

Abstract
Although the frequency and effects of neutral and nearly neutral mutations are critical to evolutionary patterns and processes governed by genetic drift, the small effects of such mutations make them difficult to study empirically. Here we present the results of a mutation-accumulation experiment designed to assess the frequencies of deleterious mutations with undetectable effects. We promoted the accumulation of spontaneous mutations by subjecting independent lineages of the RNA virus φ6 to repeated population bottlenecks of a single individual. We measured fitness following every bottleneck to obtain a complete picture of the timing and effects of the accumulated mutations with detectable effects and sequenced complete genomes to determine the number of mutations that were undetected by the fitness assays. To estimate the effects of the undetected mutations, we implemented a likelihood model developed for quantitative trait locus (QTL) data (Otto and Jones 2000) to estimate the number and effects of the undetected mutations from the measured number and effects of the detected mutations. Using this method we estimated a deleterious mutation rate of U = 0.03 and a gamma effects distribution with mean 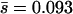 and coefficient of variation = 0.204. Although our estimates of U and
and coefficient of variation = 0.204. Although our estimates of U and 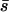 fall within the range of recent mutation rate and effect estimates in eukaryotes, the fraction of mutations with detectable effects on laboratory fitness (39%) appears to be far higher in φ6 than in eukaryotes.
fall within the range of recent mutation rate and effect estimates in eukaryotes, the fraction of mutations with detectable effects on laboratory fitness (39%) appears to be far higher in φ6 than in eukaryotes.
THE frequency with which neutral and nearly neutral mutations arise is critical to evolutionary patterns and processes that are governed by genetic drift, including the genetic basis of quantitative trait variation (Keightley and Hill 1988; Eyre-Walker et al. 2006), the molecular clock (Ohta 1977), and Muller's ratchet (Butcher 1995). However, because mutations with small effects are impossible to detect using phenotypic assays, most experiments designed to measure directly the frequency and effects of spontaneous mutations have neglected this class of mutations (but see Davies et al. 1999; Estes et al. 2004). Estimates of nearly neutral mutation rates have, therefore, been limited mostly to inferences from phylogenetic analyses of sequence data, inferences that are only as good as the assumptions they make about population demography and the neutrality of synonymous mutations.
Empirical attempts to measure the shape of the mutation effect distribution come from two sources—collections of artificially induced mutations (Elena et al. 1998; Sanjuan et al. 2004) and mutation-accumulation (MA) experiments (e.g., Mukai 1964; Wloch et al. 2001). Two recent studies—one of spontaneous and induced mutations in yeast (Wloch et al. 2001) and one of induced mutations in the RNA virus vesicular stomatitis virus (VSV) (Sanjuan et al. 2004)—offer the most direct measures of the mutation effect distribution. These studies represented substantial advances over previous MA experiments because they directly measured the effects of individual mutations. Nonetheless, they serve to illustrate the two major difficulties inherent in any attempt to measure the rate of mutations with small effects. First, the yeast study used a phenotypic assay to detect mutations and was limited to estimating the rates and effects of mutations of moderate to large effect. And second, studies that use induced mutations to overcome the first limitation may not accurately capture the properties of spontaneous mutations. The relative abundance of mutations of small and large effects was found to differ among spontaneous and induced mutations in yeast (Wloch et al. 2001). Thus, although the use of site-directed mutagenesis to induce mutations in the VSV genome allowed the detection of a substantial fraction of mutations with neutral or nearly neutral effects, it is not known whether this fraction is representative of spontaneous mutations.
We attempted to overcome both of these difficulties by conducting a traditional MA experiment with the RNA bacteriophage φ6, a model system in which it is possible to obtain complete knowledge of the mutational and fitness changes that occurred over the course of the experiment. Following the usual design of MA experiments, we promoted the accumulation of spontaneous mutations by subjecting 10 independent lineages to repeated population bottlenecks of a single individual. The extreme bottlenecks effectively eliminated selection, allowing nonlethal mutations, regardless of whether they were advantageous, deleterious, or neutral, to increase to fixation (a frequency of 100%) with approximately the same probability. We measured fitness following every bottleneck and sequenced whole genomes before and after mutation accumulation. These data provided a complete picture of the timing and effects of the accumulated mutations and allowed us to determine the relative frequencies of mutations with large, small, and nearly neutral effects on fitness.
MATERIALS AND METHODS
Strains and culture conditions:
The RNA bacteriophage φ6 used in this study is a laboratory genotype descended from the original isolate (Vidaver et al. 1973). Pseudomonas syringae pv. phaseolicola, the standard host of φ6, was obtained from the American Type Culture Collection (ATCC no. 21781). Details of diluting, filtering, culture, and storage of phage and bacteria are published (Mindich et al. 1976; Chao and Tran 1997). All phage and bacteria were grown in LC medium (5 g/liter yeast extract, 10 g/liter bactotryptone, 5 g/liter NaCl) at 25°.
Serial propagation through bottlenecks of a single phage:
The standard culture method for viruses easily allows for creating bottlenecks. Following the protocol described in (Burch and Chao 1999), phage were inoculated onto plates containing a lawn of the standard host P. phaseolicola at a sufficiently low density to ensure the formation of isolated plaques after a 24-hr incubation. Because plaques are generally initiated by a single phage, bottlenecks of a single phage were achieved by harvesting phage from a single randomly chosen plaque. Phage from this plaque were plated on a fresh lawn to obtain a new set of plaques, and the cycle was repeated 40 times in 10 independent lineages. Each bottleneck corresponds to ∼5 generations, bringing the total to 200 generations of mutation accumulation.
Fitness measures:
The fitness of individual phage genotypes was determined from measures of plaque size, as described in (Burch and Chao 2004). Phage were plated on a lawn of P. phaseolicola and incubated for 24 hr, and digital pictures were taken and used to measure the area of isolated plaques on each plate. In φ6, we have shown that under the current culture conditions log(fitness) is related to plaque area by the following linear relationship: log W = 0.044 × (plaque area) − 0.340 (R2 = 0.968, F1,7 = 213.4, P < 0.0001) (Burch and Chao 2004). Day-to-day variation in plaque size affects all genotypes equally, shifting the line up and down without affecting the slope. To ensure that day-to-day variation did not affect estimates of mutational effects (s), all of the data for an individual lineage (i.e., for a single plot in Figure 1) were collected on the same day from frozen stocks. Data for different lineages were collected on different days. Therefore, day-to-day variation explains the difference among lineages in the plaque area of the ancestral phage.
Figure 1.—

Stepwise regression analysis of fitness data. Data are mean plaque size at each bottleneck during mutation accumulation for 10 lineages labeled A–J. The illustrated regression models give the best fit to the data and were chosen from among models with different numbers of steps that either allowed both beneficial and deleterious mutations (red) or allowed only deleterious mutations (blue).
Genome isolation, RT–PCR, and sequencing:
Concentrated phage preparations were treated with DNase and RNase, then treated with 50:50 phenol:chloroform, and ethanol precipitated. The resulting RNA pellet was suspended in 50 μl TE and used as template for a reverse transcriptase reaction performed using N6 primers (first-strand cDNA synthesis kit; Amersham Biosciences, Piscataway, NJ). The product of the reverse transcriptase reaction was then amplified with φ6-specific primers by PCR, and PCR products were sequenced directly at the University of North Carolina Core Sequencing Facility using both forward and reverse primers to achieve double coverage of most genome regions. In total, we sequenced 12,381 bases (92.5%) of each genome, including all of the coding regions but missing the first and last 70–200 noncoding bases of each genome segment. Although these noncoding regions are known to contain regions of RNA 2° structure necessary for genome replication and packaging, we did not sequence them because the methods required to sequence the ends of linear genome segments are technically difficult and prone to sequencing errors.
Stepwise regression analysis of fitness trajectories:
The timing of mutational events and the effects of mutations on fitness were estimated using a forward stepwise least-squares linear regression. Our algorithm started with a model that assumed no mutations (i.e., steps that decrease or increase fitness) and iteratively added mutations in the following manner. In each iteration one additional mutation is added to the set of existing mutations. The location of the mutation is chosen in such a way that addition of a step at that location produces the largest reduction in the residual sum of squares (RSS). Mutations were added to the model until each bottleneck was associated with a mutation. At the end of this process a nested sequence of fitted models was obtained. We then chose the “best” model as the one that gave the smallest value for the Bayesian information criterion (BIC). This criterion is a popular method for model selection proposed by Schwarz (1978). The BIC balances the RSS of a model and the number of parameters involved in fitting that model. Note that with the addition of each mutation to the model there is a reduction in the RSS, but two parameters are added to the model, one for the step location and the other for the height of the added step. To reflect the underlying biological process, we also implemented a constrained version of the algorithm that allowed only deleterious mutations (i.e., steps that reduced fitness). In this case, when mutations were added to the model, we considered only locations that would result in decreasing steps.
We investigated the nature of the detection bias in the stepwise analysis by analyzing simulated MA lineages. In the simulated lineages, mutations accumulated according to a Poisson process with the observed rate U = 0.028 (Table 2; estimated from sequence data), and mutation effects were drawn randomly from a gamma distribution with the observed mean effect 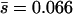 and coefficient of variation (or shape parameter) β = 0.162 (Table 2; maximum-likelihood estimate, MLE, for nDEL = 56). The simulated measure of fitness at a particular time during MA was determined by summing the effects of accumulated mutations and the experimental error, which was drawn randomly from a normal distribution with mean zero and the standard deviation observed in the real data. Our simulated data contained only deleterious mutations; therefore, we analyzed the simulated data using the constrained algorithm that allowed only decreasing steps.
and coefficient of variation (or shape parameter) β = 0.162 (Table 2; maximum-likelihood estimate, MLE, for nDEL = 56). The simulated measure of fitness at a particular time during MA was determined by summing the effects of accumulated mutations and the experimental error, which was drawn randomly from a normal distribution with mean zero and the standard deviation observed in the real data. Our simulated data contained only deleterious mutations; therefore, we analyzed the simulated data using the constrained algorithm that allowed only decreasing steps.
TABLE 2.
Comparison of analysis methods
| Estimation method | U |  |
β |
|---|---|---|---|
| Data-driven estimates of the distribution of fixed mutations | |||
Detected mutationsa (Umin, 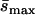 ) ) |
0.011 | 0.169 | 0.076 |
| MLEb (nDEL = 56) | 0.028 | 0.066 | 0.162 |
| MLEb (nDEL ≤ 56) | 0.017 | 0.109 | 0.116 |
| Data-driven estimates of the distribution of new mutationsc | |||
| MLE (nDEL = 56) | 0.030 | 0.093 | 0.204 |
| MLE (nDEL ≤ 56) | 0.021 | 0.142 | 0.141 |
| Model-based estimates of the distribution of fixed mutations | |||
| Bateman–Mukai | 0.035 | 0.0269 | 0d |
| Maximum likelihoode | 0.045 | 0.0207 | 0 |
Mutations whose fitness effects were detected by the stepwise regression analysis.
Estimated using the method of Otto and Jones (2000) by specifying that all of the accumulated mutations were deleterious (nDEL = 56) or allowing a fraction of the accumulated mutations to be neutral (nDEL ≤ 56).
Estimated by correcting the estimated fixed mutation distributions for the probability that mutations of various effect sizes were lost due to selection during plaque growth.
The Bateman–Mukai analysis method does not estimate Var(s). Rather, it assumes Var(s) = 0.
Estimated using the method of Keightley (1994).
We modeled the detection bias using a logistic function g(s) that describes the ratio of detected mutations to actual mutations as a function of mutation effect s. We use a logistic equation of the form
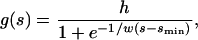 |
(1) |
where h represents the ratio of detected to actual mutations for mutations of large effects and can exceed 1 if a measurement bias causes the effects of large effect mutations to be overestimated, w describes the width of the detection threshold or the extent to which an increase in effect size leads to an increase in detectability, and smin describes the location of the detection threshold.
We simulated and analyzed 100,000 MA lineages to yield collections of both simulated and detected mutation effect sizes. We divided both collections into bins of width 0.01 and determined the ratio of detected mutations to simulated mutations for each bin. We then plotted this ratio vs. the average effect size for each bin and fitted Equation 1 to the data by determining the values of h, w, and smin that minimized the residual sum of squares over the range 0 < s < 0.3 (see Figure 2B).
Figure 2.—

Comparison of observed and simulated distributions of deleterious mutation effects. (A) Points indicate the cumulative frequency of deleterious mutations with measured effects between zero and s. Note that the measured effect of 34/56 mutations was zero because these mutations were not detected by the stepwise analysis. Lines illustrate the effects of distributions corresponding to the global MLE (nDEL = 33.4, solid line) and the constrained MLE (nDEL = 56, dashed line). (B) Detection threshold estimated from data simulated using the gamma distribution of mutation effects corresponding to the constrained MLE. (C) Probability density function of the global MLE (solid line) and the constrained MLE (dashed line). To aid the comparison, we adjusted the total area under the curves to correspond to the proportion of mutations with deleterious effects. Total area was 33.4/56 = 0.596 (global MLE) or 56/56 = 1 (constrained MLE). (D) Likelihood profile of the 22 detected deleterious mutation effects. We show the global MLE (closed circle), the constrained MLE (open circle), and contour lines corresponding to drops in ln(likelihood) of −1.92 (95% confidence region, thick line), −10, and −20 (thin lines).
Estimating the distribution of mutational effects:
We modified the maximum-likelihood approach of Otto and Jones (2000) to estimate the effects of the undetected mutations from the measured effects of the detected mutations. The approach was developed for an analogous problem in quantitative trait analysis in which only the largest quantitative trait loci (QTL) can be detected, but one hopes to estimate the number and effects of QTL that were not detected. In our scenario, the method uses maximum likelihood to estimate the distribution of effects of the accumulated mutations from the observed mutational effects si and the location and nature of the detection threshold g(s).
To calculate a likelihood of the observed mutational effects, we first determine the probability density function for detectable mutations,
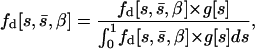 |
(2) |
where s is the effect of the mutation of interest, 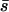 is the average mutational effect, β is the coefficient of variation of the effects distribution, and
is the average mutational effect, β is the coefficient of variation of the effects distribution, and 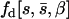 is the probability density function of the gamma distribution specified by
is the probability density function of the gamma distribution specified by 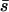 and β. If we now let the random variable S = (s1, s2, … , sn) represent the n observed mutational effects, the likelihood of obtaining these observations is
and β. If we now let the random variable S = (s1, s2, … , sn) represent the n observed mutational effects, the likelihood of obtaining these observations is
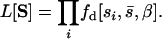 |
(3) |
Using this equation it is possible to obtain MLEs for both  and β by searching the joint parameter space or to obtain an MLE for β after specifying a fixed value for
and β by searching the joint parameter space or to obtain an MLE for β after specifying a fixed value for 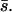 Note that the mean mutational effect
Note that the mean mutational effect 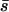 is exactly determined by the total drop in fitness due to mutation accumulation (
is exactly determined by the total drop in fitness due to mutation accumulation (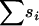 ) and the number of accumulated deleterious mutations (
) and the number of accumulated deleterious mutations ( ). That is,
). That is,  Therefore, by assuming that all of the accumulated mutations had nonzero deleterious fitness effects, we can specify
Therefore, by assuming that all of the accumulated mutations had nonzero deleterious fitness effects, we can specify  (on the basis of genome sequence data) and
(on the basis of genome sequence data) and 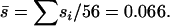
Estimating the effect of selection within the plaque:
Modifying the approach of Kibota and Lynch (1996) to fit plaque growth dynamics (which differ from bacterial colony growth dynamics), we obtained loss probabilities (Ploss) from a deterministic model that assumes that an individual phage is transferred after 24 hr of plaque growth, that phage achieve five discrete generations in 24 hr, and that wild-type and newly mutated phages have relative fecundities of 1 and 1 − s per generation, respectively. Letting q[s, t] be the frequency of mutations with a particular effect s after t generations, the initial frequency is q[s, 0] = 0. Each generation, mutation acts to increase and selection acts to decrease the frequency of deleterious mutations according to
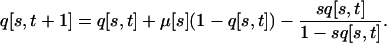 |
(4) |
We used this iterative equation to calculate q[s, 5] for each s. We specified 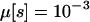 for all s, but the resulting estimates of Ploss(s) were not sensitive to this choice. The probability of loss was then calculated as
for all s, but the resulting estimates of Ploss(s) were not sensitive to this choice. The probability of loss was then calculated as
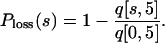 |
(5) |
Model-based analyses of mutation rate and effects:
To investigate the extent to which our analysis represented an improvement over more traditional analysis methods, we implemented two model-based methods for estimating U and s—the Bateman–Mukai (BM) method and a maximum-likelihood (ML) method. Both methods assume a model of the mutational process to estimate U and s from fitness data collected only at the start and end of mutation accumulation.
The BM method assumes that the number of mutations per lineage is Poisson distributed with parameter U and that mutations have identical effects, s. U and s are then determined from the equations
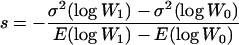 |
(6) |
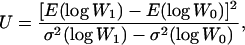 |
(7) |
where W0 and W1 are, respectively, fitness before and after mutation accumulation, E(log Wi) is the expectation, or mean, of log Wi over the 10 lineages, and 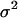 is the among-line variance. Note that our use of log fitness instead of fitness differs from the original approach of Bateman and Mukai (Bateman 1959; Mukai 1964). We discuss the advantage of analyzing means and variances of log fitness in (Burch and Chao 2004).
is the among-line variance. Note that our use of log fitness instead of fitness differs from the original approach of Bateman and Mukai (Bateman 1959; Mukai 1964). We discuss the advantage of analyzing means and variances of log fitness in (Burch and Chao 2004).
The ML approach also assumes that the number of mutations per lineage is Poisson distributed with parameter U, but assumes that mutational effects are drawn from a gamma distribution with scale and shape parameters α and β. The ML estimates for U and 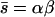 were calculated using a C program provided by Peter Keightley (Keightley 1994). The program evaluates the log likelihood of the data as a function of particular parameter combinations using numerical integration (Keightley 1994, 1998). We searched over a wide range of parameter combinations to ensure that we found the global maximum likelihood. Ninety-five percent confidence limits were defined by a
were calculated using a C program provided by Peter Keightley (Keightley 1994). The program evaluates the log likelihood of the data as a function of particular parameter combinations using numerical integration (Keightley 1994, 1998). We searched over a wide range of parameter combinations to ensure that we found the global maximum likelihood. Ninety-five percent confidence limits were defined by a  -fold drop in the log likelihood relative to its maximum value.
-fold drop in the log likelihood relative to its maximum value.
Phylogenetic estimation of the genomewide deleterious mutation rate:
Sequences of 24 φ6 relatives were downloaded from GenBank (accession nos. DQ273591 to DQ273614, L segment; DQ273639 to DQ273662, S segment). The sequences were truncated to coding regions with entire codons, with a final length of 411 nt (L) and 249 nt (S). These truncated sequences aligned perfectly with the φ6 sequences (NC003715, L; NC00371, S) with no gaps. The L and S sequence alignments contained 96 and 54 variable sites, respectively. Because reassortment (i.e., recombination) among segments is frequent (Silander et al. 2005), the L and S sequence sets were analyzed separately.
MrBayes (http://mrbayes.csit.fsu.edu/index.php) was used to reconstruct phylogenetic trees for each set of 25 sequences, using the general time-reversible model of nucleotide substitution and the gamma model of among-site rate variation (discretized gamma distribution with four categories). The algorithm was run for 2,000,000 generations, with trees sampled every 1000 generations, and reached convergence as assessed by the standard error of split frequencies. The sampled trees were used to make a consensus tree after a burn-in period of 1000, using the “allcompat” option to provide a fully resolved tree. Although some nodes were not well supported, the dN/dS analysis described below is robust to noise in the tree topology.
We used the CODEML program in PAML (version 3.15; http://abacus.gene.ucl.ac.uk/software/paml.html) to estimate the dN/dS ratios for each tree. We used the F3x4 codon model and allowed PAML to estimate the transition/transversion ratio. We tested models that allowed among-site variation in dN/dS, but these models either failed to improve the fit to the data (L segment) or did not substantially change the overall dN/dS (S segment).
RESULTS
Identification of mutations by genome sequencing:
To obtain a direct measure of the distribution of mutational effects, we used the bacteriophage φ6 to found 10 lineages that were independently propagated through 40 bottlenecks of a single phage. To obtain an exact count of the number of mutations that occurred during mutation accumulation, we sequenced the phage genotype from each lineage following the 40th population bottleneck. Sequencing 92.5% of the complete genomes (including all coding regions) revealed a total of 52 mutations. We estimate that 4 additional mutations were missed by our sequencing efforts (0.075 portion unsequenced × 52 observed mutations), bringing the total estimated mutation count to 56. The identity and molecular consequences of the 52 identified mutations are listed in Table 1. Most mutations resulted from base substitutions, although one insertion mutation was also observed. Transitions outnumbered transversions by 46 to 5, and we observed 32 nonsynonymous coding, 10 synonymous coding, and 10 noncoding mutations.
TABLE 1.
Accumulated mutations
| Lineage | Segment/nt mutationa | Gene or regionb | Functional consequence |
|---|---|---|---|
| A | S/a1378g | P9 | K13R |
| S/c2164t | P5 | A182V | |
| S/a2453g | 3′-UTR | S11L | |
| M/a804g | First IGR | ||
| L/c489t | P7 | ||
| B | L/a270g | P14 | M1V; start codon lost |
| C | S/t1867c | P5 | V83A |
| S/g2141a | P5 | Silent | |
| S/c2627t | 3′-UTR | K42R | |
| M/a491g | P10 | E51G | |
| M/t760c | First IGR | N406D | |
| M/a3660 | P13 | Silent | |
| L/a5166g | P1 | ||
| L/g5774a | P1 | ||
| D | L/a315g | P14 | T16A |
| L/t4668c | P1 | S240P | |
| L/a5296g | P1 | Y449C | |
| L/c5931t | P1 | L611F | |
| E | S/c2352t | 3′-UTR | Premature termination |
| M/3526+a | P13 | L4S | |
| L/t280c | P14 | Silent | |
| L/g835a | P7 | N56K | |
| L/c1110g | P2 | A104T | |
| L/g3252a | P4 | K197R | |
| L/a3532g | P4 | ||
| F | S/g410a | P8 | A36T |
| S/t2513c | 3′-UTR | A40T | |
| M/a3424c | Third IGR | T142M | |
| L/g387a | P14 | V355A | |
| L/c882t | P7 | N578D | |
| L/t2006c | P2 | Silent | |
| L/a2674g | P2 | Q28R | |
| L/c3441t | P4 | ||
| L/a4033g | P1 | ||
| G | S/c1438t | P9 | A33V |
| S/a1489g | P9 | Q50R | |
| S/a1744c | P5 | K42T | |
| M/c827t | First IGR | T361I | |
| M/c2556t | P3 | D39N | |
| M/g3623a | P13 | Silent | |
| L/c1524t | P2 | ||
| H | M/g2908a | P3 | Silent |
| M/c938a | First IGR | Silent | |
| M/a965t | First IGR | ||
| L/t1096c | P2 | ||
| I | S/t1266c | P12 | Silent |
| L/g1024a | P2 | A28T | |
| L/a4245g | P1 | N99D | |
| J | M/c2478t | P3 | T335I |
| L/t580c | P7 | Silent | |
| L/a4054g | P1 | Q35R | |
| L/c4586t | P1 | Silent |
Substitution mutations are indicated using the convention: ancestral nucleotide, position, mutant nucleotide. Insertion mutations are indicated using the convention: position plus added nucleotide.
Nomenclature is as follows: P1–P14, protein 1–protein 14; UTR, untranslated region; IGR, intergenic region.
We used a chi-square goodness-of-fit statistic (Sokal and Rohlf 1995) to compare the observed distribution of mutations among lineages to a Poisson distribution with parameter λ = 5.2 (52 total mutations/10 lineages) and found no significant deviation (χ2 = 3.827; d.f. = 5; P = 0.5746). This result indicates that mutations were not significantly clustered among lineages and suggests that the per generation mutation rate remained constant across the 10 lineages throughout the experiment. In addition, a comparison of the observed location of mutations on the three genome segments (small segment, 13; medium, 18; large, 26) to the random expectation given the proportion of sequenced bases in each segment (small, 0.213 proportion of sequenced bases × 52 identified mutations = 11.1; medium, 0.299 × 52 = 15.6; large, 0.487 × 52 = 25.3) demonstrated that the location of mutations did not deviate significantly from a random distribution among genome segments (χ2 = 0.1183; d.f. = 1; P = 0.7309).
Identification of mutations by their effects on fitness:
We measured the fitness of each lineage following each of the 40 single-phage bottlenecks (Figure 1). Because mutations are acquired in discrete events, the appearance of deleterious mutations should be detectable in these data as stepwise changes in fitness at discrete time points. Therefore, we attempted to identify mutational events by assessing the fit to the data of regression models containing different numbers of steps. We assessed the fit of the data to two types of regression models—models in which both beneficial and deleterious mutations were allowed and models that were constrained to contain only deleterious mutations. The best-fit models of both types are shown in Figure 1.
For the most part, models that included beneficial mutations (red lines in Figure 1) were deemed biologically unrealistic. In particular, three of four of the detected beneficial mutations occurred within one or two bottlenecks of a deleterious mutation (lineages A, B, and J), suggesting that these steps resulted from noise in the data rather than actual beneficial mutations. Therefore, it seemed biologically more plausible to assume that our data set did not contain any beneficial mutations. Models that were constrained to contain only deleterious mutations gave a good fit to the data (blue lines in Figure 1). The constrained stepwise regression analysis detected between one and four mutations in each lineage, for a combined total of 22 mutations across all 10 lineages. The distribution of effects of these mutations is shown in Figure 2A. The effects of the detected mutations ranged from smin = 0.054 to smax = 0.418, and mutations of small effect were more common than mutations of large effect.
The absence of detectable mutations with effects < ∼5% confirmed the expectation that phenotypic assays often miss mutations with the smallest effects. In our assay, it was feasible to collect only one fitness measure per bottleneck; thus, statistical power relied on obtaining fitness measures of the same genotype over many consecutive bottlenecks. In this case, limitations on our ability to detect mutations with small phenotypic effects depended on the relative magnitudes of experimental error and mutational effects and on the number of bottlenecks that occurred between mutational events.
Investigation of the detection threshold:
We investigated the nature of the detection threshold in the stepwise regression analysis by analyzing simulated data sets. We simulated the accumulation of mutations in 100,000 MA lineages using the mutation rate and effect parameters estimated from the real data. We assumed that all of the mutations that accumulated were deleterious, allowing us to fix the mutation rate parameter U = 0.028 (56 mutations/10 lineages/200 generations per lineage) and the average mutation effect  Note that the detection threshold depends on U and the shape of the effect distribution (i.e., on β) so we had to estimate the detection threshold and β simultaneously. We began with
Note that the detection threshold depends on U and the shape of the effect distribution (i.e., on β) so we had to estimate the detection threshold and β simultaneously. We began with 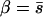 (exponential distribution) and iterated through a process of (1) estimating the detection threshold using data simulated with the current estimate of β and (2) updating the estimate of β using the current detection threshold to obtain a more accurate MLE of β (below). After a small number of iterations, the detection threshold and the MLE of β remained unchanged.
(exponential distribution) and iterated through a process of (1) estimating the detection threshold using data simulated with the current estimate of β and (2) updating the estimate of β using the current detection threshold to obtain a more accurate MLE of β (below). After a small number of iterations, the detection threshold and the MLE of β remained unchanged.
By analyzing the simulated MA lineages using the stepwise regression method, we can calculate the ratio of detected to simulated mutations for any effect size and determine the nature of the detection threshold from these data. The resulting data and detection threshold are shown in Figure 2B. Thirty-nine percent of the simulated mutations had effects that were too small to be detected by the stepwise regression analysis, but the probability of detecting a mutation increased rapidly as the effect size increased above smin = 0.059. We also noted a tendency to overestimate the effect size of mutations with large effects s > 0.08, a phenomenon that is analogous to the “Beavis effect” in QTL analyses (Beavis 1994, 1998). The best-fitting logistic equation (smin = 0.059, w = 0.0105, h = 1.17) accurately describes the nature of the detection threshold (Figure 2B), especially over the range of effect sizes in which mutations tended to fall (89% of detected mutations had effects s < 0.3).
Estimating the distribution of mutation effects:
From the genome sequencing we knew that the number of accumulated deleterious mutations was nDEL ≤ 56 and that the remaining  mutations have essentially neutral effects. To investigate the shape of the mutation effect distribution we started with the assumption that all of the accumulated mutations were deleterious; i.e., nDEL = 56. In this case, the average mutation effect was fixed at
mutations have essentially neutral effects. To investigate the shape of the mutation effect distribution we started with the assumption that all of the accumulated mutations were deleterious; i.e., nDEL = 56. In this case, the average mutation effect was fixed at 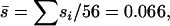 and we compared the likelihood of the observed mutation effects for gamma effects distributions with various coefficients of variation, β. The resulting MLE of β was 0.162 (95% C.I.: 0.082 ≤ β ≤ 0.457), which corresponds to the L-shaped gamma distribution shown in Figure 2, A and C (dashed lines). We also allowed nDEL < 56 and compared the likelihood of the observed mutation effects for gamma effects distribution with various
and we compared the likelihood of the observed mutation effects for gamma effects distributions with various coefficients of variation, β. The resulting MLE of β was 0.162 (95% C.I.: 0.082 ≤ β ≤ 0.457), which corresponds to the L-shaped gamma distribution shown in Figure 2, A and C (dashed lines). We also allowed nDEL < 56 and compared the likelihood of the observed mutation effects for gamma effects distribution with various  and β (likelihood profile shown in Figure 2D). The resulting MLEs were
and β (likelihood profile shown in Figure 2D). The resulting MLEs were 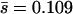 (nDEL = 33.4) and β = 0.116, but the 95% confidence limits on these estimates include the ML gamma distribution that corresponds to nDEL = 56. Figure 2A illustrates that the gamma distributions corresponding to nDEL = 56 and nDEL = 33.4 both give a good fit to the data.
(nDEL = 33.4) and β = 0.116, but the 95% confidence limits on these estimates include the ML gamma distribution that corresponds to nDEL = 56. Figure 2A illustrates that the gamma distributions corresponding to nDEL = 56 and nDEL = 33.4 both give a good fit to the data.
Estimating the effect of selection within the plaque:
Although we tried to minimize its effects, selection operating during the growth of each plaque will have caused the distribution of mutations that fixed in our experiment to deviate somewhat from the distribution we hoped to measure—the distribution of new mutations. To estimate the effects distribution of new mutations we modeled the probability that newly arisen deleterious mutations were lost due to selection during plaque growth. The resulting loss probabilities are plotted in Figure 3A. Although the probability that new mutations with small to moderate effects (i.e., s < 0.1) were lost due to selection is negligible, we observed a number of mutations with effects large enough (s > 0.2) that their probability of accumulating in our experiment was reduced by >30% relative to a neutral mutation.
Figure 3.—

Selection in the plaque biases the distribution of fixed mutations toward mutations of small effect. (A) Probability that a new mutation with effect s is lost due to selection. Probabilities were calculated using a deterministic model of plaque growth. (B) The probability density function of fixed mutations (solid line) was adjusted using the loss probability function to obtain a probability density function of new mutations (dashed line). Both probability density functions correspond to the constrained MLE in which nDEL = 56. (C) Probability density function of fixed mutations (solid line) and of new mutations (dashed line) corresponding to the global MLE in which nDEL = 33.4.
To obtain estimates of the probability density functions of new mutations, we combined the loss probability function Ploss[s] (Figure 3A) with the probability density functions of fixed mutations (estimated above; Figure 2C) as follows:
 |
(8) |
In addition, we used the loss probability function to correct the genomewide deleterious mutation rate estimate,
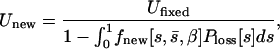 |
(9) |
where Unew and Ufixed are the rates at which mutations appear and fix in our experiments, respectively. Ufixed was estimated by the ML procedure described above.
We compare the resulting probability density functions of new vs. fixed mutations in Figure 3B (nDEL = 56) and 3C (nDEL = 33.4). Although the distributions are visually similar, in each case the distribution of new mutations contains substantially more weight in the tail of large s-values and substantially less weight at s-values near zero. The resulting distributions are no longer gamma shaped, but they are nonetheless characterized by a mean effect 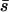 and coefficient of variation β. We report the adjusted U,
and coefficient of variation β. We report the adjusted U,  and β in Table 2. Note that the estimates of U are not much affected by selection in the plaque, but the distribution parameters
and β in Table 2. Note that the estimates of U are not much affected by selection in the plaque, but the distribution parameters  and β change substantially. For example, in the case of the distribution estimated by assuming that all of the accumulated mutations are deleterious (i.e., nDEL = 56), the mean mutational effect changes from
and β change substantially. For example, in the case of the distribution estimated by assuming that all of the accumulated mutations are deleterious (i.e., nDEL = 56), the mean mutational effect changes from 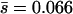 (fixed mutations) to
(fixed mutations) to 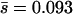 (new mutations), a difference of 41%.
(new mutations), a difference of 41%.
Traditional MA analyses:
To compare the conclusions from the current data-driven analysis to the conclusions of the traditional model-based MA analyses, we also analyzed the data using the BM approach (Bateman 1959; Mukai 1964) and the ML approach of Keightley (1994). These analyses used fitness data only from the start and finish of MA (before mutation accumulation,  and
and 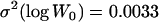 ; after mutation accumulation,
; after mutation accumulation, 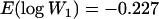 and
and  ). To estimate the rate and effects of mutations in the absence of fitness data at intermediate time points, these methods assume that mutations are acquired through a Poisson process with rate U and that mutation effects are drawn from either an equal-effects distribution (BM) or a gamma distribution with shape and scale parameters α and β (ML). Estimates of
). To estimate the rate and effects of mutations in the absence of fitness data at intermediate time points, these methods assume that mutations are acquired through a Poisson process with rate U and that mutation effects are drawn from either an equal-effects distribution (BM) or a gamma distribution with shape and scale parameters α and β (ML). Estimates of  U, and the coefficient of variation in mutation effects (β) from the data-driven analysis and from these model-based analyses are compared in Table 2. The model-based analyses both estimated mutation rates that were higher than the number of mutations that occurred (as determined from genome sequencing) and, therefore, mutational effects that were smaller than in reality. Note that the ML approach estimated an equal-effects distribution, despite the considerable variation in effect sizes exhibited by the underlying data (Figure 2A).
U, and the coefficient of variation in mutation effects (β) from the data-driven analysis and from these model-based analyses are compared in Table 2. The model-based analyses both estimated mutation rates that were higher than the number of mutations that occurred (as determined from genome sequencing) and, therefore, mutational effects that were smaller than in reality. Note that the ML approach estimated an equal-effects distribution, despite the considerable variation in effect sizes exhibited by the underlying data (Figure 2A).
Phylogenetic estimation of selective constraint at nonsynonymous sites:
An alternative way to estimate the fraction of mutations with deleterious effects is to estimate from phylogenetic data the extent of selective constraint operating on coding sequences (e.g., Eyre-Walker and Keightley 1999). By assuming that synonymous mutations are neutral, it is possible to estimate the fraction of nonsynonymous mutations that are also neutral as the dN/dS ratio. We estimated the dN/dS ratio from a collection of φ6 relatives isolated from nature (Silander et al. 2005). Phylogenetic trees were constructed from the two alignments of coding sequences in that study: 411 bp of the polymerase gene P2 and 249 bp of the nucleocapsid shell gene P8. The estimated dN/dS ratios were 0.0076 and 0.1009, respectively, combining for an average dN/dS ratio of 0.0542. In other words, very few nonsynonymous mutations evolve neutrally in φ6.
DISCUSSION
In this article we present the results of an MA experiment designed to provide a direct measure of the distribution of spontaneous mutational effects on fitness in an RNA virus. By measuring fitness following every bottleneck during mutation accumulation and sequencing whole genomes at the conclusion of the experiment we could make substantial refinements in analysis and interpretation compared to previous MA experiments. Although we were similarly constrained to assume a particular type of effects distribution (gamma), the ability to measure directly the effects of accumulated mutations with the largest effects provided both greater power to estimate the underlying distribution parameters (i.e., the mean and variance in mutational effects) and greater confidence that a gamma distribution was a reasonable assumption.
In addition, we note that our data differ somewhat from the data produced via site-directed mutagenesis of the RNA virus VSV (Sanjuan et al. 2004). Both experimental designs produced measures of the effects of mutations with the largest effects and an estimate of the relative abundances of mutations with detectable and undetectable effects on laboratory fitness. Although the shapes of the effect distributions estimated for spontaneous deleterious mutations in φ6 and for induced deleterious mutations in VSV are difficult to distinguish, the mean mutational effect and the proportion of mutations with detectable effects on laboratory fitness both differed. The mean effect of spontaneous mutations in φ6 (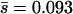 ) was lower than the mean effect of random induced nonlethal mutations in VSV (
) was lower than the mean effect of random induced nonlethal mutations in VSV (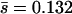 ; estimated from Table 1 in Sanjuan et al. 2004), and the proportion of spontaneous mutations in φ6 with detectable effects on laboratory fitness (39%) was lower than that in VSV (55%; estimated from Table 1 in Sanjuan et al. 2004).
; estimated from Table 1 in Sanjuan et al. 2004), and the proportion of spontaneous mutations in φ6 with detectable effects on laboratory fitness (39%) was lower than that in VSV (55%; estimated from Table 1 in Sanjuan et al. 2004).
It would be a stretch to conclude that the underlying causes of these differences are biological rather than experimental, but in both cases the direction of the difference is consistent with our a priori expectation of the difference between spontaneous and random induced mutations. Whereas the probability of sampling a particular random induced mutation does not depend on the identity of the mutation, the probability of sampling a particular spontaneous mutation can depend radically on mutation identity. In particular, transition mutations are expected to be far more likely than transversion mutations, especially in RNA viruses (in our data set transitions outnumbered transversions by 46 to 5). Furthermore, redundancy in the genetic code is expected to make the average effect of a transition smaller than the average effect of a transversion. As a result, the distribution of spontaneous mutations is expected to contain a greater proportion of small-effect mutations than the distribution of random induced mutations. In sum, the fundamental difference between the identities of spontaneous and induced mutations likely explains our lower estimates of both 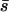 and the proportion of mutations with detectable effects on laboratory fitness.
and the proportion of mutations with detectable effects on laboratory fitness.
Shape of the mutation effect distribution:
We assessed the fit of two qualitatively different effect distributions: a continuous gamma distribution in which all mutations are deleterious but many have small effects (dashed line in Figure 2A) and a discontinuous distribution in which most mutations are deleterious with gamma-distributed effects, but other mutations are essentially neutral, i.e., have essentially no effect on fitness (solid line in Figure 2A). From a statistical perspective, the simpler model in which all mutations are deleterious is preferable. The more complex model that includes two categories of mutations—deleterious and neutral—does not significantly improve the fit to the data. However, from a biological perspective, our intuition tells us that mutation effects are likely to be divided into two categories with qualitatively different effects. In general, we expect nonsynonymous mutations to have deleterious effects and synonymous and noncoding mutations to be essentially neutral. The close match between the number of deleterious mutations predicted by the maximum-likelihood analysis (nDEL = 33.4) and the number of accumulated nonsynonymous mutations (32) supports this view.
In addition, patterns of molecular evolution in φ6 support our intuition that the distribution of mutational effects is discontinuous. dN/dS ratios in φ6 gene phylogenies are near zero (mean dN/dS = 0.05), suggesting that nonsynonymous mutations are almost universally deleterious (i.e., s > 1/Ne) and that synonymous mutations are effectively neutral (i.e., s < 1/Ne). Noncoding mutations also appear to evolve neutrally, at rates that are similar to the synonymous substitution rate (Silander et al. 2005).
We note that the deleterious mutations in the estimated discontinuous distribution are described by a gamma distribution that is not L-shaped, but very nearly exponential—the exact distribution shape that has been assumed for decades.
Comparisons to other methods and organisms:
One of the most puzzling outcomes of our analysis arises from the comparison with the traditional MA analysis methods. First, despite the extensive variation in effect sizes among the mutations in our collection, the traditional ML analysis (Keightley 1994) indicated that the fitness declines were best modeled using an equal-effects distribution. Second, the assumption made by the traditional analysis methods that mutations have identical effects is expected to cause U to be underestimated and s to be overestimated. Contrary to this expectation, the BM and ML estimates of U (and s) are higher (and lower) than our more accurate estimates. Although the BM and ML estimates differ from our estimates only by 2- to 3-fold, it is difficult to gain an intuition for why the estimate of U (that is supposed to be an underestimate) exceeds the value obtained by counting the total number of mutations by genome sequencing even though this count includes many effectively neutral mutations. We note that our data were subject to day effects that had similar effects on the initial and final fitness measures for each lineage and that the ML method uses a statistical model that includes environmental (i.e., plate) effects, but not day effects (Keightley 1994). Even so, while the presence of day effects might explain the surprising ML estimates, it cannot explain the surprising BM estimates.
A comparison of the U and s estimates obtained here in an RNA virus to previous estimates from eukaryotic organisms also yields a surprising outcome. Contrary to the a priori expectation that RNA viruses are radically different from other organisms, our estimates of U = 0.030 and s = 0.093 are both within the range of recent estimates in eukaryotes: 10−3 < U < 10−1 and 0.06 < s < 0.2 (Keightley and Caballero 1997; Fry et al. 1999; Wloch et al. 2001; Shaw et al. 2002; Joseph and Hall 2004; Baer et al. 2005). Clearly some RNA viruses have higher genomic deleterious mutation rates than φ6, but our data suggest that it is inappropriate to lump RNA viruses together into a single group that is subject to different evolutionary rules than organisms with DNA genomes.
Although estimates of the average mutation effect in φ6 and other organisms are similar, the distribution of mutation effects appears to differ. In our experiment, as in the previous study of the RNA virus VSV (Sanjuan et al. 2004), more than one-third of mutations (22/56) had detectable effects on laboratory fitness. In contrast, MA studies of the eukaryote Caenorhabditis elegans showed that only 1–4% of mutations have detectable effects on laboratory fitness (Davies et al. 1999; Estes et al. 2004). It is tempting to explain this difference by claiming that viral genomes are more compact than eukaryotic genomes or that viral genomes contain more functional RNA secondary structure (Brinton and Dispoto 1988; Brown et al. 1992; Berkhout and Schoneveld 1993). However, it is also possible that the laboratory environment more closely mimics the natural environment of viruses than it does the natural environment of C. elegans. We favor the latter explanation because most gene knockouts in φ6 either are lethal or have substantial effects on laboratory fitness (Mindich 2005), whereas most gene knockouts in eukaryotes show no detectable effect on laboratory fitness (Shoemaker et al. 1996; Winzeler et al. 1999). We suspect that viral genomes are not qualitatively different from eukaryotic genomes (except in size), but rather that measuring fitness in the laboratory has led to an underestimation of both the rate and the effects of deleterious mutations in eukaryotes.
Acknowledgments
We thank Adam Eyre-Walker, Sally Otto, Jennifer Knies, Patrick Phillips, two anonymous reviewers, and members of the Burch lab for comments that improved the experimental design and the final manuscript. In addition, we thank Steve Marron and his statistical consulting course at the University of North Carolina for providing the means to initiate this collaboration. This work was supported by a grant from the National Institutes of Health to C.L.B.
References
- Baer, C., F. Shaw, C. Steding, M. Baurngartner, A. Hawkins et al., 2005. Comparative evolutionary genetics of spontaneous mutations affecting fitness in rhabditid nematodes. Proc. Natl. Acad. Sci. USA 102: 5785–5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A. J., 1959. The viability of near-normal irradiated chromosomes. Int. J. Radiat. Biol. 1: 170–180. [Google Scholar]
- Beavis, W. D., 1994. The power and deceit of QTL experiments: lessons from comparative QTL studies. Proceedings of the Corn and Sorghum Industry Research Conference, American Seed Trade Association, Washington DC, pp. 250–266.
- Beavis, W. D., 1998. QTL analyses: power, precision, and accuracy, pp. 145–162 in Molecular Dissection of Complex Traits, edited by A. H. Patterson. CRC Press, Boca Raton, FL.
- Berkhout, B., and I. Schoneveld, 1993. Secondary structure of the HIV-2 leader RNA comprising the tRNA-primer binding site. Nucleic Acids Res. 21: 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinton, M. A., and J. H. Dispoto, 1988. Sequence and secondary structure analysis of the 5′-terminal region of flavivirus genome RNA. Virology 162: 290–299. [DOI] [PubMed] [Google Scholar]
- Brown, E. A., H. Zhang, L. H. Ping and S. M. Lemon, 1992. Secondary structure of the 5′ nontranslated regions of hepatitis C virus and pestivirus genomic RNAs. Nucleic Acids Res. 20: 5041–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, C. L., and L. Chao, 1999. Evolution by small steps and rugged landscapes in the RNA virus φ6. Genetics 151: 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, C. L., and L. Chao, 2004. Epistasis and its relationship to canalization in the RNA virus φ6. Genetics 167: 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher, D., 1995. Muller's ratchet, epistasis and mutation effects. Genetics 141: 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao, L., and T. T. Tran, 1997. The advantage of sex in the RNA virus φ6. Genetics 147: 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, E., A. Peters and P. Keightley, 1999. High frequency of cryptic deleterious mutations in Caenorhabditis elegans. Science 285: 1748–1751. [DOI] [PubMed] [Google Scholar]
- Elena, S. F., L. Ekunwe, N. Hajela, S. A. Oden and R. E. Lenski, 1998. Distribution of fitness effects caused by random insertion mutations in Escherichia coli. Genetica 102–103: 349–358. [PubMed] [Google Scholar]
- Estes, S., P. Phillips, D. Denver, W. Thomas and M. Lynch, 2004. Mutation accumulation in populations of varying size: the distribution of mutational effects for fitness correlates in Caenorhabditis elegans. Genetics 166: 1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker, A., and P. Keightley, 1999. High genomic deleterious mutation rates in hominids. Nature 397: 344–347. [DOI] [PubMed] [Google Scholar]
- Eyre-Walker, A., M. Woolfit and T. Phelps, 2006. The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173: 891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J., P. Keightley, S. Heinsohn and S. Nuzhdin, 1999. New estimates of the rates and effects of mildly deleterious mutation in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 96: 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, S., and D. Hall, 2004. Spontaneous mutations in diploid Saccharomyces cerevisiae: more beneficial than expected. Genetics 168: 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1994. The distribution of mutation effects on viability in Drosophila melanogaster. Genetics 138: 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., 1998. Inference of genome-wide mutation rates and distributions of mutation effects for fitness traits: a simulation study. Genetics 150: 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P., and A. Caballero, 1997. Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 94: 3823–3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley, P. D., and W. G. Hill, 1988. Quantitative genetic-variability maintained by mutation-stabilizing selection balance in finite popultions. Genet. Res. 52: 33–43. [DOI] [PubMed] [Google Scholar]
- Kibota, T., and M. Lynch, 1996. Estimate of the genomic mutation rate deleterious to overall fitness in E. coli. Nature 381: 694–696. [DOI] [PubMed] [Google Scholar]
- Mindich, L., 2005. Phages with segmented double-stranded RNA genomes, pp. 197–207 in The Bacteriophages, edited by R. L. Calendar. Oxford University Press, New York.
- Mindich, L., J. F. Sinclair, D. Levine and J. Cohen, 1976. Genetic studies of termperature-sensitive and nonsense mutations of bacteriophage φ6. Virology 75: 218–223. [DOI] [PubMed] [Google Scholar]
- Mukai, T., 1964. The genetic structure of natural populations of Drosophila melanogaster. I. Spontaneous mutation rate of polygenes controlling viability. Genetics 50: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta, T., 1977. Genetic-variation in multigene families. Nature 267: 515–517. [DOI] [PubMed] [Google Scholar]
- Otto, S. P., and C. D. Jones, 2000. Detecting the undetected: estimating the total number of loci underlying a quantitative trait. Genetics 156: 2093–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjuan, R., A. Moya and S. F. Elena, 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101: 8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, G., 1978. Estimating dimension of a model. Ann. Stat. 6: 461–464. [Google Scholar]
- Shaw, F., C. Geyer and R. Shaw, 2002. A comprehensive model of mutations affecting fitness and inferences for Arabidopsis thaliana. Evolution 56: 453–463. [DOI] [PubMed] [Google Scholar]
- Shoemaker, D. D., D. A. Lashkari, D. Morris, M. Mittmann and R. W. Davis, 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14: 450–456. [DOI] [PubMed] [Google Scholar]
- Silander, O. K., D. M. Weinreich, K. M. Wright, K. J. O'Keefe, C. U. Rang et al., 2005. Widespread genetic exchange among terrestrial bacteriophages. Proc. Natl. Acad. Sci. USA 102: 19009–19014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R. R., and F. J. Rohlf, 1995. Biometry. W. H. Freeman, New York.
- Vidaver, A. K., R. K. Koski and J. L. van Etten, 1973. Bacteriophage φ6: a lipid-containing virus of Pseudomonas phaseolicola. J. Virol. 11: 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson et al., 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- Wloch, D. M., K. Szafraniec, R. H. Borts and R. Korona, 2001. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 159: 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]