

Abstract
CREB is a key mediator of cAMP- and calcium-inducible transcription, where phosphorylation of serine 133 in its Kinase-Inducible Domain (KID) is often equated with transactivation. Phospho-Ser133 is required for CREB to bind the KIX domain of the coactivators CBP and p300 (CBP/p300) in vitro, although the importance of this archetype coactivator interaction for endogenous gene expression is unclear. Here, we show that the CREB interaction with KIX is necessary for only a part of cAMP-inducible transcription and CBP/p300 recruitment. Surprisingly, individual cAMP-inducible genes with CREB bound at their promoters differed in their reliance on KIX and none examined showed complete dependence. Alternatively, we found that arginine 314 (Arg314) in the CREB basic-leucine zipper (bZIP) domain contributed to CBP/p300 recruitment and KIX-independent CREB transactivation function. This implicates Transducer Of Regulated CREB (TORC), an unrelated cAMP-responsive coactivator that binds via Arg314, and which can bind CBP/p300, in these functions. Interestingly, KIX was also required for the full cAMP induction of a gene that did not require CREB. Thus, individual CREB-target gene context dictates the relative contribution of at least two different cAMP-responsive coactivation mechanisms.
Keywords: CBP, CREB, p300, TORC, transcription
Introduction
An implicit notion from studies of model factors and genes is that a transcription factor uses the same coactivation mechanism for all its target genes. Gene regulation then arises largely from synergistic or antagonistic interactions between the assemblage of DNA-bound transcription factors at the promoter or enhancer. In this scenario, transcriptional coactivators are largely utilitarian, providing enzymatic or adaptor functions to DNA-bound transcriptional activators.
There are estimated to be over 100 coactivators and co-repressors in the human genome, a number seemingly larger than necessary for ‘utilitarian' roles (Messina et al, 2004). Alternatively, the large variety of coactivators may contribute to redundancy (genetic buffering), another level of combinatorial regulation, or gene control via tissue-specific or signal-responsive coactivators (Spiegelman and Heinrich, 2004; Kasper and Brindle, 2006; Rosenfeld et al, 2006). It is less appreciated, however, that a target gene may dictate whether the presence of a recruited coactivator is required for its transcription. For example, some HIF targets recruit but do not seem to require CBP/p300 for expression, and some NF-κB targets recruit CBP/p300 when not transcribed (Leung et al, 2004; Kasper et al, 2005). That coactivators supply gene-specific functions suggests another reason for their diversity.
cAMP-responsive transcription in mammals is a paradigm for signal-dependent gene regulation (Shaywitz and Greenberg, 1999; De Cesare and Sassone-Corsi, 2000; Mayr and Montminy, 2001; Lonze and Ginty, 2002). The principal cAMP-responsive transcription factors are members of the CREB/ATF family (CREB, ATF-1, and CREMτ). These activators contain the signal-responsive Kinase-Inducible Domain (KID) activation domain, a glutamine-rich (Q) constitutive activation domain(s), and a basic-leucine zipper (bZIP) domain (Figure 1A). The bZIP chiefly provides dimerization and DNA-binding activities, but more recently it has been shown to also bind coactivators (Conkright et al, 2003). CREM isoforms that are alternatively spliced (α, β, γ, ɛ), or alternatively transcribed (the cAMP-inducible ICER), lack one or more of the activation domains, and act as repressors or context-dependent activators (Foulkes et al, 1991; Brindle et al, 1993; Molina et al, 1993). CREB is generally regarded as the predominant form in many cell types, and it is rather ubiquitously expressed in mice compared with ATF-1, and especially CREM (Bleckmann et al, 2002).
Figure 1.
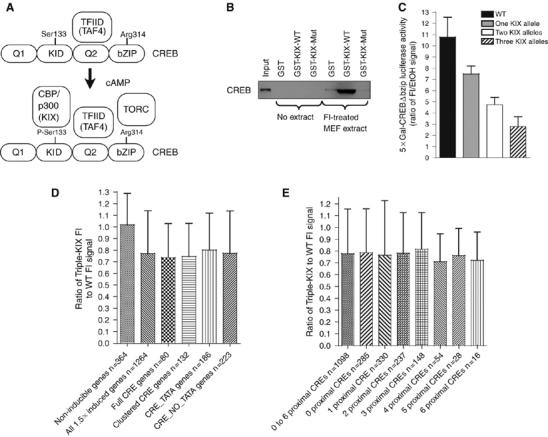
CBP/p300 KIX domain mutation leads to KIX insufficiency and moderate loss of cAMP-inducible CREB activity. (A) Current model of CREB domain (Q1, KID, Q2, bZIP) interactions with TAF4, CBP/p300, and TORC in response to cAMP. (B) GST pull-down assay with WT KIX (GST-KIX-WT) or KIX with three point mutations (GST-KIX-Mut); extracts of FI treated WT MEFs. Western blot with CREB 244 antibody. (C) Activity of Gal-CREBΔbZIP driving 5 × Gal luciferase in MEFs with one, two, or three CBPKIX and p300KIX mutant alleles (KIX allele). Shown as a ratio of FI-induced to EtOH signals (mean±s.e.m., n=2–3). (D, E) Affymetrix analysis of WT and triple-KIX mutant MEFs (n=1). Cells were treated with EtOH vehicle or FI for 90 min. Induced gene probe sets were induced at least 1.5-fold by FI in WT MEFs (n=1264). Non-inducible control gene probe sets had a difference of less than ±1% for the EtOH and FI signals in WT MEFs (D, n=364). Probe sets representing cAMP-responsive genes with different classes of promoters with CREs are indicated: canonical perfect CRE (full CRE), multiple CRE elements within 50 bp of each other (clustered CRE), with a CRE and a TATA box (CRE_TATA) or lacking a TATA box (CRE_NO_TATA) as defined by Zhang et al (2005) (D) or zero to six CREs in the region −1 to +1 kb from the transcriptional start site (E). Ratio of triple-KIX to the WT MEF FI signal (mean±s.d.).
The currently favored model for calcium- and cAMP-inducible transcription involves both constitutive and inducible recruitment of coactivators (Figure 1A). TFIID binds constitutively via its TAF4 subunit to the CREB glutamine-rich Q2 domain. TAF4 (Taf4a)-null cells are in fact deficient for CREB transactivation function (Mengus et al, 2005). cAMP or calcium signaling promotes recruitment of CBP/p300 and TORC to the Ser133-phosphorylated KID and bZIP domains of CREB, respectively (Conkright et al, 2003).
CBP (Crebbp) and p300 (Ep300) encode highly related protein acetyltransferases that possess several conserved protein-binding domains (e.g. KIX, CH1), which bind a variety of transcriptional regulators and other proteins (Goodman and Smolik, 2000). CBP/p300 are among the most highly connected proteins in the known mammalian protein interactome, with more than 340 described interaction partners (Kasper et al, 2006). Consistent with wide-reaching roles in gene regulation, null mutations of CBP or p300, or mutations in their acetyltransferase domain, are lethal in mice (Yao et al, 1998; Oike et al, 1999; Kung et al, 2000; Shikama et al, 2003). Consequently, their transcriptional roles have not been intensively studied in vivo, and relatively little is known about cAMP-responsive transcription in that context. Studies using conditional knockout alleles, however, have shown that CBP and p300 each contribute to antigen receptor signaling-responsive gene expression in T and B cells (Kasper et al, 2006; Xu et al, 2006). Knock-in mutations in the CH1 domain of CBP and p300 have revealed that they contribute to a sizeable fraction of hypoxia-inducible gene expression (Kasper et al, 2005). The remaining hypoxia-inducible transcription observed after loss of this key CBP/p300 interaction with HIF suggests that unrelated coactivators provide some redundant activity (Kasper and Brindle, 2006). One goal of this study was to test if coactivators unrelated to CBP/p300 were similarly important for the cAMP signaling pathway.
The three TORC family members, TORC1, TORC2, and TORC3 (Transducer Of Regulated CREB, or CREB-Regulated Transcription Coactivator, encoded by Crtc1, Crtc2, and Crtc3, in mice) are unrelated to CBP and p300 (Conkright et al, 2003; Iourgenko et al, 2003). They possess an N-terminal coiled-coil domain that binds the CREB bZIP, and they have a potent transactivation domain, but it is unknown if they possess enzymatic activities. TORC1 and TORC2 are sequestered in the cytoplasm until activated by cAMP or calcium, whereupon they transit to the nucleus; TORC3 is constitutively nuclear (Screaton et al, 2004).
About 300 different stimuli have been reported to cause phosphorylation of Ser133 in the KID domain of CREB, although in many instances it is unknown if CREB or its target genes are activated, particularly when the calcium or cAMP pathways are not involved (Johannessen et al, 2004). That CBP/p300 and TORC may be recruited and/or regulated differently by some of these signaling pathways has been suggested as a mechanism to yield distinctive CREB-regulated gene expression patterns (Ravnskjaer et al, 2007). In this regard, two parts of the cAMP pathway in particular are not well understood. These include the events following coactivator recruitment to CREB and the roles for gene-specific and tissue-specific elements in gene activation. In this study, cells with CBP/p300 KIX domain mutations that abrogate the interaction with the CREB KID domain enabled testing aspects of these questions.
Results
KIX domain mutations block CREB binding in vitro
We previously generated mice that carry identical point mutations in the KIX domains of CBP (CBPKIX) and p300 (p300KIX) (Kasper et al, 2002). The mutations alter three conserved residues on the surface of KIX that individually abrogate critical contacts with the CREB KID and c-Myb transactivation domains (Tyr650Ala/Ala654Gln/Tyr658Ala in CBP and Tyr630Ala/Ala634Gln/Tyr638Ala in p300) (Radhakrishnan et al, 1997; Parker et al, 1999; Kasper et al, 2002). As predicted from the analysis of single mutations (Parker et al, 1999), the three combined mutations blocked the ability of a GST-KIX fusion protein to bind CREB in nuclear extracts from primary mouse embryonic fibroblasts (MEFs) treated with forskolin and isobutyl-methylxanthine (FI; cAMP agonists that induce Ser133 phosphorylation; Figure 1B). The KIX mutation also strongly blocked CBP binding to in vitro phosphorylated GST-CREB that lacks the bZIP domain (Supplementary Figure S1).
Mice lacking a WT KIX domain die early in embryogenesis
We previously demonstrated that CREB and c-Myb transactivation function was attenuated in CBPKIX/KIX and p300KIX/KIX MEFs, but that there was no measurable defect in endogenous CREB-target gene (Fos, JunB, Crem ICER) expression, as measured by Northern blot (Kasper et al, 2002). We generated mice with KIX mutations in both CBP and p300 to test if the remaining WT KIX domains accounted for endogenous CREB-target gene transcription in CBPKIX/KIX and p300KIX/KIX MEFs. Mice on a C57BL/6 strain background that completely lack WT KIX domains (CBPKIX/KIX;p300KIX/KIX) died very early during embryogenesis, but those harboring a single WT p300 or CBP allele died by E12.5 (CBPKIX/KIX;p300+/KIX), or survived to at least day E14.5 (CBP+/KIX;p300KIX/KIX) (W Xu, unpublished data). ‘Triple-KIX' mutant MEFs could be derived from CBP+/KIX;p300KIX/KIX embryos, and these grew comparably to WT cells (W Xu, unpublished data).
We analyzed KIX mutant gene dosage on the transactivation function of a Gal-CREBΔbZIP fusion protein, which lacks the C-terminal bZIP domain and cannot bind TORC. Using multiple primary MEF isolates, we observed a decrease in cAMP-inducible Gal-CREBΔbZIP activity that was proportional to the number of mutant KIX alleles, independent of whether the KIX mutations were in CBP, p300, or both genes (i.e. three KIX mutant alleles resulted in a ∼75% reduction in transactivation function; Figure 1C). This indicates that the KIX domains of CBP and p300 function similarly in MEFs, with regard to CREB KID domain activity, and that the combined dosage of the KIX domain is important. We previously showed that the KIX mutation does not result in dominant-negative function in transient assays (Kasper et al, 2002). Thus, KIX domain insufficiency likely accounts for reduced CREB activity.
KIX domain insufficiency attenuates cAMP-inducible gene expression
Based on these results, we expected to see a reduction in endogenous CREB-target gene expression in triple-KIX mutant MEFs. However, Affymetrix microarray analysis revealed that cAMP-inducible gene expression was not as strongly affected as in the transient assays (Figure 1D). Results from 1264 probe sets on the microarray showed an at least 1.5-fold induction by cAMP in WT MEFs, for which the mean FI-dependent signal was reduced by an average of 20–30% in the triple-KIX MEFs (Supplementary Table S1; Figure 1D). Examination of control genes operationally defined as non-inducible by cAMP (signal ±1% between FI and ethanol vehicle treated WT MEFs; 364 probe sets) revealed that their expression was largely unaffected in the triple-KIX MEFs (Figure 1D). Individual cAMP-inducible genes, however, were not uniformly repressed 20–30% (Supplementary Table S1), but varied considerably in their KIX dependence (verified by quantitative real-time qRT–PCR in independent experiments; W Xu, unpublished data). These results together demonstrate that a 75% reduction in KIX domain dosage specifically reduces global cAMP-responsive gene expression by an average of 20–30%, with some genes affected more than others.
The variable effect of the KIX mutation on cAMP-inducible gene expression may predict commonalities between the similarly affected gene promoters. We examined whether particular subclasses of cAMP-inducible genes show increased sensitivity to KIX insufficiency. We assessed probe sets on the microarray representing genes defined by Zhang et al (2005) as having full-length canonical CREB-binding sites (full CREs, TGACGTCA, n=80), clustered CREs (CREs within 50 bp of each other, including TGACG/CGTCA half sites, n=132), or CRE containing genes with (n=186) or without (n=223) TATA elements (Figure 1D). These groups were not significantly different from each other, although all were significantly different from the control gene group (P<0.0001, ANOVA).
We next asked if genes with multiple promoter-proximal CREs, which may recruit CBP/p300 more efficiently because of a high local concentration of CREB, were more resistant to KIX insufficiency. Analysis of the microarray data showed, however, that cAMP-inducible genes (>1.5-fold induced in WT cells) with between zero CREs (CREs may be distal, non-canonical, or absent) and six CREs that lie between −1 and +1 kb of the transcription start site were not significantly different from each other (ANOVA, P=0.78; Figure 1E) (Zhang et al, 2005). Collectively, these results show that the cAMP-inducible gene activity in triple-KIX MEFs either relies on the remaining WT CBP protein, or that CREB (or another factor) functions by KIX-independent mechanisms.
MEFs containing only KIX mutant CBP/p300 retain partial CREB activity
To eliminate the contribution of WT CBP completely, we generated primary MEFs that contain three mutant KIX alleles for CBP and p300, and one Cre/loxP conditional knockout CBPflox allele (Figure 2A, CBPflox/KIX;p300KIX/KIX) (Kang-Decker et al, 2004). Transient expression of Cre recombinase following infection with a Cre-expressing adenovirus resulted in efficient recombination (>90%) of CBPflox (Figure 2B). Growth of the mutant CBPΔflox/KIX;p300KIX/KIX (‘triple-KIX/Δflox') and control (‘Δflox', CBP+/Δflox;p300+/+) MEFs was comparable before and after Cre-adenovirus infection (Supplementary Figure S2). Moreover, MEFs that do not express WT CBP did not have a major growth disadvantage, as the proportion of cells with the CBPΔflox allele did not decrease appreciably over a 14-day period (Figure 2B). CREB phosphorylation in response to FI was normal in the triple-KIX/Δflox MEFs (Supplementary Figure S3). The ability of endogenous CREB to stimulate luciferase reporters driven by one or four CREs in response to FI was attenuated ∼50–70% in triple-KIX/Δflox MEFs (Figure 2C and D). This remaining activity in the absence of a functional KIX domain defines a KIX-independent mechanism(s) that contributes to CREB function.
Figure 2.
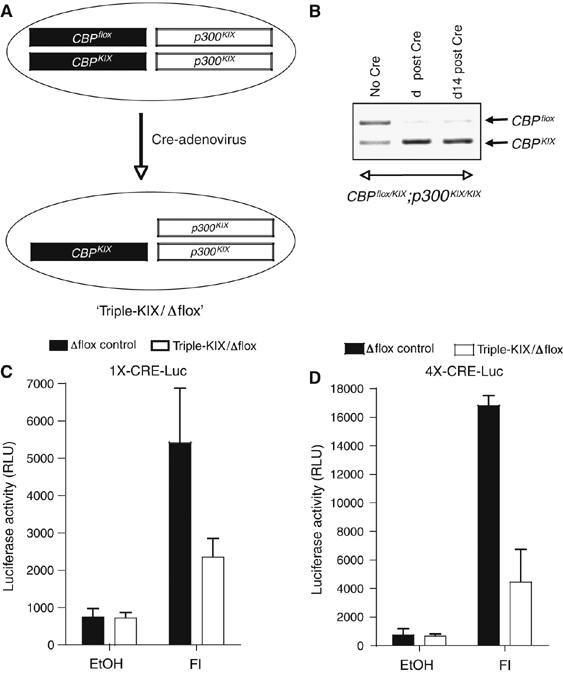
MEFs containing only KIX mutant CBP and p300 retain some CRE-responsive transactivation in response to cAMP. (A) Model of MEFs containing three CBPKIX and p300KIX alleles and one conditional allele (CBPflox). Treatment with Cre-expressing adenovirus inactivates the conditional allele (triple-KIX/Δflox MEFs). (B) Semi-quantitative PCR shows efficient deletion of CBPflox in triple-KIX/flox MEFs after treatment with Cre-expressing adenovirus. Days after infection indicated. (C, D) Luciferase reporters containing one (C) or four (D) CREs retain some FI-responsive activity in triple-KIX/Δflox MEFs (mean±s.e.m., n=9 (C) or n=6 (D)).
Individual CREB-target genes differentially require the KIX domain
We performed qRT–PCR analysis of a subset of genes identified by microarray, focusing on those with conserved CREs and with evidence for being CREB targets (Areg, Btg2, Crem ICER, Dusp1, Hlf, Junb, Nr4a3, Rgs2) (Zhang et al, 2005). Test gene mRNA, in this case, was normalized to β-actin mRNA, and then normalized to the lowest value in the entire data set of eight genes. Meta-analysis of qRT–PCR experiments that used multiple independent Δflox and triple-KIX/Δflox primary MEF isolates confirmed gene-to-gene variation in KIX dependence (Table I). Areg and Hlf were the most sensitive to KIX insufficiency (FI-dependent signal reduced ∼70–90%). Btg2, Nr4a3, and Rgs2 were more moderately affected, whereas Crem ICER, Dusp1, and Junb were largely unaffected by KIX insufficiency.
Table 1.
qRT–PCR analysis of CREB-target genes using three independent MEF isolates of each genotype (n=8–9)
| Symbol (mouse) | Name | Δflox EtOH signal (mean±s.d.) | Triple-KIX Δflox EtOH signal (mean±s.d.) | Δflox FI signal (mean±s.d.) | Triple-KIX Δflox FI signal (mean±s.d.) | Ratio of mutant FI (AVE) to control FI (AVE) signals | One-tailed t-test of FI signals |
|---|---|---|---|---|---|---|---|
| Areg | Amphiregulin | 0.02±0.01 | 0.003±0.002 | 1.92±0.81 | 0.12±0.06 | 0.063 | <0.0001 |
| Btg2 | B-cell translocation gene 2, antiproliferative | 5.43±2.16 | 4.40±1.00 | 35.35±13.04 | 25.31±17.78 | 0.72 | 0.11 |
| Crem ICER | cAMP-responsive element modulator, ICER isoform | 0.36±0.12 | 0.40±0.18 | 7.98±2.05 | 8.40±3.87 | 1.05 | 0.39 |
| Dusp1 | Dual specificity phosphatase 1 | 9.45±4.52 | 4.68±1.76 | 103.00±28.61 | 109.30±41.23 | 1.06 | 0.35 |
| Hlf | Hepatic leukemia factor | 0.08±0.08 | 0.03±0.03 | 2.17±1.52 | 0.69±0.55 | 0.32 | 0.007 |
| Junb | Jun-B oncogene | 5.03±2.57 | 4.76±1.90 | 59.72±21.79 | 74.80±57.09 | 1.25 | 0.23 |
| Nr4a3 | Nuclear receptor subfamily 4, group A, member 3 | 0.04±0.01 | 0.03±0.01 | 40.49±7.87 | 29.16±22.41 | 0.72 | 0.09 |
| Rgs2 | Regulator of G-protein signaling 2 | 1.23±0.62 | 1.79±0.70 | 64.97±18.55 | 46.25±29.27 | 0.71 | 0.06 |
| Relative expression (divided by 1000) set to the lowest real-time PCR value for this data set of eight genes. Test gene signal normalized to Actb mRNA. Note that the mean of the ratios of mutant to control FI signals is 0.74 for these eight genes, similar to the global average seen in the Affymetrix experiment in Figure 1. | |||||||
The KIX mutation attenuates but does not prevent recruitment of CBP/p300 to CREB-target genes
Prevailing models predict that the KIX mutations will block the cAMP-dependent recruitment of CBP/p300 to CREB targets. To test this, we focused on three genes (Areg, Nr4a3, and Crem ICER) that illustrate the range of KIX-dependent expression. Meta-analysis of qRT–PCR data derived from experiments independent of those in Table I confirmed that the expression of these genes represent the high, middle, and low ranges of the phenomenon (Figure 3A–C, the Gapdh mRNA-normalized expression signal was set relative to the lowest value for each gene). We next established that Areg, Nr4a3, and Crem ICER are in fact direct CREB targets, using quantitative chromatin immunoprecipitation (ChIP) assays. Robust ChIP signals were observed in WT MEFs with two different CREB-specific antisera (244, 253) but not normal rabbit serum (NRS; Figure 3D–F). The ChIP signal was also dependent on qPCR primers close to the CRE, and not on those several kilo basepairs distant (Figure 3D–F). Induction of these genes also showed CREB dependence when tested in both Creb1-null and Creb1 hypomorph MEFs (Supplementary Figure S4A–H); ATF-1 and the upregulation of activating forms of CREM with a KID domain probably contribute to the remaining expression (Supplementary Figure S4I–K) (Hummler et al, 1994). Luciferase reporters driven by the Areg, Nr4a3, and Crem ICER promoters required CRE sequences for cAMP responsiveness, further indicating that KIX-independent transcription of these genes requires CREB (Supplementary Figure S4L).
Figure 3.
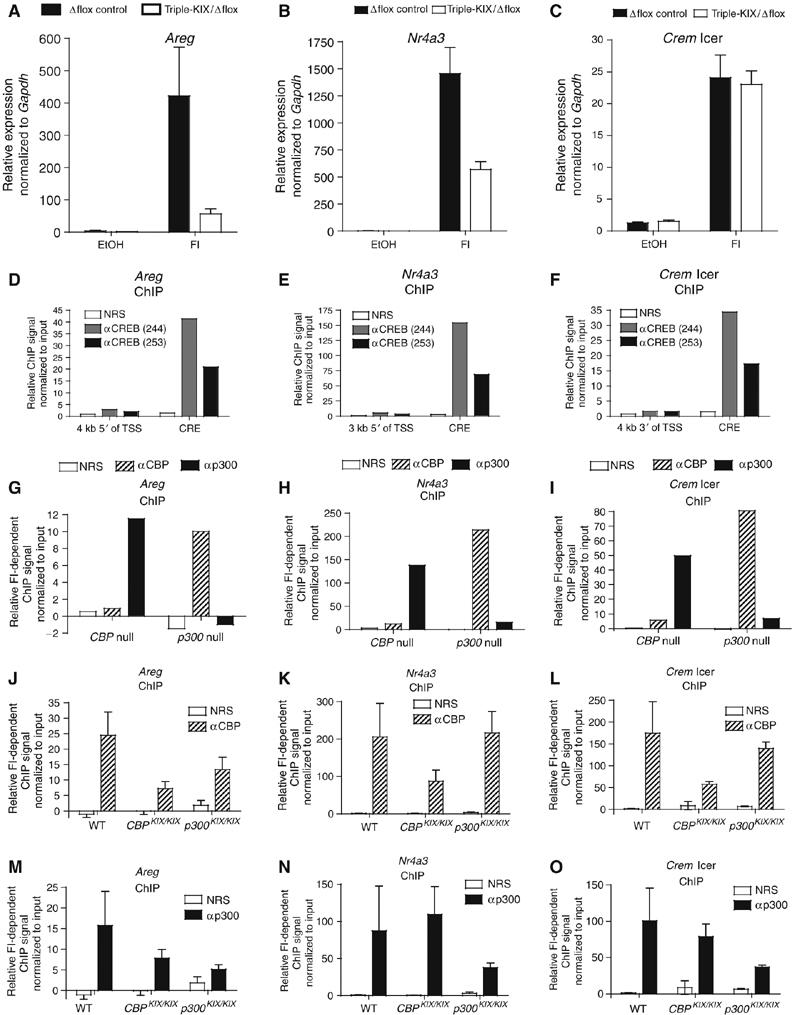
CREB-target gene expression is variably affected by the KIX mutation and does not always correlate with recruitment of mutant CBP and p300. (A–C) qRT–PCR of FI-inducible gene expression in triple-KIX/Δflox MEFs. Expression was normalized to Gapdh and the lowest value for each gene in each experiment was set to 1 (mean±s.e.m., n=4–5 representing three independent control and mutant MEF isolates). (D–O) Quantitative ChIP assays of Areg (D, G, J, M), Nr4a3 (E, H, K, N), and Crem Icer (F, I, L, O ) in WT (D–F), CBP, or p300-null (G–I), or CBPKIX/KIX and p300KIX/KIX (J–O) MEFs. Control (NRS) and specific (anti-CREB, anti-CBP, or anti-p300) antisera are indicated. Except for the CREB ChIP (D–F), FI-dependent ChIP signal was determined by subtracting the EtOH signal from the FI signal after normalizing to the input DNA signal (G–O) (mean±s.e.m., (J–O); n=2, Crem Icer (L, O); n=5, Areg (J, M); n=6, Nr4a3 (K, N)).
Next, we confirmed by ChIP that these genes were also direct CBP/p300 targets using CBP-null and p300-null MEFs. The CBP and p300 antisera were specific and showed markedly increased ChIP signals in response to FI treatment (inferred as recruitment of CBP or p300 to the CRE region), except when the protein was absent due to gene inactivation (Figure 3G–I). The residual ChIP signal observed in some instances is likely due to the incomplete floxed gene inactivation in the MEFs (∼90% efficiency; LH Kasper, unpublished data).
Finally, we determined the effect of the KIX mutation on CBP/p300 recruitment; WT, CBPKIX/KIX, and p300KIX/KIX MEFs were used; WT CBP or p300 in the mutant cells served as an internal control. Surprisingly, the KIX mutation led to only an average reduction of ∼50–70% in CBP (Figure 3J–L) and p300 (Figure 3M–O) recruitment (n=2–6 independent experiments). Thus, CREB-target genes retain an appreciable ability to recruit CBP/p300 in the absence of a functional KIX domain. However, reduced recruitment of mutant CBP/p300 did not necessarily correlate with gene expression (e.g. Crem ICER).
TORC2 can suppress the loss of KIX function
We hypothesized that KIX-dependent genes may not recruit TORC efficiently, making them more sensitive to mutant CBP/p300. We detected by ChIP, however, robust cAMP-dependent recruitment of TORC2 to Areg, Nr4a3, and Crem ICER (Figure 4A–C). We next explored if TORC2 could suppress KIX domain insufficiency in triple-KIX/Δflox MEFs. Indeed, Areg, Nr4a3, and Crem ICER cAMP-dependent expression was restored in mutant MEFs infected with retrovirus expressing TORC2 to levels equal to or greater than control cells expressing GFP alone (Figure 4D–F). Western blot showed that the virus overexpressed TORC2 (Supplementary Figure S5). TORCs are not known to possess HAT activity, so it is unlikely that TORC can suppress KIX insufficiency in that way directly, but it may help recruit CBP/p300 (Ravnskjaer et al, 2007).
Figure 4.
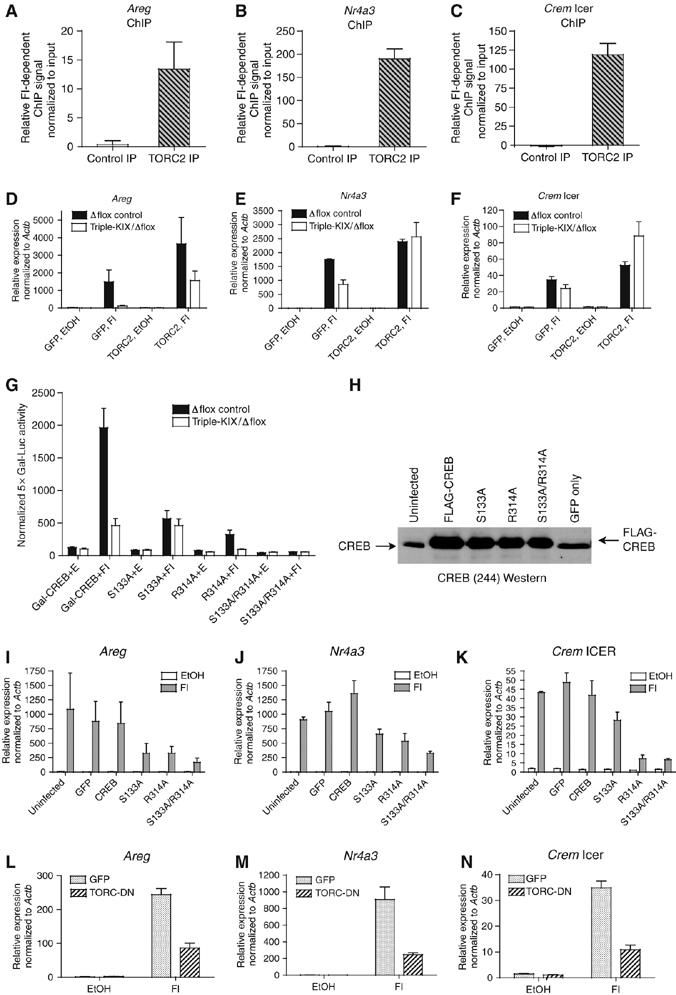
TORC has a role in KIX-independent FI-inducible CREB-target expression. (A–C) Quantitative ChIP assays in WT MEFs. Control (TORC2 antiserum+blocking peptide) and TORC2 antiserum used in IPs. FI-dependent ChIP signal was determined by subtracting the EtOH signal from the FI signal after normalizing to the input DNA signal (mean±s.e.m., n=2). (D–F) qRT–PCR of control and triple-KIX/Δflox MEFs overexpressing GFP or TORC2. (mean±s.e.m., n=5). (G) Activity of Gal-CREB and Gal-CREB mutants in Δflox control and triple-KIX/Δflox MEFs (mean±s.e.m., n=6). (H) FLAG-CREB and FLAG-CREB mutants are comparably overexpressed in MEFs. (I–K) qRT–PCR of CREB-target genes in WT MEFs overexpressing FLAG-CREB and mutants (mean±s.e.m., n=3). (L–N) qRT–PCR of CREB-target genes in WT MEFs expressing GFP or TORC-DN (mean±s.e.m., n=4). For gene expression qRT–PCR, expression was normalized to Actb and the lowest value for each gene in each experiment was set to 1 (D–F, I–N).
Arg314 in the CREB bZIP domain is essential for KIX-independent activation of CREB
If TORC is limiting under conditions of KIX insufficiency, then blocking both KIX and TORC binding to CREB should further inhibit its transactivation function. First, we confirmed that Ser133 does not interact with another region of CBP/p300, and KIX does not interact with another region of CREB. Transient transfection assays showed that Gal-CREB (full length, including the bZIP) with a Ser133 to Ala mutation (S133A) retained ∼25% of activity in control MEFs, as did WT Gal-CREB in triple-KIX/Δflox MEFs. Importantly, the effects of the KIX and S133A mutations were not additive (Figure 4G). Other CBP/p300-interacting fusion activators (Gal-Ets-1 and Gal-HIF-1α) that do not bind KIX were unaffected by the triple-KIX/Δflox mutations (Supplementary Figure S6) (Yang et al, 1998; Kasper et al, 2005).
We next mutated arginine 314 (Arg314) to Ala (Gal-CREB R314A). Mutation of Arg314 blocks TORC binding in vitro, but does not affect CREB dimerization or DNA-binding activity (the Gal4 DNA-binding domain supplies these functions for Gal-CREB) (Screaton et al, 2004). Activity of the R314A mutant was reduced ∼2-fold more than S133A in control Δflox MEFs, but R314A lost essentially all cAMP inducibility in triple-KIX/Δflox MEFs (Figure 4G). This suggests the interaction with the KIX domain (via CREB Ser133) and TORC (via CREB Arg314) accounts for all cAMP-responsive Gal-CREB transactivation function in MEFs. This finding was reinforced when S133 and R314 were simultaneously mutated, resulting in no measurable cAMP-dependent induction (Figure 4G). All Gal-CREB constructs were expressed comparably in Cos7 cells and had similar activity as in MEFs (Supplementary Figure S7). All told, this suggests that the interaction of Gal-CREB with both KIX and TORC is necessary for cAMP-inducible transactivation function.
We next tested if Ser133 and Arg314 were required for CREB-dependent activation of endogenous genes. N-terminal FLAG-tagged CREB was expressed in WT MEFs using a retrovirus that also expresses GFP; GFP+ cells were then purified by FACS (to >90%), and FLAG-CREB overexpression was confirmed by Western blot (Figure 4H). CREB was not generally limiting, as expression of WT FLAG-CREB did not strongly affect the expression of Areg, Nr4a3, and Crem ICER when compared with uninfected or GFP virus-infected MEFs (Figure 4I–K). Expression of FLAG-CREB S133A or R314A repressed Areg and Nr4a3 to similar levels (∼50–70% of WT FLAG-CREB) (Figure 4I and J). In contrast, Crem ICER was considerably more sensitive to the R314A mutant than S133A; consistent with this gene's resistance to KIX insufficiency (Figure 4K). A double mutant FLAG-CREB (S133A/R314A) repressed expression to levels greater than (Areg, Nr4a3) or equal to (Crem ICER) the most severe single mutant (Figure 4I–K). Although residual gene expression may be due to endogenous CREB or alternative activator/coactivator mechanisms, these results show that both S133 and R314 contribute to CREB function in a gene-specific manner.
A dominant-negative TORC2 inhibits cAMP-responsive CREB-target gene expression
The importance of CREB Arg314 is consistent with TORC involvement. To test this more directly, we used a dominant-negative construct consisting of TORC1 residues 1–44 of its CREB-binding domain fused to GFP (TORC-DN). A similar construct was shown by Bittinger et al (2004) to inhibit CREB activation by all three TORC proteins. TORC-DN is a reasonable alternative to knockout and knockdown approaches because all three TORC genes are expressed in MEFs (LH Kasper, unpublished data), TORC mutant mice have not been described, and siRNA approaches are highly inefficient in primary mouse cells such as MEFs. WT MEFs were efficiently transduced (>95% GFP+) with retroviruses expressing TORC-DN or GFP alone as a control. cAMP-responsive expression for Areg, Nr4a3, and Crem ICER was attenuated ∼60–75% by TORC-DN compared with control cells (Figure 4L–N); comparable to that seen with FLAG-CREB R314A (Figure 4I–K). The TORC-DN and FLAG-CREB data are consistent with TORC mediating a substantial portion of CREB-target gene activation.
The R314A mutation inhibits CBP/p300 and TORC2 recruitment to CREB targets more than S133A
To determine if the S133A and R314A mutations affect CBP/p300 and TORC2 recruitment, we performed sequential ChIP (SeqChIP) assays. SeqChIP gives information about the co-occupancy of a specific genomic region by multiple proteins and was performed by first immunoprecipitating FLAG-CREB with anti-FLAG antibody, and then eluting with FLAG peptide and subjecting the eluate to a second immunoprecipitation (IP) for CBP/p300 or TORC2 (Figure 5A) (Geisberg and Struhl, 2004). Areg gave a weak SeqChIP signal and was not tested further (LH Kasper, unpublished data). Efficient retroviral transduction (∼30–50% GFP+ cells) was established by flow cytometry or fluorescence microscopy, and FLAG-CREB expression was confirmed by Western blot (S Lerach and T Jeevan, unpublished data). Nr4a3 and Crem ICER gave strong cAMP-dependent SeqChIP signals for CBP/p300 (combined CBP and p300 IP) and TORC2 in WT MEFs transduced with FLAG-CREB virus (Figure 5B–I). SeqChIP was specific for FLAG-CREB, as the TORC2- or CBP/p300-dependent signal was undetectable in MEFs transduced with a virus expressing only GFP (Figure 5F–I).
Figure 5.
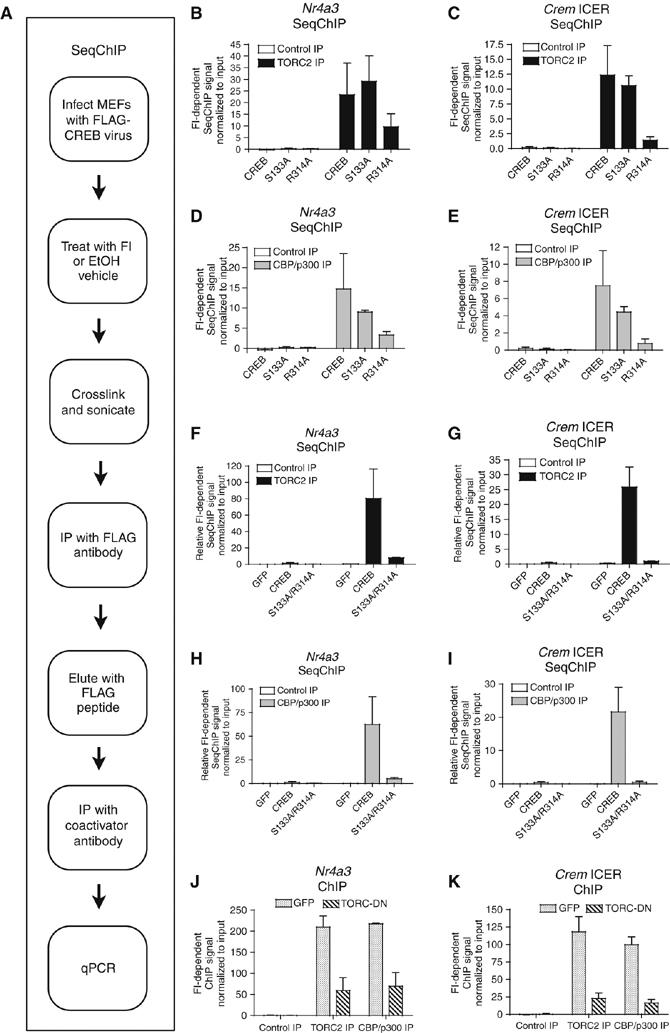
CREB R314A mutant and TORC-DN affect recruitment of TORC2 and CBP/p300 to CREB-target genes. (A) SeqChIP flow chart. (B–I) SeqChIP assays on EtOH or FI treated MEFs overexpressing FLAG-CREB, FLAG CREB mutants, or GFP. qRT–PCR of ChIP signal normalized to primary DNA input. (mean±s.e.m., n=2). (J, K) Quantitative ChIP assay on WT MEFs overexpressing GFP or TORC-DN protein. Control and specific (anti-TORC2, anti-CBP/p300) antisera are indicated (mean±s.e.m., n=2). FI-dependent ChIP signal determined by subtracting the EtOH signal from the FI signal after normalizing to the input DNA signal (B–K).
FLAG-CREB S133A was able to recruit TORC2 about as efficiently as FLAG-CREB (Figure 5B and C), but its ability to recruit CBP/p300 was modestly attenuated by ∼40% (Figure 5D and E), approximately the same as ChIP assays using KIX mutant MEFs (Figure 3). In contrast, the recruitment of TORC2 by FLAG-CREB R314A was reduced ∼60% for Nr4a3 and ∼90% for Crem ICER (Figure 5B and C). Surprisingly, CBP/p300 recruitment to FLAG-CREB R314A was more strongly affected (reduced ∼80–90%) than with the S133A mutation, indicating that Arg314 contributes to recruitment of CBP/p300 as well as TORC2 (Figure 5D and E). Moreover, the FLAG-CREB S133A (Figure 5) and KIX mutant cell ChIP data (Figure 3) reinforce the notion that the KID domain interaction with KIX is not the sole means of CBP/p300 recruitment for CREB.
CREB S133A/R314A cannot efficiently recruit CBP/p300 and TORC2 in response to cAMP
Gene expression analysis indicated that mutation of both Ser133 and Arg314 should severely reduce TORC2 and CBP/p300 recruitment. We tested this by SeqChIP using WT MEFs expressing FLAG-CREB S133A/R314A. The cAMP-dependent recruitment of TORC2 (Figure 5F and G) and CBP/p300 (Figure 5H and I) was reduced from ∼90% (Nr4a3) to ∼95% (Crem ICER) in FLAG-CREB S133A/R314A expressing MEFs, compared with FLAG-CREB. MEFs expressing GFP alone yielded only background signal (Figure 5F–I). These results correlate with the reduction in gene expression seen in FLAG-CREB S133A/R314A expressing cells (Figure 4J and K). Therefore, CREB Ser133 and Arg314 are together highly essential for CBP/p300 and TORC2 recruitment to Nr4a3 and Crem ICER and contribute to the majority of cAMP-responsive gene expression.
TORC-DN inhibits the recruitment of both TORC2 and CBP/p300 to CREB-target genes
The effect of the R314A mutation on TORC2 and CBP/p300 recruitment implies that TORC-DN should act similarly. We performed ChIP assays on WT MEFs transduced with TORC-DN or GFP-only expressing retroviruses. Both TORC2 and CBP/p300 cAMP-dependent recruitment were reduced ∼70% for Nr4a3 and ∼80% for Crem ICER by TORC-DN (Figure 5J and K). The TORC-DN GFP fusion protein was recruited directly to Nr4a3 and Crem ICER, as determined by ChIP using a GFP antibody (Supplementary Figure S8). The efficacy of TORC-DN and FLAG-CREB R314A at blocking TORC2 and CBP/p300 recruitment were comparable (compare Figure 5B–E to J and K). This suggests that TORC-DN acts mainly by occluding TORC binding to CREB, rather than by steric hindrance of CBP/p300 binding by the GFP moiety of the TORC-DN fusion. The reduced expression (Figure 4M and N) and ChIP signal (Figure 5J and K) are consistent with TORC-DN blocking the ability of TORC to bind CREB, and TORC contributing to CBP/p300 recruitment. Indeed, purified recombinant GST-TORC2 was able to bind immunopurified CBP/p300 in a far-Western blot assay (Supplementary Figure S9). Thus, TORC2 and CBP/p300 can directly interact, and this may contribute to KIX-independent recruitment of CBP/p300.
A non-canonical CREB-target gene requires another coactivation mechanism
Testing other CREB targets revealed that not all rely as much on the canonical coactivation mechanisms involving CREB-Ser133:KIX and CREB-Arg314:TORC2 (Figure 1A). Hepatic leukemia factor (Hlf) has a full-length CRE and is dependent on the KIX domain for ∼70–80% of its inducible expression (Table I; Figure 6A). Hlf is a direct target of CREB by ChIP (Figure 6B), but it did not require CREB for expression (Supplementary Figure S10A and B; a role for ATF-1 and CREMτ cannot be ruled out, however). A minimal Hlf promoter-driven reporter was constitutively active but barely induced by cAMP; mutation of the CRE did not repress activity (Supplementary Figure S10C). Thus, the role, if any, of CREB/ATF-1/CREMτ and the promoter-proximal CRE in Hlf expression is unclear. It is also uncertain if Hlf is a CBP/p300 and TORC2 target, because of weak ChIP signals, perhaps due to the high GC content of the promoter (LH Kasper, unpublished data). Hlf expression was also not rescued by TORC2 overexpression in KIX-insufficient cells (Figure 6C). Consistent with the lack of CREB dependence, overexpression of the FLAG-CREB mutants had no repressive effect on Hlf (Figure 6D), even though ChIP showed that they bound the Hlf CRE region (Supplementary Figures S10D). Thus, Hlf binds CREB, yet it relies largely on an atypical KIX-dependent mechanism that may involve another cAMP-responsive factor.
Figure 6.
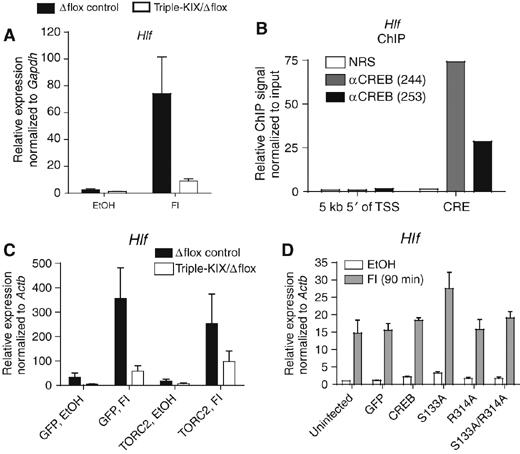
Hlf is a KIX-sensitive CREB-target gene that is not rescued by TORC2. (A) qRT–PCR of Hlf expression in Δflox control and triple-KIX/Δflox MEFs (mean±s.e.m., n=5). (B) Quantitative ChIP of Hlf in WT MEFs. Control (NRS) and CREB antisera are indicated. (C) qRT–PCR of Hlf in Δflox control and triple-KIX/Δflox MEFs overexpressing GFP or TORC2 (mean±s.e.m., n=5). (D) qRT–PCR of Hlf in WT MEFs overexpressing FLAG-CREB and mutants (mean±s.e.m., n=3).
Discussion
A recent review on coactivators states: ‘It remains to be established whether (the) order of recruitment and the enzymatic components of (coactivator) complexes for a specific transcription factor are distinct for different cohorts of regulated genes' (Rosenfeld et al, 2006). We begin to address these issues for CREB and its regulated genes in this study.
We showed that the archetype-inducible coactivation mechanism, where Ser133-phosphorylated CREB binds CBP/p300 via the KIX domain, does not account for the majority of cAMP-inducible gene expression (Radhakrishnan et al, 1997). This physical interaction with CBP/p300 KIX is often viewed as the major activating event in the induction of CREB targets (Carlezon et al, 2005; Hong et al, 2005). While the KIX:KID complex contributes significantly to cAMP-inducible transcription, it appears that TORC binding to the CREB bZIP domain is also critical. This is consistent with other recent models of CREB activation (Conkright et al, 2003; Iourgenko et al, 2003). Moreover, our findings indicate that the CREB interaction with TORC contributes to CBP/p300 recruitment (Figure 7). This observation is supported by evidence that TORC2 directly binds CBP in vitro (Supplementary Figure S9), RNAi knockdown of TORC2 in HEK293T cells reduces CBP/p300 occupancy at CREB-target genes, and RNAi knockdown of CBP reduces TORC2 recruitment (Ravnskjaer et al, 2007). Finally, we showed that CREB-target genes differentially require these distinct coactivation mechanisms.
Figure 7.
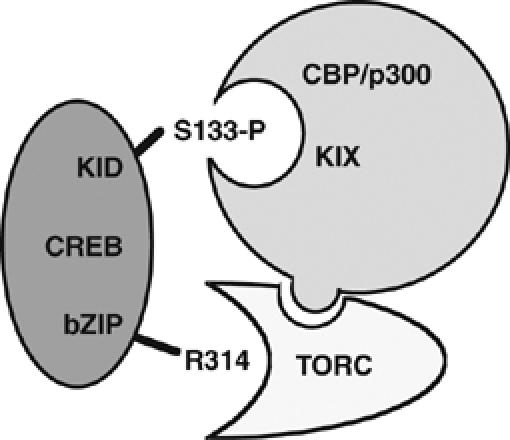
New canonical model of CREB-, CBP/p300-, and TORC cAMP-inducible interactions. Proposed direct interaction between CBP/p300 and TORC2 is shown. TAF4 is omitted for clarity.
There are at least two distinct cAMP-responsive coactivation mechanisms
CREB-target genes can be defined as having a CRE, binding CREB, and being induced by cAMP. For such genes examined here, and which showed CREB dependence for expression, there appear to be at least one KIX-dependent and one KIX-independent mechanism. The KIX-dependent mechanism was defined by sensitivity to the KIX mutation, and rescue by TORC2 overexpression (e.g. Areg, Nr4a3). Such genes were also sensitive to the CREB Ser133 and Arg314 mutations. This fits a model having the canonical CREB interactions with CBP/p300 and TORC, and where CBP/p300 and TORC directly interact (Figure 7). The KIX-independent mechanism was typified by insensitivity to KIX insufficiency (e.g. Crem ICER), or partial gene expression in the presence of the KIX mutations (e.g. Areg, Nr4a3). There were comparable effects of FLAG-CREB-R314A and TORC-DN on gene expression and CBP/p300 and TORC2 recruitment (consistent with TORC dependence). These two mechanisms may act on the same target gene, together or in part, and may not contribute equally.
Another atypical KIX-dependent mechanism was characterized by gene expression (i.e. Hlf) that was sensitive to KIX insufficiency but not the FLAG-CREB mutants. Although present at the Hlf promoter, CREB appeared to be non-essential, although it remains possible that ATF-1 and CREMτ act uniquely or redundantly at Hlf. Moreover, a luciferase reporter having a minimal Hlf promoter fragment containing the CRE was barely induced by cAMP, indicating that endogenous gene context is important. Nor could TORC2 overexpression rescue Hlf. Thus, the effect of the KIX mutation on Hlf may be indirect, although its induction by cAMP did not require new protein synthesis (W Xu and LH Kasper, unpublished data). It is also possible that Hlf mRNA is regulated by KIX in a post-transcriptional manner. Hlf clearly requires further study to determine if it uses a novel coactivator interaction with ATF-1, CREMτ, or another protein.
There were hints of another coactivation mechanism(s) that was independent of both the KID:KIX and bZIP:TORC interactions. Evidence for this is the modest gene expression (e.g. Nr4a3) observed in the presence of FLAG-CREB S133A/R314A and the remaining Hlf expression seen in KIX-deficient MEFs. Definitive assessment of all the proposed mechanisms will require cells carrying mutations in CBP/p300 KIX and the TORC genes.
Requirement for the KIX-dependent mechanism in different tissues may vary
Our studies focused on primary MEFs, so it is possible that other cell types are more (or less) dependent on KIX. The reduced expression of some CREB-target genes in the hippocampi of CBPKIX/KIX mice during memory consolidation suggests that the KIX domain is important in neurons as well (Wood et al, 2006). In contrast, CREB-target gene expression in the liver of fasting CBPKIX/KIX mice was not affected, and instead TORC2 was more critical (Koo et al, 2005). Where CBP KIX appears not to be limiting, it is important to note that WT p300 is still present.
Do dissimilar CBP/p300-dependent transcription factors share other coactivators?
There were striking similarities between the effects of the KIX mutations on cAMP-responsive transcription, and those of a CBP/p300 CH1 domain mutation on hypoxia-responsive transcription (Kasper and Brindle, 2006; Kasper et al, 2006). Both pathways use completely unrelated transcription factors (CREB and HIF), but share CBP/p300 as key coactivators, albeit via distinct molecular interactions. In both instances, loss of the main coactivator interface with the transcription factor led to a partial overall reduction in signal-dependent gene expression, the extent of which varied between individual genes. Given that TORC is not known to have a role as a HIF coactivator, this implies that at least two families of coactivators distinct from CBP/p300 can impart (partially) redundant activities to CBP/p300-interacting transcription factors. This further suggests that if CBP and p300 could both be removed from a cell that there would be a surprising amount of transcription remaining.
Individual CREB-target genes dictate coactivation mechanism requirements
Unexpectedly, individual CREB-target genes required the different cAMP-responsive coactivation mechanisms to varying degrees. This points to a direct role for DNA sequences, including base modifications, or gene-associated proteins, in determining coactivator requirements for cAMP induction. KIX-independent mechanisms may involve transactivators besides CREB in some cases, but the targets examined here all contain CREs where CREB can be detected by ChIP (Table I) (Zhang et al, 2005). Previously defined cis-acting sites in these genes, including core promoter elements, may determine differential KIX dependence. However, the presence or absence of TATA elements, or the number of promoter proximal CREs, was not an obvious indicator of KIX dependence. Ravnskjaer et al (2007) describe how TORC is only recruited to a subset of CREB-target genes, suggesting that there is a relationship between TORC recruitment and dependence on the KIX domain. DNA binding site sequence plays a role in determining which coactivators form a productive complex with NF-κB (Leung et al, 2004), so it will be interesting to see if this occurs for CREB.
The gene most sensitive to KIX insufficiency (Areg) also had the weakest expression (albeit qRT–PCR is semi-quantitative when comparing different mRNAs). Perhaps in WT cells, weak transcription is caused by an inability to efficiently recruit transcriptional machinery, including CBP/p300, rendering these genes especially susceptible to loss of KIX function. Arguing against this, Crem ICER is highly resistant to the KIX mutations, but is the third most weakly expressed gene (Table I).
A transcription factor does not use the same coactivation mechanism at all its target genes
That genes do not universally require specific subunits of global transcriptional cofactor complexes has been observed for some TAFs (O'Brien and Tjian, 2000; Chen and Manley, 2003; Shen et al, 2003; Indra et al, 2005). In Drosophila Schneider cells, MTF-1 also requires distinct Mediator coactivator subunits to drive metal-responsive transcription of different target genes (Marr et al, 2006). These examples refer to stable multi-protein complexes, however, where target-non-essential subunits might not have any ‘choice' in being recruited to a gene. This is not the case for monomeric CBP and p300, but the analogy of recruiting a ‘coactivation tool set' still applies, whether by individual or multi-component coactivators.
It is thought that signaling pathway stimulation is insufficient to activate all target genes (Barolo and Posakony, 2002), including those for CREB (Cha-Molstad et al, 2004; Zhang et al, 2005). This has generally been attributed to activator functional insufficiency and differences in cis-regulatory sequences between target genes (Barolo and Posakony, 2002). In the context of CREB and MEFs in this study, the requirement for distinct coactivation mechanisms at different target genes is not apparent unless individual coactivator interactions are disrupted. Thus, whether a coactivator is present in a cell or at a promoter is not indicative of its role. This suggests that the CREB ‘coactivation tool set' is required to integrate gene-, tissue-, and signal-specific inputs that influence target gene expression. One prediction is that the different cAMP-responsive coactivation mechanisms will help determine the subset of CREB-target genes expressed in diverse circumstances (e.g. liver versus brain, cAMP versus calcium).
Materials and methods
Antibodies
Antiserum 6923 against mouse TORC2 aa 541–554 was generated using a peptide (CHGQQPYHRPINDFS) coupled to KLH via Cys. ChIP control antisera were either NRS Sigma) or 6923 blocked with an equal volume of 1 mg/ml TORC2-541–554 peptide. CBP (A22, C20) and p300 (N15, C20) antibodies were from Santa Cruz, and were combined for ChIP (CBP A22+C20, or p300 N15+C20); all four were used for testing CBP and p300 together. CREB antisera 244 and 253 were a gift from Marc Montminy, as was 5322 (anti-phospho-CREB/ATF-1) (Groussin et al, 2000). M2 anti-FLAG monoclonal antibody and FLAG peptide were from Sigma.
Plasmids
TORC and FLAG-CREB MSCV vectors were constructed using mouse cDNA sequences. The FLAG epitope was inserted between CREB residues 3 and 4. TORC-DN contained TORC1 aa 1–44 fused in frame to GFP in pAcGFP-N1 (Clontech) and subcloned into MSCV. 4X-CRE-Luciferase was from Stratagene. Other reporter and expression plasmids were described previously; Gal-HIF-1α (HIF-1α aa 736–836), Gal-Ets1 (Ets-1 aa 2–210), Gal-CREBΔbzip and Gal-CREB contained CREB aa 1–283 and 1–341, respectively (Yang et al, 1998; Kasper et al, 2002, 2005).
Mice and cells
CBP, p300, and Creb1 mutant mice were previously described (Blendy et al, 1996; Kasper et al, 2002, 2006; Mantamadiotis et al, 2002; Kang-Decker et al, 2004). Creb1 β-isoform hypomorphic mice were from Jackson Labs. Animal experiments were approved by the St Jude Institutional Animal Care and Use Committee and performed in accordance with IACUC guidelines. MEFs from E12.5–14.5 embryos were generated and maintained as described (Kasper et al, 2005); growth rates and morphology were comparable for WT and all mutant cells. CBP, p300, and CREB-null MEFs were generated from E14.5 embryos that were homozygous for conditional alleles of CBP, p300, or Creb1. MEFs were treated in vitro with Cre-expressing adenovirus and deletion was assessed by PCR. Loss of CREB, CBP, and p300 protein was determined by Western blot and/or immunofluorescence. For Cre-expressing adenovirus infections, MEFs were incubated overnight with virus at an MOI of 100. Experiments were performed 1 to 2 weeks after adenovirus infection, after the cells had expanded greater than 100-fold, thus diluting residual WT CBP mRNA and protein. All MEF experiments were performed with primary isolates.
Cell culture and transient assays
MEFs that express FLAG-CREB, TORC2, TORC-DN, or GFP alone were generated by transduction with MSCV retrovirus that also expresses GFP from an IRES (except for TORC-DN). Transduction efficiency (GFP+ cells) was confirmed by flow cytometry or fluorescent microscopy. For gene expression studies, MEFs were >90% GFP+ (purified by FACS if necessary). MEFs were not purified for TORC-DN ChIP (∼90% GFP+) or SeqChIP with FLAG-CREB (∼30–60% GFP+). Transient transfection assays were performed as described (Kasper et al, 2002). One to two days after transfection, MEFs were treated with ethanol or FI for 6–8 h. Test gene luciferase activity was normalized to Renilla luciferase derived from cotransfected pRL-SV40 (Promega).
ChIP and SeqChIP
MEFs were serum starved overnight and then treated for 30 min with EtOH or FI, before crosslinking for 20 min with 3% formaldehyde at room temperature. Whole-cell extract preparation, sonication, IP, and quantitative PCR were previously described (Kasper et al, 2005). SeqChIP was similar to the ChIP procedure but without a preclear step (Geisberg and Struhl, 2004); FLAG-CREB was immunoprecipitated overnight with anti-FLAG antibody in the absence of added BSA and carrier DNA, washed 4 × 5 ml with low-salt ChIP buffer, then eluted with ∼100 μg/ml FLAG peptide for several hours. The eluate was immunoprecipitated with control, CBP/p300, or TORC2 antisera, as described (Kasper et al, 2005).
qRT–PCR and microarrays
For endogenous gene expression studies, MEFs were serum-starved overnight before treatment for 90 min with 10 μM forskolin plus 100 μM IBMX, or EtOH. Trizol reagent (Invitrogen) was used for RNA extraction. qRT–PCR was performed with SYBR Green dye, as described (Kasper et al, 2005). Test gene mRNA was normalized to GAPDH or β-actin mRNA. The Hartwell Center (St Jude) generated the microarray data using Affymetrix mouse genome arrays 430A. Spotfire software was used for analysis. CREB-target gene information was from Zhang et al (2005). Analyzed probe sets were scored as ‘present' in WT MEFs treated with FI.
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Acknowledgments
We thank J Partridge for comments on the manuscript, M Paktinat for flow cytometry, M Randolph for help with mouse embryos, M Montminy and S Hedrick for the GST-TORC2 plasmid and advice, T Abel and G Schütz for the Creb1 loxp mice, and M Biesen for excellent technical assistance. We thank the following facilities at St Jude: Vector Development and Production, Flow Cytometry and Cell Sorting Shared Resource, and the Animal Resource Center. The Hartwell Center at St Jude provided oligonucleotides DNA sequencing, and performed the Affymetrix experiments. This work was supported by NIH grants CA076385 and DK058199, the Cancer Center (CORE) support grant P30 CA021765, and the American Lebanese Syrian Associated Charities (ALSAC; supporter of St Jude Children's Research Hospital).
References
- Barolo S, Posakony JW (2002) Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev 16: 1167–1181 [DOI] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M (2004) Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol 14: 2156–2161 [DOI] [PubMed] [Google Scholar]
- Bleckmann SC, Blendy JA, Rudolph D, Monaghan AP, Schmid W, Schutz G (2002) Activating transcription factor 1 and CREB are important for cell survival during early mouse development. Mol Cell Biol 22: 1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blendy JA, Kaestner KH, Schmid W, Gass P, Schutz G (1996) Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J 15: 1098–1106 [PMC free article] [PubMed] [Google Scholar]
- Brindle P, Linke S, Montminy M (1993) Protein-kinase-A-dependent activator in transcription factor CREB reveals new role for CREM repressors. Nature 364: 821–824 [DOI] [PubMed] [Google Scholar]
- Carlezon WA Jr, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28: 436–445 [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S, Goodman RH (2004) Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci USA 101: 13572–13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Manley JL (2003) Core promoter elements and TAFs contribute to the diversity of transcriptional activation in vertebrates. Mol Cell Biol 23: 7350–7362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M (2003) TORCs: transducers of regulated CREB activity. Mol Cell 12: 413–423 [DOI] [PubMed] [Google Scholar]
- De Cesare D, Sassone-Corsi P (2000) Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol 64: 343–369 [DOI] [PubMed] [Google Scholar]
- Foulkes NS, Borrelli E, Sassone-Corsi P (1991) CREM gene: use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell 64: 739–749 [DOI] [PubMed] [Google Scholar]
- Geisberg JV, Struhl K (2004) Quantitative sequential chromatin immunoprecipitation, a method for analyzing co-occupancy of proteins at genomic regions in vivo. Nucleic Acids Res 32: e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553–1577 [PubMed] [Google Scholar]
- Groussin L, Massias JF, Bertagna X, Bertherat J (2000) Loss of expression of the ubiquitous transcription factor cAMP response element-binding protein (CREB) and compensatory overexpression of the activator CREMtau in the human adrenocortical cancer cell line H295R. J Clin Endocrinol Metab 85: 345–354 [DOI] [PubMed] [Google Scholar]
- Hong EJ, West AE, Greenberg ME (2005) Transcriptional control of cognitive development. Curr Opin Neurobiol 15: 21–28 [DOI] [PubMed] [Google Scholar]
- Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G (1994) Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci USA 91: 5647–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indra AK, Mohan II WS, Frontini M, Scheer E, Messaddeq N, Metzger D, Tora L (2005) TAF10 is required for the establishment of skin barrier function in foetal, but not in adult mouse epidermis. Dev Biol 285: 28–37 [DOI] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, Labow MA (2003) Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA 100: 12147–12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U (2004) What turns CREB on? Cell Signal 16: 1211–1227 [DOI] [PubMed] [Google Scholar]
- Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, Taylor WR, Brindle PK, Van Deursen JM (2004) Loss of CBP causes T cell lymphomagenesis in synergy with p27(Kip1) insufficiency. Cancer Cell 5: 177–189 [DOI] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK (2005) Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J 24: 3846–3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, Brindle PK (2002) A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 419: 738–743 [DOI] [PubMed] [Google Scholar]
- Kasper LH, Brindle PK (2006) Mammalian gene expression program resiliency: the roles of multiple coactivator mechanisms in hypoxia-responsive transcription. Cell Cycle 5: 142–146 [DOI] [PubMed] [Google Scholar]
- Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, Murray PJ, van Deursen JM, Brindle PK (2006) Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol 26: 789–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo SH, Flechner L, Qi L, Zhang X, Screaton RA, Jeffries S, Hedrick S, Xu W, Boussouar F, Brindle P, Takemori H, Montminy M (2005) The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 437: 1109–1114 [DOI] [PubMed] [Google Scholar]
- Kung AL, Rebel VI, Bronson RT, Ch'ng LE, Sieff CA, Livingston DM, Yao TP (2000) Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev 14: 272–277 [PMC free article] [PubMed] [Google Scholar]
- Leung TH, Hoffmann A, Baltimore D (2004) One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell 118: 453–464 [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605–623 [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G (2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31: 47–54 [DOI] [PubMed] [Google Scholar]
- Marr II MT, Isogai Y, Wright KJ, Tjian R (2006) Coactivator cross-talk specifies transcriptional output. Genes Dev 20: 1458–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609 [DOI] [PubMed] [Google Scholar]
- Mengus G, Fadloun A, Kobi D, Thibault C, Perletti L, Michel I, Davidson I (2005) TAF4 inactivation in embryonic fibroblasts activates TGF beta signalling and autocrine growth. EMBO J 24: 2753–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina DN, Glasscock J, Gish W, Lovett M (2004) An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res 14: 2041–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P (1993) Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell 75: 875–886 [DOI] [PubMed] [Google Scholar]
- O'Brien T, Tjian R (2000) Different functional domains of TAFII250 modulate expression of distinct subsets of mammalian genes. Proc Natl Acad Sci USA 97: 2456–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oike Y, Takakura N, Hata A, Kaname T, Akizuki M, Yamaguchi Y, Yasue H, Araki K, Yamamura K, Suda T (1999) Mice homozygous for a truncated form of CREB-binding protein exhibit defects in hematopoiesis and vasculo-angiogenesis. Blood 93: 2771–2779 [PubMed] [Google Scholar]
- Parker D, Rivera M, Zor T, Henrion-Caude A, Radhakrishnan I, Kumar A, Shapiro LH, Wright PE, Montminy M, Brindle PK (1999) Role of secondary structure in discrimination between constitutive and inducible activators. Mol Cell Biol 19: 5601–5607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE (1997) Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91: 741–752 [DOI] [PubMed] [Google Scholar]
- Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR III, Montminy M (2007) Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. [E-pub ahead of print: 3 May 2007; doi:10.1038/sj.emboj.7601715] [DOI] [PMC free article] [PubMed]
- Rosenfeld MG, Lunyak VV, Glass CK (2006) Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev 20: 1405–1428 [DOI] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR III, Takemori H, Okamoto M, Montminy M (2004) The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119: 61–74 [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861 [DOI] [PubMed] [Google Scholar]
- Shen WC, Bhaumik SR, Causton HC, Simon I, Zhu X, Jennings EG, Wang TH, Young RA, Green MR (2003) Systematic analysis of essential yeast TAFs in genome-wide transcription and preinitiation complex assembly. EMBO J 22: 3395–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikama N, Lutz W, Kretzschmar R, Sauter N, Roth JF, Marino S, Wittwer J, Scheidweiler A, Eckner R (2003) Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J 22: 5175–5185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Heinrich R (2004) Biological control through regulated transcriptional coactivators. Cell 119: 157–167 [DOI] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T (2006) A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem 13: 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Fukuyama T, Ney PA, Wang D, Rehg J, Boyd K, van Deursen JM, Brindle PK (2006) Global transcriptional coactivators CREB-binding protein and p300 are highly essential collectively but not individually in peripheral B cells. Blood 107: 4407–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Shapiro LH, Rivera M, Kumar A, Brindle PK (1998) A role for CREB binding protein and p300 transcriptional coactivators in Ets-1 transactivation functions. Mol Cell Biol 18: 2218–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TP, Oh SP, Fuchs M, Zhou ND, Ch'ng LE, Newsome D, Bronson RT, Li E, Livingston DM, Eckner R (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93: 361–372 [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M (2005) Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA 102: 4459–4464 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Table S1