

Abstract
Bidirectional signaling is an essential feature of αIIbβ3 function. The αIIb cytoplasmic domain negatively regulates β3-mediated inside-out signaling, but little is known about the regulation of αIIb-mediated outside-in signaling. We show that αIIb-mediated outside-in signaling is enhanced in platelets of a patient lacking the terminal 39 residues of the β3 cytoplasmic tail. This enhanced signaling was detected as thromboxane A2 (TxA2) production and granule secretion, and required ligand cross-linking of αIIbβ3 and platelet aggregation. This outside-in signaling was specifically inhibited by a palmitoylated version of a β3 peptide corresponding to cytoplasmic domain residues R724-R734. Unlike the palmitoylated peptide, the nonpalmitoylated β3 peptide could not cross the platelet membrane and did not inhibit this outside-in signaling. The physiologic relevance of this β3-mediated negative regulation of αIIb outside-in signaling was demonstrated in normal platelets treated with the palmitoylated peptide and a physiologic agonist. Binding of αIIbβ3 complexes to immobilized peptides demonstrated that a peptide corresponding to β3 residues R724-R734 appears to bind to an αIIb cytoplasmic domain peptide containing residues K989-D1002, but not to control peptides. These results demonstrate that αIIb-mediated outside-in signaling resulting in TxA2 production and granule secretion is negatively regulated by a sequence of residues in the membrane distal β3 cytoplasmic domain sequence RKEFAKFEEER.
Introduction
Integrins are αβ heterodimeric receptors required for numerous essential functions in metazoan cells.1 The megakaryocyte- and platelet-specific integrin αIIbβ3 is essential for normal hemostasis.2 Platelet adhesion, aggregation, and bidirectional signaling are mediated by αIIbβ3.3 Inappropriate activation of αIIbβ3 contributes significantly to cardiovascular disease,2 the leading cause of death in the Western world. Consequently, understanding the regulation of αIIbβ3 function has enormous potential for facilitating design of antiplatelet therapeutics to reduce the risk of thrombotic events. Additionally, acquisition of this understanding has far-reaching significance for cell biology because basic concepts explaining regulation of αIIbβ3 function typically are relevant to related integrins.4-6
Many integrins, including αIIbβ3, are unable to bind their ligands or signal in their low-affinity, or resting, state.1 Transformation from the resting state to the active or high-affinity state typically results from integrin-mediated inside-out signaling initiated indirectly by activation of other receptors.1 This transformation induced by inside-out signaling (cytoplasmic interactions) may affect the equilibrium between the 2 states by trapping the receptor in the active state. The active, oligomerized receptors can initiate and propagate integrin-mediated outside-in signaling. The αIIbβ3-mediated outside-in signaling elicits thromboxane A2 (TxA2) production and adenosine triphosphate (ATP) secretion that amplifies, propagates, and thereby perpetuates the signaling initiated by physiologic agonists.7 The clinical importance of TxA2 production is evident from the efficacy of aspirin as an antithrombotic agent.8 Despite important advances in our understanding of the regulation of integrin inside-out signaling, little is known about the regulation of αIIbβ3-mediated outside-in signaling1,9 even though that signaling elicits the production of clinically important autocrines.7
The mechanisms underlying the transformation of the integrin to the high-affinity state is controversial.1,10-13 But the generally accepted model of activation is described as a switchblade-like transformation from the bent, compact, highly subunit-interacting resting state into an extended, less interacting, more open active conformation.14-17
Retention of the integrin in the bent, compact resting state apparently is controlled by interaction between the membrane proximal, highly conserved regions of the cytoplasmic domains of the α and β subunits.18-21 Disruption of this interaction by mutation results in the constitutive activation of the affected αIIbβ3 heterodimers expressed in CHO and 293T cells.19,22,23 Agonist-induced physiologic disruption of this interaction appears to be caused by the binding of talin5 or other proteins1,9 to the cytoplasmic domain of β3.
Interactions between the cytoplasmic domains of αIIbβ3 also affect αIIbβ3-mediated inside-out signaling. For example, a myristoylated peptide corresponding to the αIIb cytoplasmic domain (residues K989-E1008)13 and a palmitoylated peptide (myristoylation24 and palmitoylation25 each enable the derivatized peptides to enter the cell13,25,26) corresponding to αIIb K994-D100327 prevented or greatly diminished αIIbβ3 activation induced by inside-out αIIbβ3 signaling in response to adenosine diphosphate (ADP) and epinephrine. Presumably, binding of the myristoylated or palmitoylated αIIb peptides to the cytoplasmic domain of β3 prevented β3 inside-out signaling that would have activated αIIbβ3 under normal conditions.13,27 Thus, the interaction of membrane distal regions of the cytoplasmic domains of αIIb and β3 appear to regulate αIIbβ3-mediated inside-out signaling.
Given the regulation of β3 inside-out signaling by the cytoplasmic domains of αIIbβ3, we hypothesized that these interactions might also regulate αIIb-mediated outside-in signaling. Here we show that this hypothesis is correct and that the membrane distal region of the cytoplasmic domain of β3 negatively regulates αIIb-mediated outside-in signaling.
Patient, materials, and methods
Reagents
Apyrase, prostaglandin E1 (PGE1), the peptide RGDS, protein G, and o-phenylenediamine dihydrochloride (OPD) peroxidase substrate were from Sigma-Aldrich (St Louis, MO). Purified normal human αIIbβ3 was from Enzyme Research Laboratories (South Bend, IN). The soluble extracellular domain of human αIIbβ3 lacking the transmembrane and cytoplasmic domains was a generous gift from Dr Timothy A. Springer (Harvard Medical School, Cambridge, MA). This construct has not been described in the literature, but it was made using the same technique described for the synthesis of the soluble extracellular fragment of αVβ3.16 The complete extracellular domains of the 2 subunits were fused at each C terminus to peptides that form a disulfide-linked α-helical coiled coil.16 Donkey anti–mouse antibody was from Jackson ImmunoResearch Laboratories (West Grove, PA). The monoclonal antibodies (mAbs) PT25-2,28 AP3,29 and 7E330 were generous gifts from Dr Makoto Handa (Keio University, Tokyo, Japan), Dr Peter J. Newman (The Blood Center of Southeastern Wisconsin, Milwaukee), and Dr Barry Coller (Rockefeller University, New York, NY), respectively. D3 was prepared as previously described.31 Peptides and palmitoylated peptides were synthesized and characterized by analytical high-performance liquid chromatography and time-of-flight matrix-assisted laser desorption ionization mass spectroscopy and purified, if necessary, by the Hartwell Center for Bioinformation and Biotechnology (St Jude Children's Research Hospital, Memphis, TN). Fluorescein isothiocyanate (FITC) derivatization of peptides was performed as described,32 and the derivatized peptides were purified by using Waters Sep-Pak Vac 6cc C18 column (Waters, Milford, MA). Care was taken to keep the pH neutral during the FITC derivatization process to minimize the chance of FITC-facilitated hydrolysis of the derivatized amino-terminal residues (Edman degradation).
Patient
The patient has a variant form of Glanzmann thrombasthenia designated here as VGTΔ724.4 The patient is a young man with a life-long history of enhanced bruising, mucosal bleeding, and petechiae. He has a normal platelet count but a prolonged bleeding time. The patient contains 2 distinct mutant alleles for β3; an allele that contains a transition (CGA [R] to TGA [nonsense]) mutation that results in a truncated β3 lacking all but the 8 membrane proximal cytoplasmic domain residues. The other allele sustains a deletion of β3 T1181 resulting in a frame shift and a nonsense codon corresponding to amino acid 642. So, the abnormal platelets express only αIIbβ3 complexes containing the truncated β3 subunit, which are otherwise structurally normal. These complexes are expressed at about 40% of the normal level. As a consequence of the truncation, the platelets do not aggregate to physiologic agonists because the truncated β3 subunit cannot mediate inside-out signaling that activates the altered αIIbβ3 complex.
Washed platelet preparation and aggregation
After informed consent was obtained, blood was collected from healthy donors and the variant thrombasthenic patient (with approval from the Cincinnati Children's Hospital Medical Center Institutional Review Board) into empty syringes and then transferred to polypropylene centrifuge tubes containing 100 μl/mL Whites anticoagulant (2.94% sodium citrate, 136 mM glucose, pH 6.4) 0.1 μg/mL PGE1, and 1 U/mL apyrase. Platelet-rich plasma (PRP) was prepared by differential centrifugation. Washed platelets were prepared by differential centrifugation (1100g for 10 minutes) of the PRP containing 5 mM EDTA (ethylenediaminetetraacetic acid). Platelets were resuspended into modified Tyrode solution (12 mM NaHCO3, 138 mM NaCl, 5.5 mM glucose, 2.9 mM KCl, 2 mM MgCl2, 0.42 mM NaH2PO4, 10 mM HEPES [N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid], pH 7.4). Aggregation was measured in a lumi-aggregometer (Chrono-Log, Havertown, PA) using washed platelets (300 μL) adjusted to approximately 106 platelets/μL.
Measurement of ATP secretion
ATP secretion was measured using the CHRONO-LUME reagent (Chrono-Log) as described.7 ATP secretion was used as a surrogate measure of ADP release, so the terms are used interchangeably in the text.
Measurement of TxA2 production
TxB2, a stable metabolite of TxA2, was measured as described.7 TxB2 was measured to indirectly estimate TxA2 production, so the terms are used interchangeably in the text.
Measurement of PF4 secretion
Platelet factor 4 (PF4) was released from the α-granules by the activated platelets. After washed platelets were stimulated, supernatant fractions were collected and assayed using an Asserachrom PF4 quantitative enzyme-linked immunosorbent assay (ELISA) kit (Diagnostica Stago, Parsippany, NJ), as described.33
Laser scanning confocal microscopy
Laser scanning confocal microscopy was used to visualize FITC-labeled palmitoylated peptide and FITC-labeled nonpalmitoylated peptide-treated, washed normal platelets. For this purpose, the platelets (300 μL) in Tyrode solution (containing 5 mmol/L EDTA) were incubated with 10 μmol/L final concentration of either the FITC-labeled palmitoylated peptide RKEFAKFEEER or the FITC-labeled nonpalmitoylated peptide RKEFAKFEEER, at 37°C with stirring at 1000 rpm in a Chrono-Log aggregometer for 5 minutes. Then the platelets were centrifuged at 1100g for 5 minutes, and each pellet was suspended in 300 μL Tyrode solution. These platelets were fixed by incubation in an equal volume of fixative solution (2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer containing 2% sucrose, pH 7.3) for 10 minutes at 37°C. The fixed platelets were washed 3 times with phosphate-buffered saline (PBS), and placed on plus glass slides. Successive z-axis serial sections (optical slices) of the fixed platelets were obtained using a Nikon C1 laser scanning confocal microscope (Nikon, Tokyo, Japan) with a × 60 Plan Apo 1.2 NA water objective. Representative images corresponding to the ventral surface, the interior of platelets, and the dorsal surface were selected from a optical-sectioning series of each platelet containing 30 images. Sections were taken every 0.3 μm starting from ventral surface. The color images were transformed into black-and- white images through Photoshop (Adobe System, Mountain View, CA).
Peptide binding to αIIbβ3
The ability of a peptide corresponding to residues R724-R734 of the membrane distal cytoplasmic domain of β3 to bind to the cytoplasmic domain of αIIb was tested using a microtiter well binding assay.33 For this purpose, 200 μL of 3% bovine serum albumin (BSA) in PBS, or a 5-mmol/L solution of RKEFAKFEEER (β3, R724-R734), its scrambled control EAERKFERKFE, ARAKWDTANN (β3, A735-N744) its scrambled control NNWTAAARKD, KVGFFKRNRPPLEED (αIIb, K989-D1003) and its scrambled control FPFVGNKDKRLEREP, and LSARLAF33 or its scrambled control FRALASL33 were incubated in carbonate coating buffer (1.59 g/L Na2CO3 and 2.93g/L NaHCO3, pH 9.6) in the wells of XENOBIND Covalent Binding Microwell Plates (Xenopore, Saddle Brook, NJ) at 37°C for 2 hours to immobilize the peptides. After incubation, the wells were washed 3 times with 300 μL PBS (pH 7.5, with 0.1% Tween 20). Then the wells were postcoated with albumin by using 300 μL PBS containing 3% BSA for 2 hours at 37°C. The postcoated wells were washed 3 times with 300 μL PBS. Then, 200 μLof an 8-μg/mL solution of purified αIIbβ3 or of the extracellular aspect of αIIbβ3 (truncated form of αIIbβ3 lacking the transmembrane and cytoplasmic domains of the receptor16) were incubated with or without peptides (10 μM) in the wells at 37°C for 2 hours. After washing the wells 6 times with PBS, they were incubated for 2 hours with 200 μL of a 1-μg/mL solution of AP3 in PBS containing 3% BSA at 37°C for 2 hours. After incubation, the wells were washed 6 times with PBS, then the wells were incubated with a final concentration of 0.2 μg/mL horseradish peroxidase–conjugated donkey anti–mouse antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) in PBS containing 3% BSA at 37°C for 1 hour. After washing 6 times with PBS, the plates were developed according to the manufacturer's instructions and absorbance was read at 490 nm.
Statistical analysis
Means were compared using the Student t test.
Results
LIBS-specific antibody treatment of VGTΔ724 platelets induces TxA2 production and secretion
The hypothesis that the membrane distal region of the cytoplasmic domain of β3 negatively regulates αIIb-mediated outside-in signaling was tested using human platelets that express a truncation of the membrane distal region of the cytoplasmic domain of β3. These platelets (VGTΔ724) have normal external and transmembrane domains of αIIbβ3 but lack all of the β3 cytoplasmic domain other than the 8 membrane proximal residues because of a truncation at residue 724.4 These platelets fail to aggregate in response to physiologic agonists or spread on fibrinogen.4 Although the abnormal integrin complexes are not constitutively active and cannot be activated by αIIbβ3-mediated inside-out signaling in response to thrombin and other agonists, they can be activated and induced to bind ligand by the ligand-induced binding site (LIBS)–specific mAbs D3 and LIBS6.4 The lack of activation of αIIbβ3 in these platelets in response to physiologic agonists apparently results from the inability of the truncated β3 cytoplasmic domain to mediate the inside-out signaling required to activate αIIbβ3 in response to those agonists.4,5,18,34
The VGTΔ724 platelets that lack the 39 membrane distal β3 cytoplasmic domain residues4 were treated with the LIBS-specific mAbs D331 and PT25-228 to test our hypothesis. These antibodies cause αIIbβ3 activation but do not elicit TxA2 production or ATP secretion by normal platelets.28,31 Normal and VGTΔ724 platelets were treated independently with D3 and PT25-2 in the presence of fibrinogen with stirring. Normal and VGTΔ724 platelets aggregated in the presence of fibrinogen in response to D3 and PT25-2 (Figure 1). In contrast to normal platelets, which neither underwent shape change nor produced TxA2 or secreted the contents of their storage granules, the VGTΔ724 platelets changed shape, produced TxA2, and secreted ATP and PF4 (Figure 1). The D3-induced shape change was confirmed using scanning electron microscopy (not shown). These results support the view that the cytoplasmic domain of β3 negatively regulates αIIb-mediated outside-in signaling.
Figure 1.

LIBS-specific mAb-induced signaling by platelets lacking the β3 cytoplasmic domain. (A) Sequences of the αIIb and β3 cytoplasmic domains present in normal and VGTΔ724 platelets. (B) Aggregation traces of normal and VGTΔ724 platelets treated separately with the LIBS-specific mAbs D3 (30 μg/mL) and PT25-2 (30 μg/mL) in the presence of fibrinogen (Fg; 250 μg/mL). It is noteworthy that D3 and PT25-2 each induced shape change of VGTΔ724, but not the normal platelets. (C) Graph presentation of TxA2 production as well as ATP and PF4 secretion. The error bars represent SD, n = 3. In some cases, the values of the SD were so small that the bars cannot be seen.
D3-induced TxA2 production and secretion requires fibrinogen binding and platelet aggregation
Blockade of fibrinogen binding by the peptide RGDS35 or the anti-αIIbβ3 mAb 7E330 (both agents block fibrinogen binding) in the presence of D3 eliminated shape change, aggregation, and TxA2 production and ATP secretion (Figure 2). Also, D3 or fibrinogen alone did not cause TxA2 production or ATP secretion (not shown). These results demonstrate that the D3-induced signaling measured here requires ligand cross-linking of the receptors. This is evident because RGDS treatment of platelets enhances D3 binding,31 but unlike fibrinogen, RGDS cannot cross-link the receptors.
Figure 2.

αIIb-mediated outside-in signaling requires fibrinogen binding and aggregation. (A) Aggregation traces of VGTΔ724 platelets treated with D3 (30 μg/mL) in the presence of Fg (250 μg/mL), with or without the peptide RGDS (1 mM) or the mAb 7E3 (10 μg/mL) (RGDS and 7E3 prevent Fg binding to αIIbβ3). (B) D3 plus Fg-induced TxA2 production and ATP secretion by VGTΔ724 platelets treated with substances that inhibit Fg binding to αIIbβ3. (C) Aggregation traces of (450 μL) normal and VGTΔ724 platelets in response to D3 (30 μg/mL) plus Fg (250 μg/mL) without stirring. (D) TxA2 production by normal and VGTΔ724 platelets was measured at zero time and every 2 minutes for 8 minutes; the final concentration of secreted ATP also was measured. Data shown here were from 3 experiments. The error bars represent SD, n = 3. In some cases, the values of the SD were so small that the bars cannot be seen.
Furthermore, platelet aggregation appears to be required for the D3 plus fibrinogen-induced signaling characterized here because treatment of the VGTΔ724 platelets with D3 and fibrinogen in the absence of stirring did not elicit a substantial level of TxA2 production or ATP secretion (Figure 2). This conclusion is supported by the observation that a 2-fold dilution of platelets resulted in approximately a 4-fold decrease of TxA2 production and about a 3-fold decrease of ATP secretion (the dilution experiments were repeated 3 times), rather than the 2-fold decrease in TxA2 production and ATP secretion that should have occurred if aggregation was not required for signaling (Figure 2). However, our data do not exclude the occurrence of a low level of signaling in the VGTΔ724 platelets that is elicited by ligand cross-linking of the receptors but is enhanced exponentially by aggregation.
D3-induced signaling is not Fcγ receptor IIA–dependent
The signaling that occurred in response to D3 or PT25-2 plus fibrinogen presumably might have resulted from either αIIbβ3 outside-in signaling or Fc receptor–mediated signaling in response to clustered antibodies. Accordingly, the role of the Fc receptor (FcγRIIA) in D3 plus fibrinogen-induced signaling by VGTΔ724 platelets was evaluated by the following experiments. Normal and VGTΔ724 platelets were treated with D3 and a donkey anti–mouse IgG1κ immunoglobulin (D3 is an IgG1κ immunoglobulin) to cause Fc-dependent TxA2 production and ATP secretion.36 This treatment of the platelets was repeated in the presence of protein G, which binds to the Fc domain of IgG37 and thereby prevents Fc receptor–dependent signaling. Cross-linking of D3 with the anti-IgG1κ immunoglobulin caused shape change of and TxA2 production and ATP secretion by both normal and VGTΔ724 platelets, but these responses were eliminated by the protein G. In contrast, the signaling caused by D3 plus fibrinogen treatment of the VGTΔ724 platelets was not affected by protein G (Figure 3). These results demonstrate that the signaling induced by D3 and presumably PT25-2 was not Fc receptor (FcγRIIA) dependent because it was not blocked by protein G and therefore support the view that the LIBS-specific antibody-induced TxA2 production and ATP secretion resulted from αIIb3-mediated outside-in signaling.
Figure 3.

The signaling induced by the LIBS-specific antibodies is not Fc receptor (FcγRIIA)–dependent. (A) Aggregation traces of normal and VGTΔ724 platelets treated with D3 (30 μg/mL) plus Fg (250 μg/mL) in the presence of protein G (15 μg/mL). (B) Aggregation traces of normal and VGTΔ724 platelets treated with D3 (30 μg/mL) plus donkey anti–mouse polyclonal antibody (Ab; 30 μg/mL) with or without protein G. (C) TxA2 production (top) and ATP secretion (bottom) by normal and VGTΔ724 platelets in response to different treatments which inhibit or enhance Fc receptor–mediated signaling. Data shown here were from 3 experiments. Error bars represent SD, n = 3. In some cases, the values of the SD were so small that the bars cannot be seen.
A palmitoylated peptide containing the β3 sequence R724-R734 inhibits D3-induced signaling in VGTΔ724 platelets
The results presented support the view that the membrane distal region of the β3 cytoplasmic domain negatively regulates αIIb outside-in signaling. If the mutual interactions between the membrane distal regions of the cytoplasmic domains of αIIbβ3 regulate both αIIb and β3 outside-in signaling, a palmitoylated peptide (palmitoylation of the peptide enables it to enter the platelets25,26) corresponding to an appropriate region of the β3 cytoplasmic domain missing in the VGTΔ724 platelets might be expected to inhibit the αIIb-mediated outside-in signaling induced by D3 plus fibrinogen. Accordingly we tested 2 peptides, one containing β3 sequence RKEFAKFEEER, corresponding to residues R724-R734, the other containing the β3 sequence ARAKWDTANN, corresponding to residues A735-N744. Residues R724-R734 correspond to the N-terminal 11 amino acids of the β3 cytoplasmic domain segment missing in the VGTΔ724 platelets (Figure 4). Residues A735-N744 correspond to the C-terminal 10 amino acids contiguous with RKEFAKFEEER. The β3 segment represented by these peptides was selected as a potential regulator of αIIb signaling because it is part of the membrane distal region of the β3 cytoplasmic domain missing in VGTΔ724 platelets, and because this region does not participate in maintaining αIIbβ3 complexes in the resting configuration18-23 as is evident from the fact that the αIIbβ3 expressed on VGTΔ724 platelets is in the resting configuration.4
Figure 4.

The β3 palmitoylated peptide pRKEFAKFEEER (pR724-R734) negatively regulates αIIb-mediated outside-in signaling. (A) Sequences of the αIIb and β3 cytoplasmic domains present in normal platelets. (B) Aggregation traces of VGTΔ724 platelets treated with D3 (30 μg/mL) in the presence of Fg (250 μg/mL) with or without the β3 palmitoylated peptide p-R724-R734 (pRKEFAKFEEER, 10 μM), which corresponds to the region of β3 underlined in panel A; this sequence is part of the cytoplasmic domain missing in VGTΔ724 platelets. The peptides pEAERKFERKFE (p control; 10 μM), a scrambled, palmitoylated version of pRKEFAKFEEER, a nonpalmitoylated form of the peptide R724-R734 (RKEFAKFEEER, 10 μM), and the palmitoylated β3 peptide p-A735-N744 (pARAKWDTANN) were used as controls. In contrast to the control peptides, pRKEFAKFEEER eliminated secretion-induced shape change by D3 plus Fg-treated VGTΔ724 platelets. (C) TxA2 production (top) as well as ATP (middle) and PF4 secretion (bottom) by VGTΔ724 platelets treated with D3 (30 μg/mL) in the presence of Fg (250 μg/mL) with or without p-R724-R734 (10 μM), or 10 μM of the control peptides. There are no significant differences (P < .05) between the values of D3 plus Fg, D3 plus Fg plus pEAERKFERKFE or pARAKWDTANN-treated platelets for TxA2 production and secretion of ATP and PF4. The error bars represent SD, n = 4. In some cases, the values of the SD were so small that the bars cannot be seen.
The results of these tests support the view that a region of the membrane distal region of β3 can negatively regulate αIIb outside-in signaling. A palmitoylated derivative of the peptide R724-R734 (pRKEFAKFEEER), but not the nonpalmitoylated peptide RKEFAKFEEER, or a scrambled control peptide containing the same amino acid composition, but a different sequence (pEAERKFERKFE), or pARAKWDTANN inhibited D3 plus fibrinogen-induced shape change or TxA2 production and ATP secretion by VGTΔ724 platelets (Figure 4). These results demonstrate that a peptide corresponding to a specific region of the β3 cytoplasmic domain missing in the VGTΔ724 platelets can inhibit D3 plus fibrinogen-induced signaling in those platelets. The conclusion that the inhibition of the αIIb-mediated signaling results from pRKEFAKFEEER acting inside the platelets is supported by the results of a laser confocal microscopy study (Figure 5). In this study, visualization of platelets treated with either FITC-derivatized palmitoylated or nonpalmitoylated RKEFAKFEEER demonstrates that palmitoylation renders the FITC-conjugated peptide platelet permeable (Figure 5).
Figure 5.
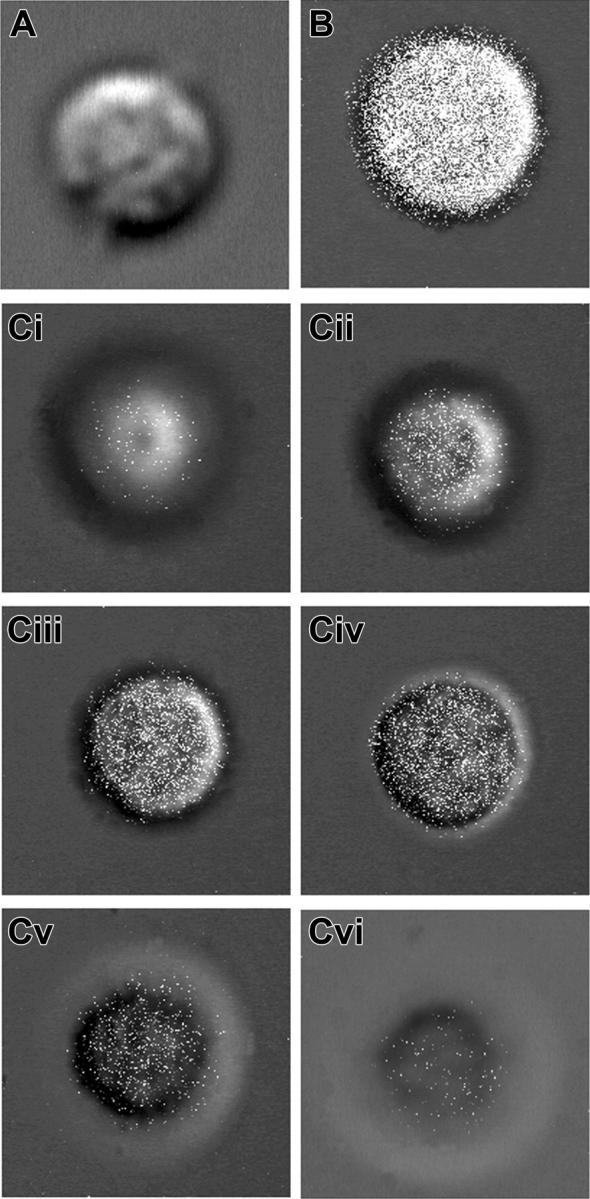
The β3 palmitoylated peptide pRKEFAKFEEER but not the nonpalmitoylated version RKEFAKFEEER is platelet permeable. (A) A confocal projection image composed of optical slice images of a single representative normal platelet treated with the FITC-derivatized, nonpalmitoylated peptide RKEFAKFEEE. Images of optical slices of the platelet treated with this peptide revealed no label (not shown). (B) A confocal projection image composed of the optical slice images of single representative normal human platelet treated with the FITC-derivatized, palmitoylated peptide pRKEFAKFEEER. (C) Six representative noncontiguous consecutive optical slice images of this platelet are labeled i-vi. (i) An image of the ventral surface; (ii-v) images of the interior of the platelets; (vi) an image of the dorsal surface of the platelet. The white points in the images represent the FITC fluorescence. These images demonstrate that palmitoylated RKEFAKFEEER, but not nonpalmitoylated RKEFAKFEEER, is platelet permeable and therefore can enter the platelets and reside in their interior.
RKEFAKFEEER binds to a cytoplasmic domain of αIIb
A hypothetical explanation for the inhibition of the αIIb outside-in signaling in the VGTΔ724 platelets demonstrated in Figure 4 is that it results from the direct interaction of the membrane distal cytoplasmic domains of β3 and αIIb. As part of a test of this hypothesis, the binding of the membrane distal cytoplasmic domain of β3 to αIIbβ3 was tested using a microtiter well binding assay.32 The β3 peptide RKEFAKFEEER or control peptides were immobilized via their amino termini to the wells of XENOBIND plates. Intact αIIbβ3 complexes or complexes of truncated αIIbβ3 corresponding to the extracellular aspect of the receptor16 were tested for the ability to bind to the immobilized peptides. Intact αIIbβ3 bound to RKEFAKFEEER, LSARLAF,33 an agonist peptide that binds to αIIbβ3 and causes outside-in signaling,38 but not to the β3 peptide ARAKWDTANN or to the scrambled control peptides EAERKFERKFE, NNWTAARKD, and FRALSAL (scrambled versions of RKEFAKFEEER, ARAKWDTANN, and LSARLAF, respectively; Figure 6). In contrast, complexes of truncated αIIbβ3 bound only to LSARLAF, not to RKEFAKFEEER. These results demonstrate that RKEFAKFEEER binds to a cytoplasmic or transmembrane domain of αIIbβ3 but not to the extracellular aspect of αIIbβ3.
Figure 6.

The β3 cytoplasmic domain peptide RKEFAKFEEER binds to the cytoplasmic domain of αIIb. The details of the methodology for the binding experiments are described in “Patient, materials, methods.” (A) Normal αIIbβ3 bound to the peptides RKEFAKFEEER and LSARLAF, but not to the β3 peptide ARAKWDTANN, or the scrambled control peptides EAERKFERKFE, NNWTAAARKD, and FRALASL. In contrast, truncated αIIbβ3 did not bind to RKEFAKFEEER; it bound only to LSARLAF. (B) Normal αIIbβ3 bound to the αIIb cytoplasmic domain peptide KVGFFKRNRPPLEED but not to the scrambled version FPFVGNKDKRLEREP. Truncated αIIbβ3 did not bind to KVGFFKRNRPPLEED or FPFVGNKDKRLEREP. The β3 peptide RKEFAKFEEER but not its scrambled control version (Control 1) inhibited the binding of αIIbβ3 to immobilized KVGFFKRNRPPLEED. Conversely, KVGFFKRNRPPLEED, but not the scrambled control version (Control 3) inhibited the binding of αIIbβ3 to RKEFAKFEEER. The simplest interpretation of these data is that RKEFAKFEEER or a sequence therein can bind to the cytoplasmic domain of αIIb. The data represent the results of 3 experiments. The error bars represent SD, n = 3. In some cases, the values of the SD were so small that the bars cannot be seen.
The binding assay was also used to demonstrate that the β3 peptide RKEFAKFEEER binds to the cytoplasmic domain of αIIb. This was done by incubating an αIIb peptide with the αIIbβ3 during the binding assay. The objective was to learn if the αIIb peptide could prevent the binding of αIIbβ3 to the β3 peptide. The αIIb peptide KVGFFKRNRPPLEED, which corresponds to αIIb cytoplasmic domain residues K989-D1003, was tested in this assay. Binding of αIIbβ3 to RKEFAKFEEER was inhibited by KVGFFKRNRPPLEED, but not by the corresponding scrambled control peptide FPFVGNKDKRLEREP. Also, αIIbβ3 bound to the immobilized αIIb peptide KVGFFKRNRPPLEED, but not to the immobilized scrambled control peptide FPFVGNKDKRLEREP (Figure 6). Truncated αIIbβ3 did not bind to the αIIb peptide. In the reciprocal experiment, the β3 peptide RKEFAKFEEER, but not the scrambled version EAERKFERKFE, inhibited the binding of αIIbβ3 to KVGFFKRNRPPLEED. Assuming the absence of allosteric effects, these results demonstrate that a membrane distal region of the cytoplasmic domain of β3, RKEFAKFEEER, or a sequence therein appears to be able to bind to the cytoplasmic domain of αIIb.
αIIbβ3 outside-in signaling induced by a physiologic agonist in normal platelets is also regulated negatively by pRKEFAKFEEER
The results demonstrate that the β3 sequence RKEFAKFEEER can negatively regulate αIIb-mediated outside-in signaling in VGTΔ724 platelets, but they do not address the issue of the relevance of that regulation to αIIbβ3 outside-in signaling initiated by a physiologic agonist in normal platelets. That issue was resolved by evaluating the ability of the palmitoylated β3 peptide pRKEFAKFEEER to inhibit αIIbβ3-mediated outside-in signaling induced in normal platelets by γ-thrombin. Washed control platelets were treated with a level of γ-thrombin that caused secretion-dependent aggregation. Under those conditions, most of the TxA2 production and ATP secretion that occurred in response to γ-thrombin was dependent on aggregation and the aggregation was dependent on aggregation-driven secretion (Figure 7). This relationship is evident from the ability of the antihuman αIIbβ3 mAb 7E3 to inhibit the aggregation of (Figure 7A), and about 80% of the TxA2 production and ATP secretion by the γ-thrombin–stimulated platelets (Figure 7B). Therefore, the majority of the TxA2 production and ATP secretion that occurred in response to this level of γ-thrombin was dependent on αIIbβ3 outside-in signaling.
Figure 7.

Palmitoylated peptide pR724-R734 (pRKEFAKFEEER) inhibited low-level γ-thrombin–induced αIIbβ3-mediated outside-in signaling and its associated TxA2 production and ATP secretion. (A) Aggregation traces of normal platelets treated with low-level γ-thrombin (5 nM) in the presence of 7E3 (10 μg/mL), the palmitoylated peptide p-R724-R734 (pRKEFAKFEEER, 10 μM) with or without Fg (250 μg/mL), a scrambled, palmitoylated control peptide (pEAERKFERKFE, 10 μM), a nonpalmitoylated version of peptide R724-R734, (RKEFAKFEEER, 10 μM), or the palmitoylated peptide pA735-N744 (pARAKWDTANN, 10 μM). In contrast to pARAKWDTANN and the control peptides, 7E3 and pRKEFAKFEEER inhibited aggregation induced by low-level γ-thrombin. Exogenous Fg restored aggregation to platelets treated with pRKEFAKFEEER. (B) TxA2 production (left) and ATP secretion (right) by normal platelets treated with γ-thrombin alone or in the presence of 7E3 (10 μg/mL), pRKEFAKFEEER (10 μM) with or without Fg, or the control peptides (10 μM). There are no significant differences (P < .05) between the values of 7E3 and pRKEFAKFEEER with or without Fg-treated platelets for TxA2 production and ATP secretion. The error bars represent SD, n = 3.
The results of this investigation were unambiguous; pRKEFAKFEEER, but not the palmitoylated scrambled control peptide pEAERKFERKFE, the nonpalmitoylated β3 peptide RKEFAKFEEER, or the palmitoylated β3 peptide pARAKWDTANN inhibited aggregation (Figure 7A), TxA2 production, or ATP secretion (Figure 7B).
As a control to demonstrate that the β3 peptide was not eliminating inside-out signaling, exogenous fibrinogen was added to the γ-thrombin, pRKEFAKFEEER-treated platelets. The expectation was that if pRKEFAKFEEER eliminated inside-out signaling, exogenous fibrinogen would not mediate aggregation. In contrast, if pRKEFAKFEEER did not eliminate the inside-out signaling, the exogenous fibrinogen should support a low level of platelet aggregation similar to that supported by D3 plus fibrinogen. Furthermore, the aggregation facilitated by the exogenous fibrinogen should not stimulate TxA2 production and ATP secretion over and above the level stimulated by γ-thrombin in the presence of the mAb 7E3 because the outside-in signaling would be inhibited by pRKEFAKFEEER. In accordance with these expectations, aggregation of γ-thrombin–stimulated platelets was not prevented by pRKEFAKFEEER in the presence of fibrinogen, but was barely discernible in the absence of fibrinogen (Figure 7A). Also, TxA2 and ATP secretion were not enhanced by aggregation mediated by exogenous fibrinogen in the presence of pRKEFAKFEEER (Figure 7B). Therefore, it is clear that αIIbβ3 was activated by treatment with a low level of γ-thrombin, but an insufficient level of fibrinogen was secreted to support easily discernible aggregation in the absence of outside-in signaling. These results demonstrate that the regulation of integrin-mediated signaling documented here is not unique to the VGTΔ724 platelets or to αIIbβ3-mediated signaling induced by D3 or PT25-2 and fibrinogen (Figure 7).
Discussion
The results presented here extend our understanding of one aspect of the regulation of αIIbβ3-mediated bidirectional signaling by the cytoplasmic domains of αII and β3. Previous work had established that inside-out signaling requires the membrane distal region of β34,5,18,34 and that the membrane distal region of αIIb negatively regulates inside-out signaling that appears to be mediated by β3.13,27 However, little had been established about the regulation of αIIb outside-in signaling.1,9 Our results provide the new insight that the cytoplasmic domain of β3 can negatively regulate αIIbβ3-mediated outside-in signaling in normal platelets stimulated with a physiologic agonist.
The data presented in Figures 1 and 2 demonstrate that treatment of VGTΔ724, but not normal platelets, with the LIBS-specific mAbs D3 or PT25-2 in the presence of fibrinogen caused shape change, TxA2 production, and ATP secretion. This difference in behavior occurred even though both types of platelets aggregated in response to the antibodies in the presence of fibrinogen. Furthermore, cross-linking of αIIbβ3 by the ligand apparently is necessary for the signaling induced by treatment with the antibodies because although the binding of the peptide RGDS to αIIbβ3 is enhanced by D3 treatment of the platelets,31 it did not cause shape change, TxA2 production, or ATP secretion by VGTΔ724 platelets. However although necessary, cross-linking by ligand apparently is not sufficient to enable D3 to elicit the signaling described here because treatment of VGTΔ724 platelets with D3 plus fibrinogen in the absence of stirring did not elicit substantial signaling. The apparent requirement for aggregation for this signaling was confirmed by a dilution experiment (Figure 2). So, both receptor cross-linking and platelet aggregation appear to be required to elicit the signaling characterized here in the VGTΔ724 platelets.
In accordance with our hypothesis that the membrane distal region of the cytoplasmic domain of β3 negatively regulates αIIb outside-in signaling, a palmitoylated peptide corresponding to residues R724-R734 of β3 inhibited D3 plus fibrinogen-induced signaling by VGTΔ724 platelets (Figure 4). The specificity of inhibition by pRKEFAKFEEER was demonstrated by the inability of pARAKWDTANN, a palmitoylated peptide corresponding to the contiguous 10 C-terminal β3 residues A735-N744, to inhibit D3 plus fibrinogen-induced signaling by VGTΔ724 platelets or to bind to αIIbβ3.
Importantly, the negative regulation of αIIb outside-in signaling demonstrated by the data presented here is not limited to signaling by VGTΔ724 platelets. The broad physiologic significance of this regulation is demonstrated by the data in Figure 7; as with VGTΔ724 platelets, pRKEFAKFEEER, but not the control peptides, inhibited αIIbβ3 outside-in signaling induced in normal platelets by γ-thrombin. So, the negative regulation of αIIb signaling by the membrane distal region of the β3 cytoplasmic domain apparently is a normal aspect of the regulation of αIIbβ3-mediated bidirectional signaling in platelets.
The conclusion that pRKEFAKFEEER is working inside the platelets rather than externally is supported by the inability of nonpalmitoylated RKEFAKFEEER to inhibit either the D3 plus fibrinogen or the γ-thrombin–induced αIIb outside-in signaling (Figures 4 and 7) and by our demonstration that FITC-labeled pRKEFAKFEEER, but not FITC-labeled nonpalmitoylated RKEFAKFEEER, could be visualized by confocal microscopy in platelets treated independently with these peptides (Figure 5).
The results of microtiter well binding assays also support the model that pRKEFAKFEEER inhibits αIIb signaling by binding to the cytoplasmic domain of αIIb rather than by interacting with the extracellular aspect of αIIbβ3. Those assays demonstrated that the β3 peptide RKEFAKFEEER apparently can bind to the cytoplasmic domain of αIIb but not the extracellular aspect of αIIbβ3 (Figure 6). The results of the binding experiment in combination with the signaling behavior of the VGTΔ724 platelets are consistent with the model that the membrane distal region of the cytoplasmic domain of β3 can regulate αIIb outside-in signaling by directly binding to the cytoplasmic aspect of αIIb.
A number of unanswered questions were raised by our observations. For example, the inability of D3 or PT25-2 plus fibrinogen-mediated aggregation to elicit the outside-in signaling characterized here in normal platelets remains unexplained. That behavior contrasts with the ability of αIIbβ3-mediated outside-in signaling in normal platelets stimulated with a low level of γ-thrombin to enhance TxA2 production and granule secretion (Figure 7). Even more perplexing is the observation that although receptor activation and aggregation are required for D3 plus fibrinogen to elicit signaling in the VGTΔ724 platelets, they are not necessarily sufficient to elicit signaling in those platelets. This is evident because treatment of those platelets with the reducing agent dithiothreitol plus fibrinogen39 resulted in platelet aggregation, but did not elicit the signaling documented here (not shown). The reason for this unexpected behavior is not known. Further work is required to resolve these issues.
In summary and conclusion, the data presented here demonstrate that a region within the membrane distal cytoplasmic domain of β3 can bind to the cytoplasmic domain of αIIb and in that manner negatively regulate αIIb-mediated outside-in signaling in platelets.
Acknowledgments
We thank Dr Makota Handa,28 Peter J. Newman,29 and Barry S. Coller30 for the generous gifts of PT25-2, AP3, and 7E3, respectively; Drs Timothy Springer, Junichi Takagi,16 and Aideen Mulligen for the truncated version of αIIbβ3; Sharon Frase of the University of Memphis Integrated Microscopy Center for her assistance with the microscopy; and Ann Becker for her expert technical assistance.
Prepublished online as Blood First Edition Paper, February 8, 2005; DOI 10.1182/blood-2004-07-2718.
Supported in part by National Institutes of Health grants HL63216 and P30CA21765 and the W. Harry Feinstone Center for Genomic Research.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110: 673-687. [DOI] [PubMed] [Google Scholar]
- 2.Lefkovits J, Plow EF, Topol EJ. Platelet glycoprotein IIb/IIIa receptors in cardiovascular medicine. N Engl J Med. 1995;332: 1553-1559. [DOI] [PubMed] [Google Scholar]
- 3.Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91: 2645-2657. [PubMed] [Google Scholar]
- 4.Wang R, Shattil SJ, Ambruso DR, Newman PJ. Truncation of the cytoplasmic domain of beta3 in a variant form of Glanzmann thrombasthenia abrogates signaling through the integrin alpha(IIb)-beta3 complex. J Clin Invest. 1997;100: 2393-2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tadokoro S, Shattil SJ, Eto K, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. 2003;302: 103-106. [DOI] [PubMed] [Google Scholar]
- 6.Hibbs ML, Xu H, Stacker SA, Springer TA. Regulation of adhesion of ICAM-1 by the cytoplasmic domain of LFA-1 integrin beta subunit. Science. 1991;251: 1611-1613. [DOI] [PubMed] [Google Scholar]
- 7.Cho MJ, Liu J, Pestina TI, et al. The roles of alpha IIb beta 3-mediated outside-in signal transduction, thromboxane A2, and adenosine diphosphate in collagen-induced platelet aggregation. Blood. 2003;101: 2646-2651. [DOI] [PubMed] [Google Scholar]
- 8.FitzGerald GA. Mechanisms of platelet activation: thromboxane A2 as an amplifying signal for other agonists. Am J Cardiol. 1991;68: 11B-15B. [DOI] [PubMed] [Google Scholar]
- 9.Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2004;2: 0726-0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hynes RO. Changing partners. Science. 2003;300: 755-756. [DOI] [PubMed] [Google Scholar]
- 11.Arnaout MA, Goodman SL, Xiong JP. Coming to grips with integrin binding to ligands. Curr Opin Cell Biol. 2002;14: 641-651. [DOI] [PubMed] [Google Scholar]
- 12.Calzada MJ, Alvarez MV, Gonzalez-Rodriguez J. Agonist-specific structural rearrangements of integrin alpha IIbbeta 3: confirmation of the bent conformation in platelets at rest and after activation. J Biol Chem. 2002;277: 39899-39908. [DOI] [PubMed] [Google Scholar]
- 13.Vinogradova O, Haas T, Plow EF, Qin J. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci U S A. 2000;97: 1450-1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong JP, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science. 2001;294: 339-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beglova N, Blacklow SC, Takagi J, Springer TA. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat Struct Biol. 2002;9: 282-287. [DOI] [PubMed] [Google Scholar]
- 16.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110: 599-611. [DOI] [PubMed] [Google Scholar]
- 17.Weisel JW, Nagaswami C, Vilaire G, Bennett JS. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J Biol Chem. 1992;267: 16637-16643. [PubMed] [Google Scholar]
- 18.O'Toole TE, Katagiri Y, Faull RJ, et al. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124: 1047-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hughes PE, Diaz-Gonzalez F, Leong L, et al. Breaking the integrin hinge: a defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271: 6571-6574. [DOI] [PubMed] [Google Scholar]
- 20.Lu C, Takagi J, Springer TA. Association of the membrane proximal regions of the alpha and beta subunit cytoplasmic domains constrains an integrin in the inactive state. J Biol Chem. 2001;276: 14642-14648. [DOI] [PubMed] [Google Scholar]
- 21.Vinogradova O, Velyvis A, Velyviene A, et al. A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell. 2002;110: 587-597. [DOI] [PubMed] [Google Scholar]
- 22.Li R, Mitra N, Gratkowski H, et al. Activation of integrin alphaIIbbeta3 by modulation of transmembrane helix associations. Science. 2003;300: 795-798. [DOI] [PubMed] [Google Scholar]
- 23.Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2: 0776-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Losonczi JA, Prestegard JH. Nuclear magnetic resonance characterization of the myristoylated, N-terminal fragment of ADP-ribosylation factor 1 in a magnetically oriented membrane array. Biochemistry. 1998;37: 706-716. [DOI] [PubMed] [Google Scholar]
- 25.Brucher KH, Garten W, Klenk HD, Shaw E, Radsak K. Inhibition of endoproteolytic cleavage of cytomegalovirus (HCMV) glycoprotein B by palmitoyl-peptidyl-chloromethyl ketone. Virology. 1990;178: 617-620. [DOI] [PubMed] [Google Scholar]
- 26.Stephens G, O'Luanaigh N, Reilly D, et al. A sequence within the cytoplasmic tail of GpIIb independently activates platelet aggregation and thromboxane synthesis. J Biol Chem. 1998;273: 20317-20322. [DOI] [PubMed] [Google Scholar]
- 27.Ginsberg MH, Yaspan B, Forsyth J, Ulmer TS, Campbell ID, Slepak M. A membrane-distal segment of the integrin alpha IIb cytoplasmic domain regulates integrin activation. J Biol Chem. 2001;276: 22514-22521. [DOI] [PubMed] [Google Scholar]
- 28.Tokuhira M, Handa M, Kamata T, et al. A novel regulatory epitope defined by a murine monoclonal antibody to the platelet GPIIb-IIIa complex (alpha IIb beta 3 integrin). Thromb Haemost. 1996;76: 1038-1046. [PubMed] [Google Scholar]
- 29.Newman PJ, Allen RW, Kahn RA, Kunicki TJ. Quantitation of membrane glycoprotein IIIa on intact human platelets using the monoclonal antibody, AP3. Blood. 1985;65: 227-232. [PubMed] [Google Scholar]
- 30.Coller BS. A new murine monoclonal antibody reports an activation-dependent change in the conformation and/or microenvironment of the platelet glycoprotein IIb/IIIa complex. J Clin Invest. 1985;76: 101-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kouns WC, Wall CD, White MM, Fox CF, Jennings LK. A conformation-dependent epitope of human platelet glycoprotein IIIa. J Biol Chem. 1990;265: 20594-205601. [PubMed] [Google Scholar]
- 32.Anderson ME, Siahaan TJ. Mechanism of binding and internalization of ICAM-1-derived cyclic peptides by LFA-1 on the surface of T cells: a potential method for targeted drug delivery. Pharm Res. 2003;20: 1523-1532. [DOI] [PubMed] [Google Scholar]
- 33.Derrick JM, Taylor DB, Loudon RG, Gartner TK. The peptide LSARLAF causes platelet secretion and aggregation by directly activating the integrin alphaIIbbeta3. Biochem J. 1997;325: 309-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen YP, Djaffar I, Pidard D, et al. Ser-752→Pro mutation in the cytoplasmic domain of integrin beta 3 subunit and defective activation of platelet integrin alpha IIb beta 3 (glycoprotein IIb-IIIa) in a variant of Glanzmann thrombasthenia. Proc Natl Acad Sci U S A. 1992;89: 10169-10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gartner TK, Bennett JS. The tetrapeptide analogue of the cell attachment site of fibronectin inhibits platelet aggregation and fibrinogen binding to activated platelets. J Biol Chem. 1985;260: 11891-11894. [PubMed] [Google Scholar]
- 36.Anderson GP, Anderson CL. Signal transduction by the platelet Fc receptor. Blood. 1990;76: 1165-1172. [PubMed] [Google Scholar]
- 37.Derrick JP, Wigley DB. The third IgG-binding domain from streptococcal protein G: an analysis by X-ray crystallography of the structure alone and in a complex with Fab. J Mol Biol. 1994;243: 906-918. [DOI] [PubMed] [Google Scholar]
- 38.Cho MJ, Liu J, Pestina TI, Steward SA, Jackson CW, Gartner TK. AlphaIIbbeta3-mediated outside-in signaling induced by the agonist peptide LSARLAF utilizes ADP and thromboxane A2 receptors to cause alpha-granule secretion by platelets. J Thromb Haemost. 2003;1: 363-373. [DOI] [PubMed] [Google Scholar]
- 39.Zucker MB, Masiello NC. Platelet aggregation caused by dithiothreitol. Thromb Haemost. 1984;51: 119-124. [PubMed] [Google Scholar]