

Abstract
The symmetric island model with D demes and equal migration rates is often chosen for the investigation of the consequences of population subdivision. Here we show that a stepping-stone model has a more pronounced effect on the genealogy of a sample. For samples from a small geographical region commonly used in genetic studies of humans and Drosophila, there is a shift of the frequency spectrum that decreases the number of low-frequency-derived alleles and skews the distribution of statistics of Tajima, Fu and Li, and Fay and Wu. Stepping-stone spatial structure also changes the two-locus sampling distribution and increases both linkage disequilibrium and the probability that two sites are perfectly correlated. This may cause a false prediction of cold spots of recombination and may confuse haplotype tests that compute probabilities on the basis of a homogeneously mixing population.
HOMOGENEOUSLY mixing populations of constant size are a convenient setting to develop the theory of population genetics. However, when one wants to understand patterns observed in data, one must consider the effects of population growth, bottlenecks, and population subdivision. When the consequences of population subdivision are investigated, the symmetric island model with D demes and equal migration rates is the usual choice, and the case of two demes is especially popular. The island model is easy to analyze mathematically due to the fact that if two lineages are not in the same deme, then their relative location is not important. However, this has the unrealistic consequence that after one migration event, the lineage is distributed uniformly over the species range.
An alternative approach to modeling spatial structure that does not suffer from this defect is the stepping-stone model. In this article, we investigate the consequences of modeling space as a two-dimensional stepping-stone model in which there is an L × L grid of colonies and migration only to neighboring colonies. The migration scheme is very simple; however, it results in what Wright called isolation by distance. In other words, it takes a number of migration events for the lineages to spread across the system. As we will see, this feature, which is certainly present in Drosophila and early human populations, causes a dramatic change in the coalescence structure of lineages.
The reason for this is intuitively clear. At small times the lineages have not had a chance to spread across the population, so the effective population size is reduced. The coalescence rate is increased, reducing the number of low-frequency-derived alleles, skewing the site frequency spectrum, and increasing linkage disequilibrium. These effects occur in the island model as well; two lineages sampled from one deme have an increased coalescence rate until one of them migrates, at which point they behave like a random sample from the overall population. In contrast, as we later explain, in the stepping-stone model the effective population size increases roughly linearly in time.
The main point of this article is to argue that spatial structure in the form of the stepping-stone model has a different effect from the symmetric island model and can have a much greater impact on genealogies, so it should also be considered when one wants to assess the impact of spatial structure on estimation procedures or statistical tests. We begin by reviewing theoretical results of Cox and Durrett (2002) and Zähle et al. (2005) for the coalescence time of a sample of size n and contrast these results with the corresponding facts about the symmetric island model. The strange nonlinear time scaling needed to reduce genealogies in the stepping-stone model to Kingman's coalescent indicates that there is a strong effect on commonly used statistics, but the exact nature of the changes is difficult to analyze mathematically. Because of this, we turn to simulations to demonstrate the effect of stepping-stone spatial structure on the decay of linkage disequilibrium, the site frequency spectrum, and the distribution of test statistics based on the site frequency spectrum.
THEORETICAL RESULTS
Wakeley, with various coauthors, has investigated the island model when the number of demes is large. Let N be the number of diploid individuals per colony. Wakeley (1998) has shown that for a scattered sample in which we get at most one sequence from each deme, in the limit as 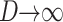 the genealogy of a sample of size n is the same as that of a homogenously mixing population of size
the genealogy of a sample of size n is the same as that of a homogenously mixing population of size
 |
(1) |
This formula for Ne is the same as the one of Nei and Takahata (1993).
The reason for the simplification for large D is easy to understand. At most times all lineages are in different demes, their actual locations are irrelevant, and the coalescence times will have the lack of memory property that characterizes the exponential. If we sample n chromosomes from one deme then there is an initial period called the “scattering phase,” which ends when all of the surviving lineages are in different demes. This initial phase is short compared to the coalescence time, so it is equivalent to a random reduction in the sample size. For more details, see p. 1864 of Wakeley (1999).
Lessard and Wakeley (2004) have recently extended this analysis to the two-locus ancestral graph in a subdivided population. Wakeley and Lessard (2003) have applied these results to the study of linkage disequilibrium (LD) in humans. They found that their model with a large number of demes fitted the data for humans well (see Figure 2 on p. 1049 of Wakeley and Lessard 2003), in contrast to Reich et al. (2002), who did not get a good fit from the two-island model.
Figure 2.—



Sample data sets for (a) a homogeneously mixing population, (b) an island model with 4Nm = 1, and (c) a stepping-stone model with 4Nm = 10. Asterisks mark nucleotides that are different from the ancestral state. The numbers at the right indicate how many times each haplotype was observed. Each square in the triangular plot represents the r2-value for a pair of SNPs. r2 was calculated for all pairs of SNPs and shaded according to the magnitude. The color scale used was a gradation from white to black, with white representing r2 = 0 and black representing r2 = 1.
The stepping-stone model has been extensively studied since its introduction by Kimura in the 1950s. There have been many important contributions by Kimura and Weiss, Malécot, Maruyama, Nagylaki, Crow and Aoki, Slatkin, and others. Here we focus on recent results of Cox and Durrett (2002) and Zähle et al. (2005), referring the reader to the 2002 article for more on the historical development.
Cox and Durrett (2002) investigated the Moran model in which each individual is replaced at rate 1, replacement comes from a different deme with the migration probability m (called ν by Cox and Durrett), and the probability a migrant to x comes from y is q(y − x), where q is a probability distribution with finite range that has the same symmetries as the two-dimensional lattice. This symmetry assumption implies that the two coordinates are uncorrelated and have the same variance σ2. The grid of colonies is an L × L square and the difference y − x is computed modulo L; i.e., we have periodic boundary conditions that identify opposite edges of the square. This assumption is a mathematical convenience that has been used in many previous studies, but is not necessary. The proofs in Cox and Durrett (2002) and Zähle et al. (2005) extend easily to a flat universe with migration out of the system suppressed or reflected back in, and the qualitative behavior is the same.
Theorem 4 of Cox and Durrett (2002) shows that if we pick any two chromosomes and  as L → ∞ then the coalescence time divided by
as L → ∞ then the coalescence time divided by 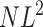 converges to an exponential distribution with mean 1. In words, if the per colony migration rate, Nm, is much larger than log L the system is essentially homogeneously mixing. This is a strong migration limit that corresponds to results of Nagylaki (1980) and Notohara (1993) for the island model. It should also be compared to the remark of Kimura and Maruyama (1971, pp. 125–126) that “marked local differentiation of gene frequencies can occur if
converges to an exponential distribution with mean 1. In words, if the per colony migration rate, Nm, is much larger than log L the system is essentially homogeneously mixing. This is a strong migration limit that corresponds to results of Nagylaki (1980) and Notohara (1993) for the island model. It should also be compared to the remark of Kimura and Maruyama (1971, pp. 125–126) that “marked local differentiation of gene frequencies can occur if  ” while “if
” while “if 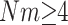 the whole population tends to behave as a panmictic unit.” As we can see from the mathematical result, the cutoff between the two behaviors is not constant somewhere between 1 and 4, but depends on the number of demes and increases like log L.
the whole population tends to behave as a panmictic unit.” As we can see from the mathematical result, the cutoff between the two behaviors is not constant somewhere between 1 and 4, but depends on the number of demes and increases like log L.
To state results for the case in which the local population size N is not much larger than log L, we need the following rescaled migration rate:
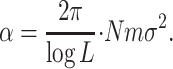 |
This definition may look mysterious but as we will see α is the natural rescaled migration rate for the stepping-stone model, which is the analog of M = 4Nm for the island model. Note that in the stepping-stone model the variance σ2 joins the product Nm to give the composite parameter that describes the strength of migration, but in contrast to the island model this quantity is divided by log L. The 2π that comes from the central limit theorem is included to make the next formula simple.
Theorem 5 of Cox and Durrett (2002) shows that two chromosomes sampled at random from the population have a coalescence time that is asymptotically, as L → ∞, exponential with mean
 |
This result is similar to one found by Barton et al. (2002) in a model with a grid of colonies with the local allele frequencies modeled by diffusion processes. As stated in Equation 9 of Charlesworth et al. (2003) if the density of individuals ρ = 1 the mean coalescence time of two individuals is
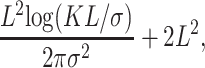 |
where K is a constant. The second term and the K/σ inside the logarithm are there to make the approximation more accurate for small L. They are not important as L → ∞, so they should be dropped when comparing with our asymptotic result, but even with this there remains the difference of the factor of 1 + α. It is difficult to say what causes this difference since the result cited by Charlesworth et al. (2003) and attributed to Barton et al. (2002) does not appear in that article.
Theorem 2 in Zähle et al. (2005) extends the result for the coalescence time of two chromosomes by showing that a sample of n chromosomes chosen at random from the population has the same genealogy as a sample of size n from a homogeneously mixing population of size
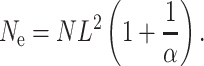 |
(2) |
Note that this has the same form as Ne in the island model when the number of demes D = L2 and the scaled migration rate M = α. To illustrate the use of these formulas, consider our 10 × 10 grid with migration to nearest neighbors so L = 10 and  . If the local population size is N = 25 and we choose m = 0.1 so that 4Nm = 1 then α = π(0.25)/log(10) = 0.341 and Ne = 2500(1.341)/0.341 = 9829 vs. the actual population size of 2500.
. If the local population size is N = 25 and we choose m = 0.1 so that 4Nm = 1 then α = π(0.25)/log(10) = 0.341 and Ne = 2500(1.341)/0.341 = 9829 vs. the actual population size of 2500.
In many genetic studies, samples are not chosen at random from the population as a whole. For example, one of the samples in Sabeti
et al. (2002) consists of 73 Beni individuals who are civil servants in Benin City, Nigeria. To capture this type of local sample in our framework, we assume that the n chromosomes are sampled at random from a Lβ × Lβ square of colonies. Theorem 3 in Zähle
et al. (2005) shows that we get the ordinary coalescent after a nonlinear time change in which times  with
with 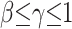 correspond to time
correspond to time 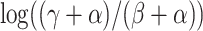 in the coalescent, and then time proceeds at the usual linear rate for a population with the Ne given in (2). Here, we have changed Zähle
et al.'s (2005) 2m in the denominator to 4πmσ2. This does not affect the limit theorem, but as we will see, it makes for a better approximation.
in the coalescent, and then time proceeds at the usual linear rate for a population with the Ne given in (2). Here, we have changed Zähle
et al.'s (2005) 2m in the denominator to 4πmσ2. This does not affect the limit theorem, but as we will see, it makes for a better approximation.
To help explain the time change, we note that the coalescence rate at time s =  [that is,
[that is, 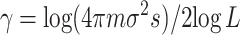 ] is
] is
 |
To make a connection with simulation results in Figure 4 of Wilkins (2004) we have graphed 1 over the coalescence rate vs. time s in Figure 1. This shows that the coalescence occurs much more rapidly in the initial stages of the stepping-stone model compared to a homogeneously mixing population. The reason for graphing 1 over the rate is that this quantity is almost linear in time. This can be seen intuitively by noting that at time s, the central limit theorem says that lineages will be spread over a region with radius of order  and hence area of order s, so the effective population size is of order s.
and hence area of order s, so the effective population size is of order s.
Figure 4.—



Comparison of the distribution of r2 for a homogeneously mixing population and a stepping-stone local sample with 4Nm = 10 for distances (a) 10–11 kb, (b) 30–31 kb, and (c) 50–51 kb.
Figure 1.—

Plot of 1 over the coalescence rate for our concrete example. L = 10, N = 25, m = 0.1, 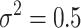 , and β = 0.2. The first phase ends at time
, and β = 0.2. The first phase ends at time 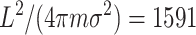 . After that time the value is
. After that time the value is 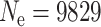 . This graph is similar to simulation results presented in Figure 4 of Wilkins (2004), except that he uses a logarithmic timescale that turns the straight line into an exponential.
. This graph is similar to simulation results presented in Figure 4 of Wilkins (2004), except that he uses a logarithmic timescale that turns the straight line into an exponential.
The remarks in the previous paragraph apply to times s =  with γ ≤ 1. At times ≥L2/4πmσ2 the lineages have had time to spread across the entire space. At this point the coalescence time, which is of order
with γ ≤ 1. At times ≥L2/4πmσ2 the lineages have had time to spread across the entire space. At this point the coalescence time, which is of order  , is much larger than the time, of order L2/mσ2, needed for the random walk on an L × L square that jumps at rate 2m and has variance σ2 to equilibrate in the uniform distribution (Cox and Durrett 2002). Thus the relative positions of lineages are unimportant and the system behaves as if it were homogeneously mixing. Since we have changed Zähle
et al.'s (2005) 2m in the denominator to 4πmσ2, 1 over the coalescence rate increases until it becomes constant in the second regime, and the transition is continuous.
, is much larger than the time, of order L2/mσ2, needed for the random walk on an L × L square that jumps at rate 2m and has variance σ2 to equilibrate in the uniform distribution (Cox and Durrett 2002). Thus the relative positions of lineages are unimportant and the system behaves as if it were homogeneously mixing. Since we have changed Zähle
et al.'s (2005) 2m in the denominator to 4πmσ2, 1 over the coalescence rate increases until it becomes constant in the second regime, and the transition is continuous.
Zähle et al. (2005) were able to compute various quantities for samples of size two under the infinite-sites model including the expected number of pairwise differences and the probability for no coalescence before recombination for two loci with a per generation recombination probability r. They showed that the latter quantity decayed more slowly in the stepping-stone model compared to a homogenously mixing population, but it is hard to relate this to commonly used measures of linkage disequilibrium.
The limit theorem of Zähle et al. (2005) is difficult to use for computations because the coalescence rate changes in time. For the ordinary coalescent one can easily compute the correlation between coalescence times at two loci for samples of size two by considering whether recombination or coalescence occurs first. One can find this result of Griffiths (1981) explained on pp. 80–83 of Durrett (2002). However, in the situation of Zähle et al. (2005) one must also consider the time at which the event occurs, and one can no longer obtain the answer by solving three equations in three unknowns. Thus, to investigate the effect of stepping-stone population structure on samples of size n > 2, we turn to simulations.
METHODS
Model simulation parameters and sampling schemes:
We simulated coalescent models with constant recombination and mutation rates across the locus in homogeneously mixing, island, and stepping-stone models using Hudson's ms program (Hudson 2002). We fix the physical size of our locus to 100 kb and set both the mutation and the recombination rate to be 10−8/nucleotide/generation. Our sample size is fixed for all models at n = 40 chromosomes. In the spatial simulations there are 100 demes and m is the probability that the new individual is a new migrant. In the stepping-stone model, space is a 10 × 10 grid with periodic boundary conditions and with migration to the four nearest neighbors with equal probabilities.
To try to minimize the differences between the spatial structures, we use Ne formulas from Nei and Takahata (1993) and Zähle et al. (2005) given earlier as (1) and (2) to determine the number of diploids per colony, N, so that the computed Ne for the population is ∼10,000. Table 1 lists the scaled migration rate (4Nm), number of diploids per colony (N), and computed effective population sizes (Ne). The definition of effective population size we use here is one-half the average coalescence time of two lineages, so this makes the mean number of pairwise differences the same.
TABLE 1.
Computed effective population size and scaled migration rates for island and stepping-stone models with our simulation parameters
| Model | 4Nm | N | Computed Ne |
|---|---|---|---|
| Island | 1 | 50 | 10,000 |
| Island | 3 | 75 | 10,000 |
| Island | 10 | 91 | 10,100 |
| Stepping stone | 1 | 25 | 9,829 |
| Stepping stone | 3 | 51 | 10,083 |
| Stepping stone | 10 | 77 | 9,957 |
To understand the effect of sampling on the statistics, we employ two different sampling schemes for the island and stepping-stone models: (i) Chromosomes are randomly selected from the population (random sampling) and (ii) 40 chromosomes are sampled from one colony in the stepping-stone model or from one deme in the island model (local sampling). The second sampling strategy corresponds to sampling from one population.
Decay of linkage disequilibrium:
Following Pritchard and Przeworski (2001), we compute the square of the correlation coefficient, which for two loci with two alleles A and a and B and b is
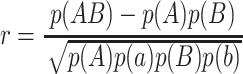 |
for all pairs of segregating sites for which the minor allele frequency is at least 0.2. To average results over the replications, we create bins of size 1000 (0.01 times the length of our region) on the basis of interSNP distance and average the r2 observations in each bin. The number of simulations used to compute averages was 400,000. In addition to investigating the mean values of r2, we examine the distribution of r2-values for distances in the bins [0.1, 0.11], [0.3, 0.31], and [0.5, 0.51], where the distance is measured relative to the length of the locus. Thus we are examining r2 for loci separated by ∼10, 30, and 50 kb. The number of simulations used to determine the distribution of r2 was 350,000.
Site frequency spectrum:
Since the alignment and the ancestral state are known, we can compute for each SNP the observed number of chromosomes i (1 ≤ i ≤ 39) that have the mutant nucleotide. This number is then divided by the total number of segregating sites from all 350,000 replications, to get the site frequency distribution.
SNP density:
We choose our physical sequence length to be 10 kb. The number of segregating sites for 350,000 simulations was tabulated and normalized.
Tajima's (1989) D-statistic and Fay and Wu's (2000) H are calculated for each replication using Hudson's “sample_stats” program, which is included with the ms program. The median, 2.5, and 97.5 percentiles are computed over 350,000 simulations.
RESULTS
To give a visualization of the impact of spatial structure on genetic data, Figure 2 gives sample outcomes for a homogeneously mixing population, an island model local sample with 4Nm = 1, and a stepping-stone local sample with 4Nm = 10. Note that there are more SNPs and many more haplotypes in the homogeneously mixing population compared to the two spatial samples.
Decay of linkage disequilibrium:
As Figure 3 shows, when 4Nm = 1, there is a large difference in the rate of decay of r2 between the homogeneously mixing and the migration models. As expected, the stepping-stone model has considerably more LD than the island model. When 4Nm = 1, the stepping-stone local sample has r2 ≈ 0.9 at a distance of 100 kb, which is considerably larger than values observed in the human genome, but of course our universe is only a 10 × 10 array of colonies. The random samples have a faster decay of r2 than the local samples, but in contrast to the theoretical results quoted above, their behavior is not the same as that of the homogeneously mixing population. One reason for this is that the sample size is n = 40, so n2 = 1600 is much larger than the number of demes, 100, and the assumption of the limit theorem is not justified. A simple calculation for 40 lineages and 100 demes shows that the probability that all lineages will be in separate demes is 0.000122. Using a Poisson approximation with mean 0.4 for the number of lineages in a deme, we see that on the average 5.26 demes will have 2 lineages.
Figure 3.—



Decay of r2 for (a) 4Nm = 1, (b) 4Nm = 3, and (c) 4Nm = 10. In a–c, the expectation for a homogeneously mixing population is given by a line. Island model results are graphed with squares and stepping-stone results with triangles, with solid symbols for random samples and open symbols for local samples.
When 4Nm = 3, values of r2 are somewhat reduced. As expected they are smallest for the random samples and largest for the local samples. When 4Nm = 10, the random samples are close to the homogeneously mixing case, but the curves for local samples are well above the homogeneously mixing decay curve.
Distribution of r2:
Figure 4 compares the distribution of r2 for a homogeneously mixing population and a stepping-stone local sample with 4Nm = 10, for SNPs separated by 10–11, 30–31, or 50–51 kb. Not only is the mean of r2 larger in the stepping-stone model, but also there is a significant probability that r2 = 1, even for SNPs separated by 50–51 kb.
Site frequency spectrum:
As Figure 5 shows, in the case of a random sample, the site frequency spectrum for the island and the stepping-stone model with 4Nm = 1 behaves like a homogeneously mixing population. That is, as Fu (1995) has shown, the probability that k members of the sample have the mutant nucleotide is c/k, where c is a constant that makes the probabilities sum to 1. This is the behavior expected on the basis of the theoretical results for random samples discussed earlier, which show that the genealogy of a random sample converges to that of Kingman's coalescent.
Figure 5.—




Site frequency spectra for (a) random samples with 4Nm = 1 and local samples with (b) 4Nm = 1, (c) 4Nm = 3, and (d) 4Nm = 10. Symbols are as described in the Figure 3 legend.
For the local samples, the site frequency spectra differ from the prediction for a homogeneously mixing population. We see from Figure 5, b–d, that both models have a significant reduction in the proportion of singletons, even when 4Nm = 10. This occurs because there is a greater initial coalescence rate due to the fact that the lineages are sampled from only one subpopulation. The reduction of singletons presents problems for the use of Fu and Li's (1993) D-statistic, which looks for an excess in the frequency of such mutations compared to the neutral expectation.
For 4Nm = 1 and 4Nm = 3, both models exhibit an excess of intermediate- and high-frequency-derived nucleotides compared to the homogeneously mixing model. When 4Nm = 10, the discrepancy at the high-frequency end is almost gone but there are significant differences for rare alleles.
SNP density:
Figure 6 shows that for random samples the SNP density from both migration models match closely the one for a homogeneously mixing population, even when 4Nm = 1. For local samples the SNP density is changed in the spatial models (see Figure 6, b–d) with the number of SNPs shifted toward smaller values and the shift more pronounced for the stepping-stone model than for the island model. Note that when 4Nm = 1, >35% of our 10-kb segments have zero SNPs in the stepping-stone model, compared to 5% for the island model, while this almost never happens in a homogeneously mixing population. The skew in the distribution persists in local samples from both spatial models when 4Nm = 3 and even when 4Nm =10.
Figure 6.—




SNP density under (a) random sampling with 4Nm = 1 and local samples with (b) 4Nm = 1, (c) 4Nm = 3, and (d) 4Nm = 10. Symbols are as described in the Figure 3 legend.
Tajima's D is a statistic that is constructed by subtracting estimates of the scaled mutation rate based on the number of pairwise differences and the number of segregating sites. These components are related to the last two quantities we have investigated, so we should not expect much difference for a random sample, but a much larger one for a local sample. The results reported in Figure 7 follow this pattern. The medians for the random samples are close to the homogeneously mixing values. However, there are significant changes in the upper and lower cutoffs for the local samples and in most cases a dramatic shift toward positive values as shown by the changes in the median and the upper cutoff. This agrees with our previous observation that there is an excess of mutations at intermediate frequencies. In the stepping-stone local samples, and to a lesser extent in the island local samples, this shift is accompanied by an increase in variability that causes the lower tail cutoffs to decrease. This is unfortunate for researchers looking for negative values of Tajima's D as indications of positive selection. This use of Tajima's D has also been shown to be misleading in bottlenecked populations (Thornton and Jensen 2007).
Figure 7.—

Medians and 95% intervals for Tajima's D-statistic for our spatial structures and sampling schemes.
Fay and Wu's H is a statistic constructed by subtracting estimates of the scaled mutation rate based on the number of pairwise differences and another based on the homozygosity of derived variants. The difference is normalized so that the H-statistic has variance 1. The homozygosity of derived variants is influenced most by variants at intermediate and high frequencies, respectively. See pp. 1406 and 1408 of Fay and Wu (2000). Since the most notable affect of spatial structure is to decrease the number of low-frequency alleles, it is not surprising to see in Figure 8 that there are no systematic changes in the median of the H-statistic, but the major effect is to increase the standard deviation and to expand the interval between the two cutoffs, which can result in spurious rejections of the neutral model.
Figure 8.—

Medians and 95% intervals for Fay and Wu's H for our spatial structures and sampling schemes.
DISCUSSION
The spatial distribution of individuals in a local sample in the island model or stepping-stone model causes coalescence to occur more rapidly in the early stages of the genealogy of a local sample. This shifts the site frequency spectrum from rare alleles toward those of intermediate frequency, and we have shown through simulation that this alters the distribution of test statistics of Tajima (1989), Fu and Li (1993), and Fay and Wu (2000). Here we have contrasted a local sample from one population with a sample chosen randomly from the entire population and shown that the local sample will have fewer alleles. In a different direction, Ptak and Przeworski (2002) have shown that taking small samples from a large number of geographic locations can increase the number of rare alleles.
Our simulation results have shown that stepping-stone population structure produces a slow decay of linkage disequilibrium and dramatically increases the probability of perfect correlation; that is, r2 = 1. This change in the two-locus sampling distribution may cause trouble for likelihood methods, such as the ones McVean et al. (2004) have used to estimate recombination rates in humans. Low recombination rates could be assigned to intervals between markers with high correlation, when in reality this is due to spatial structure. In making the last comment, we are not suggesting that when spatial structure is taken into account, all cold spots will suddenly become warm. However, the qualitative picture of recombination rate variability may be significantly changed.
For a concrete example where spatial structure may have contributed to erroneously rejecting neutrality, we consider results of Hamblin and Aquadro (1996) for the glucose dehydrogenase gene based on a sample of 11 Drosophila simulans collected in 1984 in Raleigh, North Carolina. The only test that suggested a pattern of nonneutral evolution was Fu and Li's test. In that case observing one singleton of 26 segregating sites had a probability of P < 0.05 in a two-tailed test, but might not be significant if one took into account that local sampling could itself reduce the number of singleton mutations.
The effect of local population sampling in humans, particularly in European or North American populations may not be as dramatic, since as if one goes back a few dozen generations the lineages disperse over a wide area. However, it could be more problematic when a population has occupied a small area for a long period of time. Sabeti et al. (2002) sampled 73 Beni from Benin City, Nigeria, and sequenced a region around the G6PD locus. To argue that selection had acted on this locus they examined the decay of the extended haplotype homozygosity (EHH), i.e., the probability that two randomly chosen chromosomes carrying the same core haplotype were identical by descent up to that point. To evaluate the likelihood of the observed data, they used Hudson's ms program to simulate homogeneously mixing populations of constant size, with exponential growth, a bottleneck, or a two-island model. However, as we have shown, stepping-stone spatial structure can dramatically reduce the decay of linkage disequilibrium.
We are not the first to have suggested that spatial structure of the human population may have played a role in the spurious detection of positive selection. Mekel-Bobrov et al. (2005) and Evans et al. (2005) showed in studies of abnormal spindle-like microcephaly-associated (ASPM) and microcephalin genes that the presence of haplotypes with unusually large frequencies was caused by positive selection. Currat et al. (2006) argued that human demographic models with structure followed by population growth could explain the observed haplotype frequency patterns. In reply, Mekel-Bobrov et al. (2006) argued that the bottleneck in Currat et al.'s model was unrealistically long and narrow.
While one can debate the impact of spatial structure on genealogies that cannot be directly observed, there is clear evidence that some allele frequencies show pronounced spatial structure: for example, lactase persistence (Bersaglieri et al. 2004; Tishkoff et al. 2007), the CCR5-Δ32 deletion that leads to strong resistance against HIV-1 (Stephens et al. 1998), and the Duffy blood group (Hamblin et al. 2002). One final observation that argues for the importance of spatial structure in shaping patterns of variability is that of Rosenberg et al. (2002), who have shown that with information about a large number of microsatellite loci, one can classify most of the 1056 individuals in a sample from 52 populations into their correct geographical regions.
In the other direction, one might argue that patterns of nucleotide variability are shaped over longer timescales than microsatellites, so only recent mutations will show the effects of population structure. However, this article has clearly shown that the “isolation by distance” in the stepping-stone model has a profound effect on patterns of variability, even when Nm is not much larger than 1, so it should also be considered when one wants to assess the impact of population subdivision on estimation procedures or statistical tests.
Acknowledgments
We thank Jeff Jensen, Nadia Singh, and two anonymous referees for a large number of helpful suggestions. Both authors have been partially supported by a joint National Institute of General Medical Sciences/National Science Foundation grant (DMS 0201037).
References
- Bersaglieri, T., P. C. Sabeti, N. Patterson, T. Vanderploeg, S. F. Schaffner et al., 2004. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 74 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., F. DePaulis and A. M. Etheridge, 2002. Neutral evolution in a spatially continuous population. Theor. Popul. Biol. 61 31–48. [DOI] [PubMed] [Google Scholar]
- Charlesworth, B., D. Charlesworth and N. H. Barton, 2003. The effects of genetic and geographic structure on neutral variation. Annu. Rev. Ecol. Evol. Syst. 34 99–125. [Google Scholar]
- Cox, J. T., and R. Durrett, 2002. The stepping stone model: new formulas expose old myths. Ann. Appl. Probab. 12 1348–1377. [Google Scholar]
- Currat, M., L. Excoffier, W. Maddison, S. P. Otto, N. Ray et al., 2006. Comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens” and “Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans” Science 313 172a. [DOI] [PubMed] [Google Scholar]
- Durrett, R., 2002. Probability Models for DNA Sequence Evolution. Springer, New York.
- Evans, P. D., S. L. Gilbert, N. Mekel-Bobrov, E. J. Vallender, J. R. Anderson et al., 2005. Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans. Science 309 1717–1720. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. X., 1995. Statistical properties of segregating sites. Theor. Popul. Biol. 48 172–197. [DOI] [PubMed] [Google Scholar]
- Fu, Y. X., and W. H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, R. C., 1981. Neutral two-locus multiple allele models with recombination. Theor. Popul. Biol. 19 169–186. [Google Scholar]
- Hamblin, M. T., and C. F. Aquadro, 1996. High nucleotide sequence variation in a region of low recombination in Drosophila simulans is consistent with the background selection model. Mol. Biol. Evol. 13 1133–1140. [DOI] [PubMed] [Google Scholar]
- Hamblin, M. T., E. E. Thompson and A. DiRienzo, 2002. Complex signatures of natural selection at the Duffy blood group locus. Am. J. Hum. Genet. 70 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R., 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Kimura, M., and T. Maruyama, 1971. Patterns of neutral polymorphism in a geographically structured population. Genet. Res. 18 125–131. [DOI] [PubMed] [Google Scholar]
- Lessard, S., and J. Wakeley, 2004. The two-locus ancestral graph in a subdivided population: convergence as the number of demes grow in the island model. J. Math. Biol. 48 275–292. [DOI] [PubMed] [Google Scholar]
- McVean, G. A. T., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine scale structure of recombination rate variation in the human genome. Science 304 581–584. [DOI] [PubMed] [Google Scholar]
- Mekel-Bobrov, N., S. L. Gilbert, P. D. Evans, E. J. Vallender, J. R. Anderson et al., 2005. Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens. Science 309 1720–1722. [DOI] [PubMed] [Google Scholar]
- Mekel-Bobrov, N., P. D. Evans, S. L. Gilbert, E. J. Vallender, R. R. Hudson et al., 2006. Response to comment on “Ongoing adaptive evolution of ASPM, a brain size determinant in Homo sapiens” and “Microcephalin, a gene regulating brain size, continues to evolve adaptively in humans”. Science 313 172b. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 1980. The strong-migration limit in geographically structured populations. J. Math. Biol. 9 101–114. [DOI] [PubMed] [Google Scholar]
- Nei, M., and N. Takahata, 1993. Effective population size, genetic diversity, and coalescence time in subdivided populations. J. Mol. Evol. 37 240–244. [DOI] [PubMed] [Google Scholar]
- Notohara, M., 1993. The strong-migration limit for the genealogical process in geographically structured populations. J. Math. Biol. 31 115–122. [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak, S. E., and M. Przeworski, 2002. Evidence for population growth in humans is confounded by fine-scale population structure. Trends Genet. 18 559–563. [DOI] [PubMed] [Google Scholar]
- Reich, D. E., S. F. Schaffner, M. J. Daly, G. McVean, J. C. Mullikin et al., 2002. Human genome sequence variation and the influence of gene history, mutation, and recombination. Nat. Genet. 32 135–142. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A., J. K. Pritchard, J. L. Weber, H. M. Cann, K. K. Kidd et al., 2002. Genetic structure of human populations. Science 298 2381–2385. [DOI] [PubMed] [Google Scholar]
- Sabeti, P. C., D. E. Reich, J. M. Higgins, H. Z. P. Levine, D. J. Richter et al., 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419 832–837. [DOI] [PubMed] [Google Scholar]
- Stephens, L. C., D. E. Reich, D. B. Goldstein, H. D. Shin, M. W. Smith et al., 1998. Dating the origin of the CCR5-Δ32 AIDS-resistance allele by the coalescence of haplotypes. Am. J. Hum. Genet. 62 1507–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, K. R., and J. D. Jensen, 2007. Controlling the false positive rate in multi-locus genome scans for selection. Genetics 175 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishkoff, S. A., F. A. Reed, A. Ranciaro, B. F. Voight, C. C. Babbit et al., 2007. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 39 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., 1998. Segregating sites in Wright's island model. Theor. Popul. Biol. 53 166–174. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 1999. Nonequilibrium migration in human history. Genetics 153 1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley, J., and S. Lessard, 2003. Theory of the effects of population structure and sampling on patterns of linkage disequilibrium applied to genomic data from humans. Genetics 164 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, J., 2004. A separation-of-timescales approach to the coalescent in a continuous population. J. Math. Biol. 31 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zähle, I., J. T. Cox and R. Durrett, 2005. The stepping stone model, II. Genealogies and the infinite sites model. Ann. Appl. Probab. 15 671–699. [Google Scholar]