

Abstract
A number of hormones and growth factors stimulate gene expression by promoting the phosphorylation of CREB (P-CREB), thereby enhancing its association with the histone acetylase paralogs p300 and CBP (CBP/p300). Relative to cAMP, stress signals trigger comparable amounts of CREB phosphorylation, but have minimal effects on CRE-dependent transcription. Here, we show that the latent cytoplasmic coactivator TORC2 mediates target gene activation in response to cAMP signaling by associating with CBP/p300 and increasing its recruitment to a subset of CREB target genes. TORC2 is not activated in response to stress signals, however; and in its absence, P-CREB is unable to stimulate CRE-dependent transcription, due to a block in CBP recruitment. The effect of TORC2 on CBP/p300 promoter occupancy appears pivotal because a gain of function mutant CREB polypeptide with increased affinity for CBP restored CRE-mediated transcription in cells exposed to stress signals. Taken together, these results indicate that TORC2 is one of the long sought after cofactors that mediates the differential effects of cAMP and stress pathways on CREB target gene expression.
Keywords: cAMP, CBP, CREB, p300, TORC2
Introduction
Most of the nearly 1000 transcription factors (TFs) in the mammalian genome are thought to be regulated in some manner by phosphorylation (Holmberg et al, 2002). Wedged between the signaling machinery and transcriptional apparatus, TFs employ different strategies to transmit signaling information to relevant target genes (Gardner and Montminy, 2005). Regulatory phosphorylation sites are usually found within or adjacent to transactivation domains, where they modulate TF activity through nuclear shuttling, DNA binding, or coactivator recruitment. Despite significant progress in understanding the biochemical and structural mechanisms underlying these events, however, key issues in transcriptional specificity remain unresolved. How do extracellular signals with apparently equal effects on TF phosphorylation elicit different transcriptional responses?
The transcription factor CREB is activated by a number of growth factors and hormones that trigger its phosphorylation at Ser133 and association with the coactivator paralogs CBP and p300 (Shaywitz and Greenberg, 1999; De Cesare and Sassone-Corsi, 2000; Impey and Goodman, 2001; Mayr and Montminy, 2001; Lonze and Ginty, 2002). Although they trigger CREB phosphorylation with comparable stoichiometry and kinetics, growth factor and stress pathways are ineffective in promoting transcription through a CREB binding site relative to cAMP agonists (Fisch et al, 1987; Bonni et al, 1995; Brindle et al, 1995).
Recently, a new family of CREB coactivators called transducers of regulated CREB (TORCs) (Conkright et al, 2003; Iourgenko et al, 2003; Bittinger et al, 2004; Screaton et al, 2004) has been found to increase the expression of cAMP responsive genes. Under basal conditions, TORCs are sequestered in the cytoplasm through phosphorylation by the salt inducible kinase 2 (SIK2) and other members of the AMPK family of Ser/Thr kinases (Koo et al, 2005; Shaw et al, 2005; Katoh et al, 2006). Following exposure to cAMP, TORC2 (CRTC2) is dephosphorylated and translocated to the nucleus, where it associates with CREB and increases target gene expression.
Although cAMP stimulates both CREB and TORC2 activities by phosphorylation and dephosphorylation, respectively, the extent to which these pathways overlap is unclear. Indeed, whether TORC2 is modulated as well by stress and growth factors has not been investigated. Here, we show that TORC2 functions as a switch that promotes the selective expression of cAMP but not stress inducible genes through cooperative interactions with CBP/p300. Following exposure to cAMP, TORC2 was dephosphorylated and translocated to the nucleus, where it stimulated transcription by associating directly with CBP/p300 and enhancing its occupancy over relevant CREB target genes. Indeed, CBP/p300 was also required for recruitment of TORC2 following exposure to cAMP, indicating that the TORC2:CBP/p300 complex has reciprocal effects on the recruitment of both proteins to the promoter. Taken together, these results demonstrate how cooperative interactions between two coactivators lead to the expression of specific genes via a ubiquitous transcription factor.
Results
We compared the relative effects of cAMP agonist and the phorbol ester TPA on CREB activity. Exposure of HEK293T cells to forskolin (FSK) or TPA increased amounts of P-CREB to the same extent (Figure 1A, top), but TPA had no effect on a CRE-luciferase reporter relative to FSK, which induced reporter activity by 20-fold in transient transfection assays (Figure 1A, bottom). In gene profiling studies of HEK293T cells, FSK and TPA triggered distinct subsets of cellular genes that contain cAMP response elements, are occupied by CREB in vivo and are downregulated following RNAi-mediated knockdown of CREB (Zhang et al, 2005) (Figure 1B and C; Supplementary Figures 1 and 2 and Supplementary Table 1).
Figure 1.
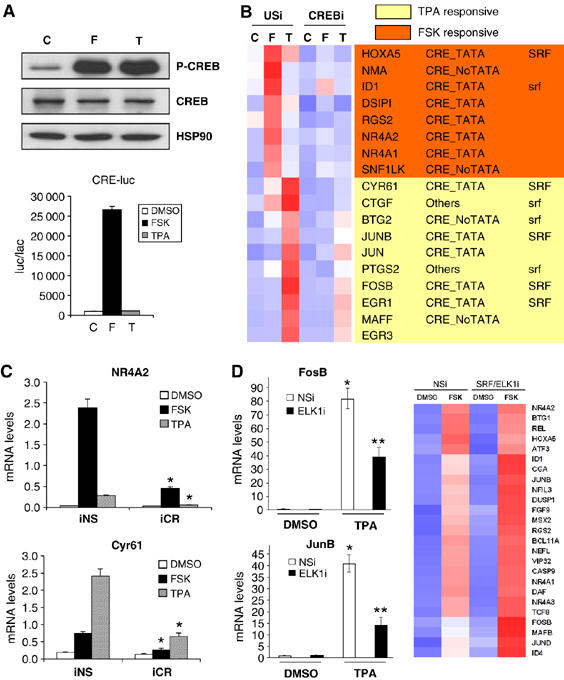
cAMP and stress signals trigger distinct CREB-regulated gene programs. (A) Relative effect of FSK(10 μM, 20 min) and TPA (20 nM, 20 min) on amounts of phospho (Ser133) CREB (P-CREB) (top) and on CRE-luciferase (CRE-luc) reporter activity in HEK293T cells (bottom). (B) Heat map from gene profiling assay of HEK293T cells exposed to FSK or TPA. Effect of CREB knockdown with CREB RNAi shown relative to unspecific (US) RNAi expression vector. Effect of CREB RNAi on amounts of CREB protein shown in Supplementary Figure 1. Presence of conserved (mouse, rat, human) CRE and TATA box (CRE-TATA/ CRE-noTATA); SRF binding sites that are conserved in mouse, rat and human orthologs (SRF) or present only in human (srf) indicated. Gene profiling data provided in Supplementary Table 1. (C) Q-PCR analysis of genes induced selectively by FSK (NR4A2) or TPA (Cyr61) in gene profiling assay. Effect of CREB and nonspecific RNAi (iCR, iNS) on mRNA levels shown. Treatment with FSK, TPA or vehicle as indicated (*significantly different from iNS (P<0.05); n=3). (D) Left: Q-PCR assay showing effect of ELK1 RNAi on mRNA amounts for stress-inducible (FosB, JunB) genes under basal conditions (DMSO) and in response to TPA. Effect of ELK1 RNAi on amounts of ELK1 protein shown in Supplementary Figure 1. Right: heat map from gene profiling assay showing effect of nonspecific or SRF+ELK1 RNAis on expression of CREB target genes in HEK293T cells exposed to FSK as indicated (* and ** differ significantly (P<0.05) and from DMSO (two-way ANOVA; n=3)). Gene profiling data included in Supplementary Table 1.
CREB has been found to stimulate a number of growth factor and stress responsive genes by cooperating with the serum response factor (SRF) and the ternary complex factor TCF/ELK1 (Kitabayashi et al, 1993; Perez-Albuerne et al, 1993; Bonni et al, 1995; Ramanan et al, 2005). Consistent with these findings, most of the TPA inducible CREB target genes also contained SRF binding sites but cAMP inducible targets did not (Figure 1B). Moreover, inhibiting SRF/ELK1 activity, either by deletion of SRF binding sites or by RNAi-mediated knockdown of ELK1 and SRF, disrupted target gene activation in response to TPA (Figure 1D; Supplementary Figures 1, 3 and 4). By contrast, knockdown of ELK1 and SRF actually increased target gene activation by FSK, suggesting that SRF and ELK1 exert counter-regulatory effects on cAMP-dependent transcription (Figure 1D, right; Supplementary Table 1).
Having seen that stress signals are less active in stimulating CRE-dependent transcription compared with cAMP, we performed a mutagenesis screen to identify CREB polypeptides that are capable of rescuing CRE reporter activity under these conditions. Out of 200 independent mutants, we found two CREB polypeptides (Ser142Phe, Ser142Leu) that stimulated transcription from a CREB binding site comparably in response to either FSK or TPA (Figure 2A; Supplementary Figure 5). In gene profiling studies of HEK293T cells exposed to TPA, Ser142Leu CREB also increased the expression of 20 target genes relative to wild-type CREB (Figure 2B; Supplementary Table 1).
Figure 2.
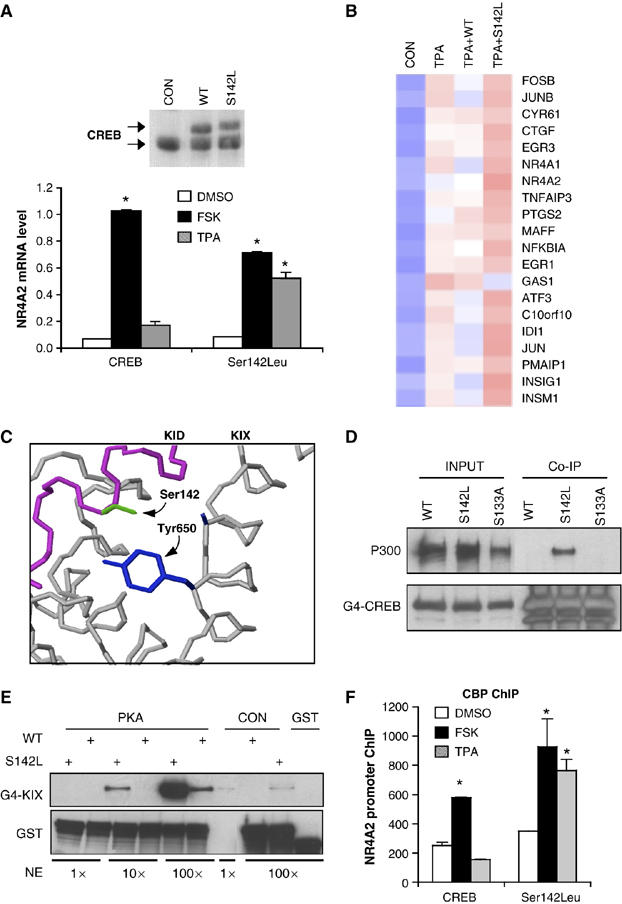
A gain-of-function CREB mutant with increased affinity for CBP/p300 stimulates CRE-dependent transcription comparably in response to cAMP and stress signals. (A) Top: Western blot assay amounts of endogenous and retroviral epitope-tagged wild-type (WT) or Ser142Leu (S142L) mutant CREB in infected HEK293T cells. Bottom: Q-PCR assay comparing effects of FSK and TPA on NR4A2 mRNA levels in HEK293T cells expressing WT and Ser142Leu mutant CREB (*significantly different from DMSO (P<0.05); n=2). (B) Heat map showing relative effects of WT and S142L mutant CREB on target gene expression compared to control (untransfected) HEK293T cells following exposure to TPA. Blue color indicates low-level expression and red indicates high mRNA expression. Gene profiling data included in Supplementary Table 1. (C) Ser142 in CREB is directed toward a hydrophobic pocket in the KIX domain of CBP. Partial structure of the KID/KIX complex showing relative position of Ser142 in CREB pointing toward a hydrophobic pocket containing Tyr650 in KIX. (D) Western blot assay showing amounts of endogenous p300 recovered from IPs of WT, phosphorylation-defective (S133A), and S142L mutant GAL4-CREB (aa 1–283) polypeptides, lacking the TORC2 binding domain (bZIP; aa 283–341) in HEK293T cells cotransfected with PKA expression plasmid. Input amounts of GAL4-CREB fusion proteins are shown. (E) Pull-down assays showing recovery of a CBP KIX domain polypeptide (G4-KIX) from nuclear extracts following incubation with glutathione–sepharose beads containing WT or Ser142Leu mutant GST-CREB (aa 1–283) proteins which lack the TORC2 interacting bZIP domain (aa 283–341). Effect of PKA phosphorylation of GST-CREB on KIX binding is indicated. Incubation with increasing amounts of nuclear extract (1 × , 10 × , 100 ×) is shown. (F) ChIP assay showing effect of WT and Ser142Leu CREB on recruitment of CBP to the NR4A2 promoter following treatment with FSK or TPA. (*significantly different (P<0.05) from DMSO control; n=2).
The NMR structure of the CREB:CBP complex, using relevant interaction domains called KID and KIX, indicates that Ser142 projects into a hydrophobic pocket in KIX (Radhakrishnan et al, 1997), and hydrophobic substitutions at this site would be predicted to stabilize the CREB:CBP interaction (Figure 2C). Supporting this idea, Ser142Leu CREB polypeptides were found to associate with p300 with 5- to 10-fold higher affinity compared with wild-type CREB by co-immunoprecipitation (co-IP) and GST pull-down assays (Figure 2D and E). As with wild-type CREB, phosphorylation of Ser142Leu CREB at Ser133 was also required for CBP binding and transcriptional activation (Figure 2E; Supplementary Figure 6). Indeed, wild-type and Ser142Leu mutant CREB proteins were also phosphorylated at Ser133 to the same extent in cells exposed to FSK or TPA (Supplementary Figure 7).
Based on its higher affinity for CBP/p300, Ser142Leu mutant CREB would be predicted to increase CBP/p300 recruitment to the promoter in response to stress signals. Following exposure to FSK, amounts of CBP over the NR4A2 promoter were increased by three-fold in cells expressing either wild-type or Ser142LeuCREB by chromatin immunoprecipitation assay (ChIP; Figure 2F). Exposure to TPA actually decreased CBP occupancy somewhat in cells expressing wild-type CREB, but it increased CBP recruitment three-fold in cells expressing Ser142Leu (Figure 2F). Taken together, these results support the notion that the recruitment of CBP is limiting for the induction of target genes by P-CREB in response to stress signals.
Selective induction of TORC2 by cAMP
The ability of cAMP but not stress signals to stimulate CBP recruitment and CRE-dependent transcription points to the potential involvement of a second cAMP-regulated cofactor in this process. In line with this notion, the TORC family of latent cytoplasmic coactivators has been found to increase the expression of CREB target genes following their dephosphorylation and nuclear translocation in response to cAMP (Screaton et al, 2004), although the relative effects of stress signals on TORC activity are unknown.
Amounts of TORC2 mRNA were 3- to 5-fold higher in HEK293T cells compared with other family members (TORC1 and TORC3; Supplementary Figure 8), prompting us to evaluate effects of cAMP and stress pathways on TORC2 shuttling and phosphorylation. Under basal conditions, endogenous and Flag epitope-tagged TORC2 (F-TORC2) proteins were primarily localized in the cytoplasm (Figure 3A; Supplementary Figure 9). Exposure to FSK triggered the inhibitory phosphorylation of the TORC2 kinase SIK2 at Ser587 (Screaton et al, 2004), leading to the subsequent dephosphorylation and nuclear translocation of TORC2 (Figure 3A top, middle and bottom; Supplementary Figures 9 and 10). By contrast, exposure to TPA had negligible effects on amounts of phospho-Ser587 SIK2 as well as on TORC2 phosphorylation or localization. Pointing to a general effect on the TORC family, exposure to FSK also promoted the dephosphorylation of other TORC family members (TORC1, TORC3) but TPA did not (Supplementary Figure 11).
Figure 3.
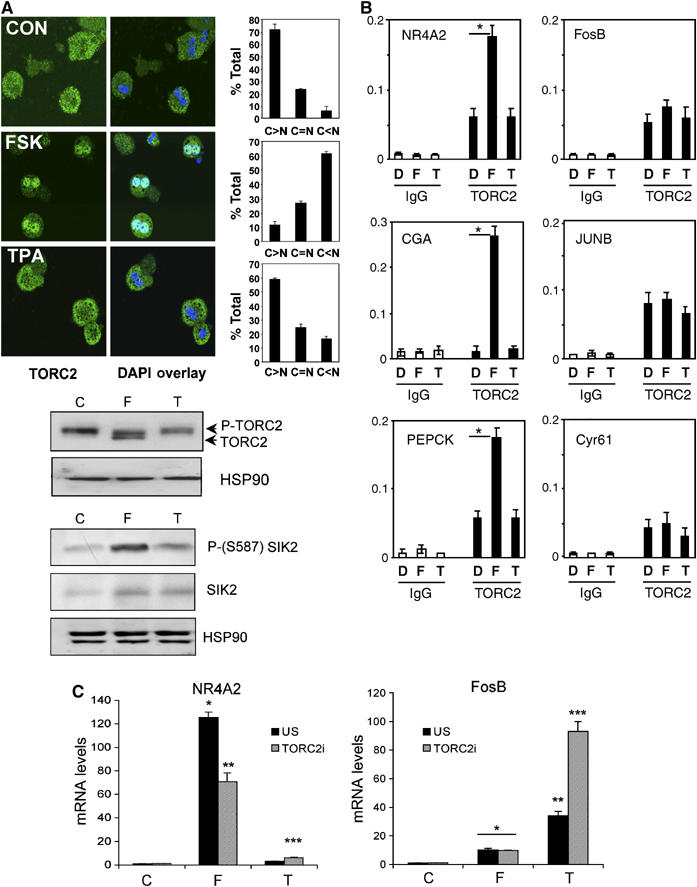
The coactivator TORC2 mediates the selective induction of cAMP but not stress responsive CREB target genes. (A) Relative effects of TPA and FSK on subcellular localization and phosphorylation status of TORC2. Top: immunofluorescence analysis of endogenous TORC2 protein in cultured primary mouse hepatocytes exposed for 30 min to FSK (10 μM), TPA (20 nM) or DMSO vehicle. DAPI staining shows double nuclei, which are characteristic of hepatocytes (Kachi and French, 1994). Right: bar graphs showing proportion of cells with nuclear (N>C), cytoplasmic (N<C) or ubiquitous (N=C) staining (out of 100 cells counted on each of two different slides for each condition). Middle: Western blot assay showing amounts of phospho- and dephospho-TORC2 in cells exposed to FSK, TPA or control vehicle (20 min). Phospho- and unphospho-TORC2 bands are indicated. Bottom: Western blot showing amounts of total and phospho (Ser587) SIK2 in HEK293T cells exposed to FSK, TPA or control vehicle (20 min). (B) ChIP assay of HEK293T cells showing relative effects of TPA (T) and FSK (F) compared with DMSO vehicle (D) on recruitment of endogenous TORC2 to cAMP responsive (left; NR4A2, CGA, PEPCK) or TPA inducible (right; FosB, JunB, Cyr61) CREB target genes (*significantly different from DMSO (P<0.05); n=2). ChIPs with control IgG are shown. Results are representative of three independent experiments. (C) Effect of TORC2 (TORC2i) or nonspecific (US) RNAi on amounts of mRNA for cAMP inducible (NR4A2) and TPA responsive (FosB) CREB target genes (*, **, and *** differ significantly from each other (P<0.05) and from control (C) (two-way ANOVA; n=3)).
Following its nuclear entry, TORC2 has been shown to stimulate cellular gene expression by associating with CREB on relevant promoters (Screaton et al, 2004; Koo et al, 2005). Consistent with this idea, exposure of HEK293T cells to FSK stimulated TORC2 recruitment to cAMP responsive (NR4A2, CGA, PEPCK) but not stress responsive (FosB, JunB, Cyr61) genes (Figure 3B). Amounts of TORC1 protein in HEK293T cells were too low to monitor promoter occupancy, but exposure to FSK also increased recruitment of TORC3 selectively to cAMP responsive genes, whereas exposure to TPA did not (Supplementary Figure 12).
Consistent with results from ChIP studies, RNAi-mediated knockdown of TORC2 reduced the expression of these cAMP inducible genes, but actually increased the expression of the stress responsive genes in cells exposed to TPA (Figure 3C). Taken together, these results indicate that members of the TORC family are selectively activated by cAMP but not stress signals. In addition to mediating induction of cAMP responsive genes, TORCs may also interfere with the expression of stress responsive CREB target genes.
TORC2 promotes CBP recruitment to the promoter
Having seen that TORC2 is required for the expression of cAMP responsive genes, we tested whether TORC2 activation is also sufficient in this regard. To address this question, we used the Ser/Thr kinase inhibitor staurosporine (STS), which has been shown to block SIK2 activity (Katoh et al, 2006). At low doses (10–30 nM), STS promoted TORC2 dephosphorylation in HEK293T cells without increasing amounts of P-CREB (Figure 4A). Consistent with these effects, exposure to STS increased the expression of cAMP responsive (NR4A2, CGA) but not stress inducible genes (FosB, Cyr61, JunB) (Figure 4B; Supplementary Figure 13); these effects were blocked by RNAi-mediated knockdown of TORC2 (Supplementary Figure 13).
Figure 4.
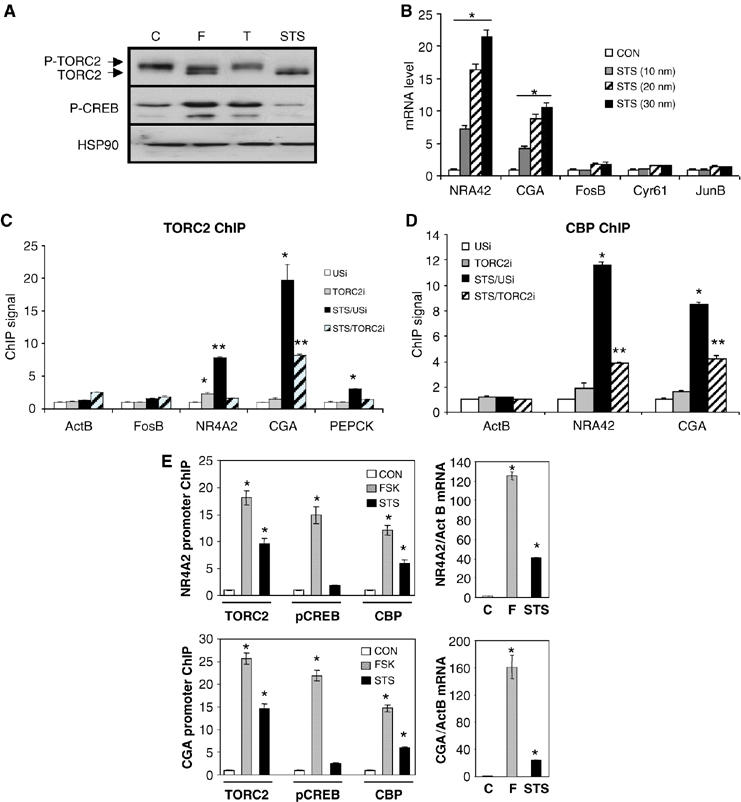
TORC2 dephosphorylation is sufficient to promote CBP recruitment and transcription of cAMP responsive CREB target genes. (A) Western blot assay showing amounts of phospho- and unphospho-TORC2 compared with P-CREB in HEK293T cells exposed to cAMP agonist (F: 10 μM), TPA (T: 20 nM) or staurosporine (STS: 30 nM) for 1 h. Amounts of HSP90 shown for comparison. (B) Q-PCR assay showing effects of STS on mRNA levels for cAMP responsive (NR4A2, CGA) or TPA inducible (FosB, Cyr61, JunB) genes in HEK293T cells exposed to STS (0, 10, 20, 30 nM) for 2 h (* for each gene significantly different from DMSO control (P<0.05); n=3). (C) ChIP assay showing effect of STS (30 nM) on recruitment of TORC2 to cAMP responsive (NR4A2, CGA, PEPCK), TPA-responsive (FosB), or constitutively active (ActB) promoters. Effect of unspecific (USi) and TORC2 RNAi on TORC2 occupancy is indicated (* and ** for each gene differ significantly from each other (P<0.05) and from USi (two-way ANOVA; n=2)). (D) Effect of STS (30 nM) on recruitment of CBP to cAMP responsive (NR4A2 and CGA) or control (ActB) promoters. Effect of unspecific and TORC2 RNAi on CBP occupancy is indicated. (E) Left: ChIP assay showing relative effects of FSK, STS (30 nM) and DMSO vehicle (30 min stimulation) on occupancy of TORC2, P-CREB and CBP over cAMP responsive NR4A2 and CGA genes. Right: Q-PCR analysis of NR4A2 and CGA mRNA levels from HEK293T cells following stimulation with FSK or STS (*significantly different from DMSO control (P<0.05); n=3).
In line with these changes in cellular gene expression, exposure to STS also stimulated recruitment of TORC2 to cAMP responsive (NR4A2, CGA, PEPCK) but not stress inducible (FosB) or constitutive (ActB) promoters (Figure 4C–E). Remarkably, amounts of CBP associated with NR4A2 and CGA promoters also increased 8- to 10-fold in response to STS by ChIP assay, and these effects were blocked by RNAi-mediated knockdown of TORC2 (Figure 4D). Highlighting the importance of CREB phosphorylation, STS was significantly less potent than FSK in stimulating CBP recruitment as well as target gene expression (Figure 4E). Taken together, these results indicate that TORC2 dephosphorylation is indeed sufficient for transcriptional activation, but that CREB phosphorylation also contributes significantly in this regard.
We explored the potential connection between CBP and TORC2 further by comparing the relative effects of cAMP on recruitment of these coactivators to a CREB target gene. Amounts of TORC2 over the cAMP responsive CGA promoter increased as early as 10 min after FSK stimulation, followed by increases in P-CREB (15 min) and lastly CBP (20 min; Figure 5A) by ChIP assay.
Figure 5.
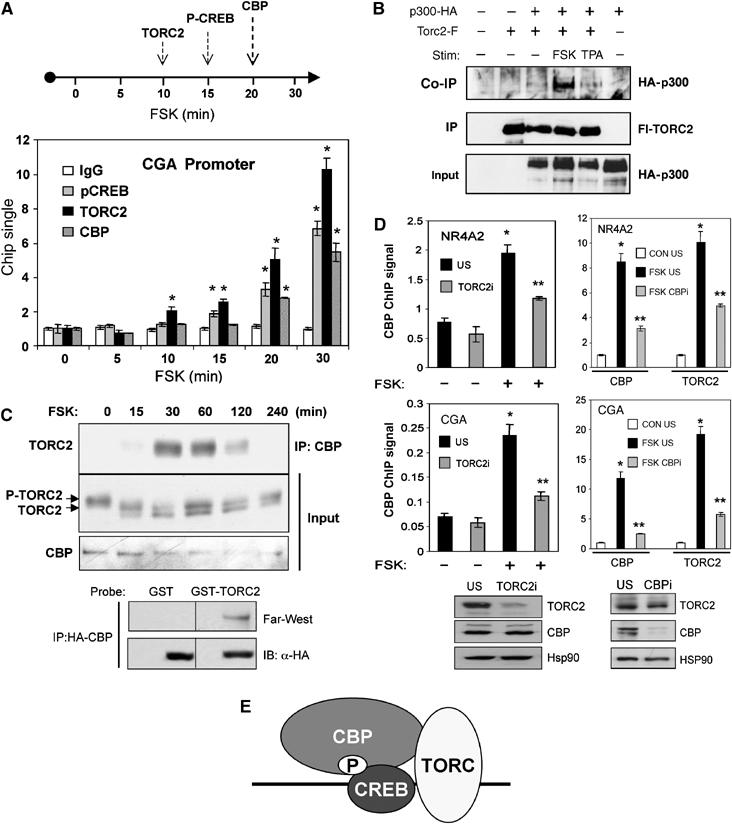
TORC2 is required for recruitment of CBP/p300 to cAMP responsive promoters. (A) ChIP assay of HEK293T cells exposed to FSK for the times indicated. Amounts of P-CREB, TORC2 and CBP associated with the cAMP inducible CGA promoter at each time point shown (*significantly different from t=0; P<0.05; n=2). Time line showing relative kinetics of TORC2, P-CREB and CBP recruitment to the CBP promoter indicated above. (B) Western blot assay of immunoprecipitates from Fl-TORC2 showing amounts of HA-tagged p300 associated with TORC2 following exposure of HEK293T cells to FSK (F: 10 μM) or TPA (T: 100 nM) for 1 h. Input levels of Fl-TORC2 and HA-p300 are shown. (C) Top: effect of FSK on the association of endogenous TORC2 with CBP in primary mouse hepatocytes. Western blot assay showing amounts of TORC2 in IPs of CBP recovered from cells following exposure to FSK for times indicated (in minutes). Input: amounts of endogenous phospho and unphospho-TORC2 and CBP proteins are shown. Bottom: Far-Western blot assay of HA-CBP immunoprecipitates prepared from HEK293T cells. Total amount of CBP in each IP is indicated. Blots were incubated with recombinant GST-TORC2 or GST proteins, and then probed with anti-GST antiserum. (D) Effect of TORC2 RNAi (left) and CBP RNAi (right) compared to nonspecific RNAi (US) on amounts of CBP and TORC2 associated with the cAMP responsive NR4A2 and CGA promoters under basal conditions and following FSK stimulation (* and ** for each gene differ significantly from each other (P<0.05) and from USi (two-way ANOVA; n=2)). Bottom: Western blots showing effects of each RNAi on amounts of TORC2 and CBP proteins. (E) cAMP stimulates CREB target gene expression through cooperative interactions between CBP and TORC2.
The delayed kinetics of CBP recruitment relative to TORC2 suggested that TORC2 may stabilize CBP occupancy over the promoter following cAMP stimulation. Supporting this idea, we identified CBP and p300 in immunoprecipitates of F-TORC2 with significant sequence coverage (CBP: 5%, p300: 5.2%; Supplementary Figure 14) by MS/MS analysis. We confirmed the TORC2:CBP interaction using epitope-tagged TORC2 and CBP/p300 proteins. Indeed, HA-p300 was recovered from immunoprecipitates of Flag-tagged TORC2 (Fl-TORC2) prepared from HEK293T cells exposed to FSK but not to TPA (Figure 5B).
Mirroring the kinetics of CBP recruitment in ChIP assays, exposure of primary cultured mouse hepatocytes to FSK also triggered the association of endogenous dephosphorylated and therefore active TORC2 with endogenous CBP after 30–60 min (Figure 5C). The TORC2:CBP interaction appeared direct; in Far-Western blot assays, recombinant GST-TORC2 protein was found to associate with full-length HA-CBP protein that had been expressed and immunoprecipitated from HEK293T cells (Figure 5C, bottom).
Having seen that cAMP triggers an interaction between CBP and TORC2, we tested whether these coactivators exert reciprocal effects on promoter occupancy. Exposure to FSK triggered recruitment of both CBP and TORC2 to cAMP responsive NR4A2 and CGA genes 5- to 10-fold by ChIP assay (Figure 5D). RNAi-mediated knockdown of TORC2 decreased amounts of CBP associated with both promoters (Figure 5D, left). Conversely, knockdown of CBP also reduced TORC2 occupancy (Figure 5D, right), indicating that cAMP but not stress signals trigger CRE-dependent transcription, in part, through the assembly of a stable CBP:TORC2 coactivator complex over specific genes (Figure 5E).
Discussion
The importance of Ser133 phosphorylation in stimulating CREB activity has been appreciated for almost 20 years. Up to 300 different signals have been shown to trigger CREB phosphorylation, often with comparable stoichiometry and kinetics (Johannessen et al, 2004a). Biochemical and structural studies have suggested that CREB phosphorylation may be sufficient for CBP recruitment (Radhakrishnan et al, 1997; Asahara et al, 2001).
Based on the discrepancy between CREB phosphorylation and CRE-dependent transcription in response to stress and mitogenic signals, however, we and others have proposed that a second cAMP-regulated event triggers cellular gene expression by increasing the CREB:CBP interaction (Brindle et al, 1993; Bonni et al, 1995). Further support for this idea emerged from studies of knock-in mice with mutations in CBP/p300 that block the interaction with P-CREB yet show only modest changes either in CBP/p300 recruitment or in cAMP-dependent transcription (Kasper et al, 2002; Xu et al, 2006).
We found that TORC2 contributes significantly to the relative potency of cAMP compared with stress signals on CREB-dependent transcription. TORC2 is dephosphorylated and shuttled to the nucleus, where it increases gene expression by associating with CBP in response to cAMP but not TPA. Other TORC family members perform somewhat redundant functions in this regard (Takemori et al, 2007), as TORC1 and TORC3 were also preferentially activated by cAMP but not stress signals.
We observed that TORC2 enhances CREB activity in part by stabilizing CBP occupancy over the promoter. Using mutant CREB polypeptides that are defective in either CBP or TORC2 binding, Xu et al (2006) also found evidence for reciprocity between these coactivators at the level of promoter recruitment. As a result, exposure to TPA did not promote CBP recruitment to promoters with only CREB binding sites due to a block in TORC2 translocation. The ability of a gain-of-function CREB mutant (Ser142Leu) with increased affinity for CBP to bypass this block supports the idea that TORC2 functions in a similar capacity. Notably, the coactivator MLL has also been found to increase the CREB:CBP interaction by associating with CBP (Ernst et al, 2001; Goto et al, 2002). Although its role in cAMP or stress signaling is unclear, we imagine that MLL may similarly modulate CREB target gene expression in a different setting.
Other CREB coactivators such as ACT (Fimia et al, 2000) and TAF4 (Saluja et al, 1998; Asahara et al, 2001) may also regulate CREB activity in response to extracellular signals (Johannessen et al, 2004b). Indeed, TAF4 appears to be critical for CREB target gene activation in response to cAMP (Mengus et al, 2005), although the degree to which the CREB:TAF4 interaction is modulated by cellular signals is unclear. TAF4, ACT, and perhaps other effectors may further stabilize the assembly of coactivator complexes over relevant CREB target genes in specific cell types.
In addition to the cooperative effects of CBP and TORC2, we also observed inhibitory interactions that contribute to the selective induction of CREB target genes by cAMP. TORC2 was recruited to only a subset of cAMP but not TPA inducible promoters, which are nevertheless comparably occupied by CREB. Although the underlying mechanism is unclear, knockdown studies suggest that SRF and ELK1 may function as TORC2 inhibitors. Notably, TCF/ELK1 has been shown to disrupt smooth muscle gene expression by competing with the coactivator myocardin for binding to SRF (Wang et al, 2004). Future studies should reveal whether ELK1 and SRF also inhibit cAMP-dependent transcription by associating with either CREB or TORC2.
Gene-specific effects may also contribute to the selective pattern of TORC2 recruitment (Xu et al, 2006). Because TORC2 associates with the CREB bZIP DNA binding and dimerization domains (Screaton et al, 2004), the sequence or orientation of CREB binding sites may well modulate CREB:TORC2 complex formation. In a recent study, for example, TORCs were found to have variable effects on the expression of steroidogenic CREB target genes in adrenocortical cells (Takemori et al, 2007). Relevant to this point, the NF-kB family of activators has been shown to recruit distinct coactivators to different genes depending on the sequence of NF-kB binding sites (Leung et al, 2004). Future comparisons of CREB binding sites from cAMP and stress responsive promoters may provide further insight in this regard.
Taken together, our studies explain, in part, how CREB triggers expression of different programs in response to signals that have comparable effects on amounts of P-CREB. The importance of a second cAMP-regulated effector, TORC2, for CBP recruitment and target gene activation illustrates a two-hit mechanism that allows to discriminate between cAMP and stress signals. Future studies should illuminate the process by which cAMP promotes TORC recruitment to only a subset of CREB occupied genes.
Materials and methods
Cell culture
HEK293T cells were maintained in Dulbecco's modified MEM with 10% fetal calf serum and exposed to FSK (10 μM), TPA (20 nM), or STS (30 nM), unless indicated otherwise. Cells were passaged every second day and maintained at 30–80% confluence(Mayr et al, 2001; Conkright et al, 2003). Primary hepatocytes were isolated as described previously (Dentin et al, 2004) and kept in Medium 199 with 2 mM GlutaMAX-I.
Plasmids and oligos
The U6 promoter-shTORC2, -shCREB and U6 promoter-shNon-specific RNAi cassette were generated from the parental pBSU6 plasmids (Conkright et al, 2003) and cloned into the NheI site of the pLenti-CMV-GFP plasmid (gift from I Verma). HEK293T cells were used along with the appropriate lentivirus construct and viral packaging plasmids to generate virus as described previously (Tiscornia et al, 2003). Viral supernatants were used to infect HEK293T cells and efficient knockdown of TORC2 was verified 3 days post-infection using Western analysis. Fl-TORC2, HA-tagged CBP plasmids and U6 promoter-shCREB have been previously described (Koo et al, 2005). RNAi oligos targeted against CREB and CBP were from Qiagen and ELK1 and SRF RNAis were from Invitrogen. Nucleotide sequences for Q-PCR primers and RNAi oligos are included in Supplementary data.
Bioinformatics
Gene profiling studies and SRF binding site analyses were performed as previously described (Zhang et al, 2005).
Immunoprecipitations and Western analysis
Antibodies against CBP (A-22), ELK1 (I-20), HSP90 (H-114), GST (Z-5) and anti-HA were from Santa Cruz. The anti-Flag antibody was purchased from Sigma. TORC2, CREB, pCREB and SIK2 antisera have been previously described (Conkright et al, 2003; Screaton et al, 2004). Rabbit polyclonal antibodies were generated against synthetic peptides for TORC1 (aa 504–530), TORC3 (aa 414–432) as previously described (Hagiwara et al, 1993). For co-IP experiments, HEK293 cells were transfected with the appropriate expression vectors using Lipofectamine 2000 (Invitrogen). Thirty-six hours post-transfection, cell were treated with either DMSO or FSK (10 μM) for 30 min, washed once with PBS and resuspended in Triton lysis buffer (20 mM Tris–HCl, pH 7.4, 137 mM NaCl, 2 mM EDTA, 1% Triton X-100, 10% glycerol, 0.5 mM dithiothreitol and protease inhibitors). Lysates were prepared by sonication on ice for 10 s followed by centrifugation (10 min at 13 000 r.p.m.) to clear debris. Immunoprecipitations (IPs) were performed overnight at 4°C with 1–5 μg of antibody and the conjugate was collected with 50 μl protein A or protein G (Sigma) beads 50% slurry.
Quantitative PCR
HEK293T cells were stimulated with FSK, TPA or STS for 2 h unless otherwise indicated. Total RNA was extracted from HEK293T cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was prepared by reverse transcription of 1 μg total RNA using Superscript II enzyme primed by random hexamers. cDNAs were amplified using the SYBR green PCR kit and an ABI PRISM 7700 sequence detector (Applied Biosystems). Relative messenger RNA expression levels were quantified using standard curves and data were normalized to -actin expression in the corresponding sample. Primer sequences are provided as Supplementary data.
Mass spectrometry
MS/MS and MUDPIT analyses were performed as previously described (Qi et al, 2006).
Immunofluorescence microscopy
Primary hepatocytes were seeded on coverslips and treated for 1 h with FSK (10 μM), TPA (20 nM) or vehicle. Cells were fixed with 4% PFA and stained with the rabbit polyclonal TORC2 antibody and Cy3-conjugated donkey anti-rabbit secondary antibody from Jackson Labs. Coverslips were mounted on glass slides using Vectashield (from Vector Laboratories) containing DAPI. Confocal imaging was performed on a Leica TCS SP2 AOBS confocal microscope.
ChIP assays
HEK293T cells were plated in 15 cm dishes and grown to approximately 80–90% confluence. Cells were then treated with FSK, TPA or STS dissolved in DMSO. Cells were crosslinked on the plates with 1% formaldehyde and chromatin prepared essentially as described (Screaton et al, 2004). For CREB, P-CREB and TORC2 IPs, rabbit polyclonal antibody raised against respective antigens (Conkright et al, 2003) was used along with rabbit IgG for negative controls. For CBP IP, a rabbit polyclonal CBP antibody A-22 (Santa Cruz Biotechnology, sc-369) was used. After removing crosslinks, DNA was extracted using phenol–chloroform and ethanol precipitated. CREB target promoters were quantified using SYBR green real-time PCR. All signals were quantified using standard curves and normalized to input chromatin signals.
Statistical analysis
Statistical analysis was performed using the Student's t-test. Where appropriate, analysis of variance (ANOVA) was used. All experiments were performed in duplicate (ChIP) or triplicate (mRNA experiments and transient transfections) and all data are representative of at least three independent experiments.
Supplementary Material
Supplementary Figures
Supplementary Figure Legends
Supplementary Data
Acknowledgments
We thank Stephanie Lerach and Susan Hedrick for technical assistance. These studies were supported by NIH grants RO1 GM37828 (MM) and GM62437 (PC). MM is supported by the Keickhefer Fdn and YL is supported by a grant from the Hillblom Fdn. KR is supported by the Danish Medical Research Council.
References
- Asahara H, Santoso B, Du K, Cole P, Montminy M (2001) Chromatin dependent cooperativity between constitutive and inducible activation domains in CREB. Mol Cell Biol 21: 7892–7900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M (2004) Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol 14: 2156–2161 [DOI] [PubMed] [Google Scholar]
- Bonni A, Ginty D, Dudek H, Greenberg M (1995) Serine 133 phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci 6: 168–183 [DOI] [PubMed] [Google Scholar]
- Brindle P, Linke S, Montminy M (1993) Analysis of a PK-A dependent activator in CREB reveals a new role for the CREM Family of repressors. Nature 364: 821–824 [DOI] [PubMed] [Google Scholar]
- Brindle P, Nakajima T, Montminy M (1995) Multiple protein kinase A-regulated events are required for transcriptional induction by cAMP. Proc Natl Acad Sci USA 92: 10521–10525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12: 413–423 [DOI] [PubMed] [Google Scholar]
- De Cesare D, Sassone-Corsi P (2000) Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol 64: 343–369 [DOI] [PubMed] [Google Scholar]
- Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, Magnuson MA, Girard J, Postic C (2004) Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 279: 20314–20326 [DOI] [PubMed] [Google Scholar]
- Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ (2001) MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol 21: 2249–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, Sassone-Corsi P (2000) A family of LIM-only transcriptional coactivators: tissue-specific expression and selective activation of CREB and CREM. Mol Cell Biol 20: 8613–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisch TM, Prywes R, Roeder RG (1987) c-fos sequence necessary for basal expression and induction by epidermal growth factor, 12-O-tetradecanoyl phorbol-13-acetate and the calcium ionophore. Mol Cell Biol 7: 3490–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner KH, Montminy M (2005) Can you hear me now? Regulating transcriptional activators by phosphorylation. Sci STKE 2005: pe44. [DOI] [PubMed] [Google Scholar]
- Goto NK, Zor T, Martinez-Yamout M, Dyson HJ, Wright PE (2002) Cooperativity in transcription factor binding to the coactivator CREB-binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem 277: 43168–43174 [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy MR (1993) Coupling of hormonal stimulation and transcription via cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol 13: 4852–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg CI, Tran SE, Eriksson JE, Sistonen L (2002) Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci 27: 619–627 [DOI] [PubMed] [Google Scholar]
- Impey S, Goodman RH (2001) CREB signaling—timing is everything. Sci STKE 2001: PE1. [DOI] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, Labow MA (2003) Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA 100: 12147–12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U (2004a) What turns CREB on? Cell Signal 16: 1211–1227 [DOI] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Seternes OM, Johansen B, Moens U (2004b) Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain. Cell Signal 16: 1187–1199 [DOI] [PubMed] [Google Scholar]
- Kachi K, French SW (1994) The connection between the nuclei of binucleated hepatocytes: an ultrastructural study. J Submicrosc Cytol Pathol 26: 163–172 [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, Brindle PK (2002) A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 419: 738–743 [DOI] [PubMed] [Google Scholar]
- Katoh Y, Takemori H, Lin X, Tamura M, MMuraoka Satoh T, Tsuchiya Y, Min L, Doi J, Miyauchi A, Witters L, Nakamura H, Okamoto M (2006) Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J 273: 2730–2748 [DOI] [PubMed] [Google Scholar]
- Kitabayashi I, Kawakami Z, Matsuoka T, Chiu R, Gachelin G, Yokoyama K (1993) Two cis-regulatory elements that mediate different signaling pathways for serum-dependent activation of the junB gene. J Biol Chem 268: 14482–14489 [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437: 1109–1111 [DOI] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D (2004) One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell 118: 453–464 [DOI] [PubMed] [Google Scholar]
- Lonze B, Ginty D (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001) Tanscriptional regulation by the phosphorylation dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609 [DOI] [PubMed] [Google Scholar]
- Mayr B, Canetierri L, Montminy M (2001) Distinct effects of cAMP and mitogenic signals on CBP recruitment impart specificity to target gene activation via CREB. Proc Natl Acad Sci USA 98: 10936–10941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengus G, Fadloun A, Kobi D, Thibault C, Perletti L, Michel I, Davidson I (2005) TAF4 inactivation in embryonic fibroblasts activates TGF beta signalling and autocrine growth. EMBO J 24: 2753–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Albuerne ED, Schatteman G, Sanders LK, Nathans D (1993) Transcriptional regulatory elements downstream of the JunB gene. Proc Natl Acad Sci USA 90: 11960–11964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L, Heredia JE, Altarejos JY, Screaton R, Goebel N, Niessen S, Macleod IX, Liew CW, Kulkarni RN, Bain J, Newgard C, Nelson M, Evans RM, Yates J, Montminy M (2006) TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science 312: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE (1997) Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91: 741–752 [DOI] [PubMed] [Google Scholar]
- Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schutz G, Linden DJ, Ginty DD (2005) SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat Neurosci 8: 759–767 [DOI] [PubMed] [Google Scholar]
- Saluja D, Vassallo M, Tanese N (1998) Distinct subdomains of human TAFII130 are required for interactions with glutamine-rich transcriptional activators. Mol Cell Biol 18: 5734–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR III, Takemori H, Okamoto M, Montminy M (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119: 61–74 [DOI] [PubMed] [Google Scholar]
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of. Science 310: 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861 [DOI] [PubMed] [Google Scholar]
- Takemori H, Kanematsu M, Kajimura J, Hatano O, Katoh Y, Lin XZ, Min L, Yamazaki T, Doi J, Okamoto M (2007) Dephosphorylation of TORC initiates expression of the StAR gene. Mol Cell Endocrinol 265–266: 196–204 [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Ikawa M, Verma IM (2003) A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA 100: 1844–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A, Olson EN (2004) Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428: 185–189 [DOI] [PubMed] [Google Scholar]
- Xu W, Kasper L, Lerach S, Jeevan T, Brindle P (2006) CREB-target genes dictate usage of at least three distinct cAMP-responsive coactivation mechanisms. Submitted [DOI] [PMC free article] [PubMed]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA 102: 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figure Legends
Supplementary Data