constructing a background phenotype vector. The use of a weighted nonparametric HAM analysis clearly further improved the cis-eQTL enrichment metric. Furthermore, this method resulted in a notable decrease in the horizontal bands in the eQTL map that were indicative of nonspecific association to background genetic structure.
Finally, we have also applied the concept of cis-eQTL enrichment to evaluate the effect of a two-factor ANOVA model in our HAM method. The incorporation of sex effects in regression models for eQTL analysis has been previously described in a recombinant inbred mouse population (Wang et al. 2006). Here, we showed that when gene expression data for both males and females were available, cis-eQTL enrichment was substantially improved relative to one-factor ANOVA models. We also performed a similar comparison of two-factor ANOVA in a second eQTL study across 23 strains and found similar results (data not shown). Although not all phenotypes will benefit from treating sex as a second factor in association analysis, the analysis of thousands of phenotypes in parallel indicated that there is an overall benefit. Incorporating the sex effect into the statistical model clearly improved the cis-eQTL enrichment relative to one-factor models. Nevertheless, the relative benefit vs. the increased cost of additional phenotyping will likely vary depending on the specific experiment and phenotype being studied.
In summary, these results demonstrated that the use of the HAM algorithm in the MDP has sufficient statistical power to identify enrichment in both the cis-eQTL band and trans-eQTL bands. Moreover, we demonstrated that enrichment of these eQTL patterns is a suitable metric to calculate relative measures of power for genomewide association mapping. We used this principle to design and optimize a novel method of accounting for population structure in the mapping population and to evaluate a two-factor ANOVA variant of our HAM method. Finally, we created a web-based tool for performing HAM analyses using the weighted bootstrap method at http://snpster.gnf.org.
Acknowledgments
The authors thank Michael Cooke and Richard Glynne for helpful comments and suggestions. Research was supported in part by National Institutes of Health awards U01 MH61915 (J.S.T.) and RO3 DK62056 (J.B.). J.S.T. is an Investigator in the Howard Hughes Medical Institute.
Microarray data from this article have been deposited in the GEO database at the NCBI under accession no. GSE5961.
References
- Beck, J. A., S. Lloyd, M. Hafezparast, M. Lennon-Pierce, J. T. Eppig et al., 2000. Genealogies of mouse inbred strains. Nat. Genet. 24: 23–25. [DOI] [PubMed] [Google Scholar]
- Bogue, M. A., and S. C. Grubb, 2004. The Mouse Phenome Project. Genetica 122: 71–74. [DOI] [PubMed] [Google Scholar]
- Brem, R. B., and L. Kruglyak, 2005. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc. Natl. Acad. Sci. USA 102: 1572–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bystrykh, L., E. Weersing, B. Dontje, S. Sutton, M. T. Pletcher et al., 2005. Uncovering regulatory pathways that affect hematopoietic stem cell function using ‘genetical genomics’. Nat. Genet. 37: 225–232. [DOI] [PubMed] [Google Scholar]
- Cervino, A. C., G. Li, S. Edwards, J. Zhu, C. Laurie et al., 2005. Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 86: 505–517. [DOI] [PubMed] [Google Scholar]
- Chesler, E. J., S. L. Rodriguez-Zas and J. S. Mogil, 2001. In silico mapping of mouse quantitative trait loci. Science 294: 2423. [DOI] [PubMed] [Google Scholar]
- Chesler, E. J., L. Lu, S. Shou, Y. Qu, J. Gu et al., 2005. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat. Genet. 37: 233–242. [DOI] [PubMed] [Google Scholar]
- Chesler, E. J., L. Bystrykh, G. de Haan, M. P. Cooke, A. Su et al., 2006. Reply to “Normalization procedures and detection of linkage signal in genetical-genomics experiments”. Nat. Genet. 38: 856–858. [DOI] [PubMed] [Google Scholar]
- Cheung, V. G., R. S. Spielman, K. G. Ewens, T. M. Weber, M. Morley et al., 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437: 1365–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G. A., D. C. Airey, H. Allayee, J. M. Angel, A. D. Attie et al., 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat. Genet. 36: 1133–1137. [DOI] [PubMed] [Google Scholar]
- Darvasi, A., 2001. In silico mapping of mouse quantitative trait loci. Science 294: 2423. [PubMed] [Google Scholar]
- Darvasi, A., and M. Soller, 1997. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 27: 125–132. [DOI] [PubMed] [Google Scholar]
- Flint, J., W. Valdar, S. Shifman and R. Mott, 2005. Strategies for mapping and cloning quantitative trait genes in rodents. Nat. Rev. Genet. 6: 271–286. [DOI] [PubMed] [Google Scholar]
- Grupe, A., S. Germer, J. Usuka, D. Aud, J. K. Belknap et al., 2001. In silico mapping of complex disease-related traits in mice. Science 292: 1915–1918. [DOI] [PubMed] [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Hubner, N., C. A. Wallace, H. Zimdahl, E. Petretto, H. Schulz et al., 2005. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat. Genet. 37: 243–253. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, G., J. Wang, J. Guo, J. Allard, J. Cheng et al., 2004. In silico genetics: identification of a functional element regulating H2-Ealpha gene expression. Science 306: 690–695. [DOI] [PubMed] [Google Scholar]
- McClurg, P., M. T. Pletcher, T. Wiltshire and A. I. Su, 2006. Comparative analysis of haplotype association mapping algorithms. BMC Bioinform. 7: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher, M. T., P. McClurg, S. Batalov, A. I. Su, S. W. Barnes et al., 2004. Use of a dense single nucleotide polymorphism map for in silico mapping in the mouse. PLoS Biol. 2: e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. a Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens, N. A. Rosenberg and P. Donnelly, 2000. b Association mapping in structured populations. Am. J. Hum. Genet. 67: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Soller, M., A. Genizi and T. Brody, 1976. On the power of experimental designs for the detection of linkage between marker loci and quantitative loci in crosses between inbred lines. Theor. Appl. Genet. 47: 35–39. [DOI] [PubMed] [Google Scholar]
- Stranger, B. E., M. S. Forrest, A. G. Clark, M. J. Minichiello, S. Deutsch et al., 2005. Genome-wide associations of gene expression variation in humans. PLoS Genet. 1: e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, A. I., T. Wiltshire, S. Batalov, H. Lapp, K. A. Ching et al., 2004. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 101: 6062–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toomajian, C., T. T. Hu, M. J. Aranzana, C. Lister, C. Tang et al., 2006. A nonparametric test reveals selection for rapid flowering in the Arabidopsis genome. PLoS Biol. 4: e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdar, W., L. C. Solberg, D. Gauguier, S. Burnett, P. Klenerman et al., 2006. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat. Genet. 38: 879–887. [DOI] [PubMed] [Google Scholar]
- Wang, S., N. Yehya, E. E. Schadt, H. Wang, T. A. Drake et al., 2006. Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet. 2: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltshire, T., M. T. Pletcher, S. Batalov, S. W. Barnes, L. M. Tarantino et al., 2003. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc. Natl. Acad. Sci. USA 100: 3380–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z., R. A. Irizarry, R. Gentleman, F. M. Murillo and F. Spencer, 2004. A model based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99: 909–917. [Google Scholar]